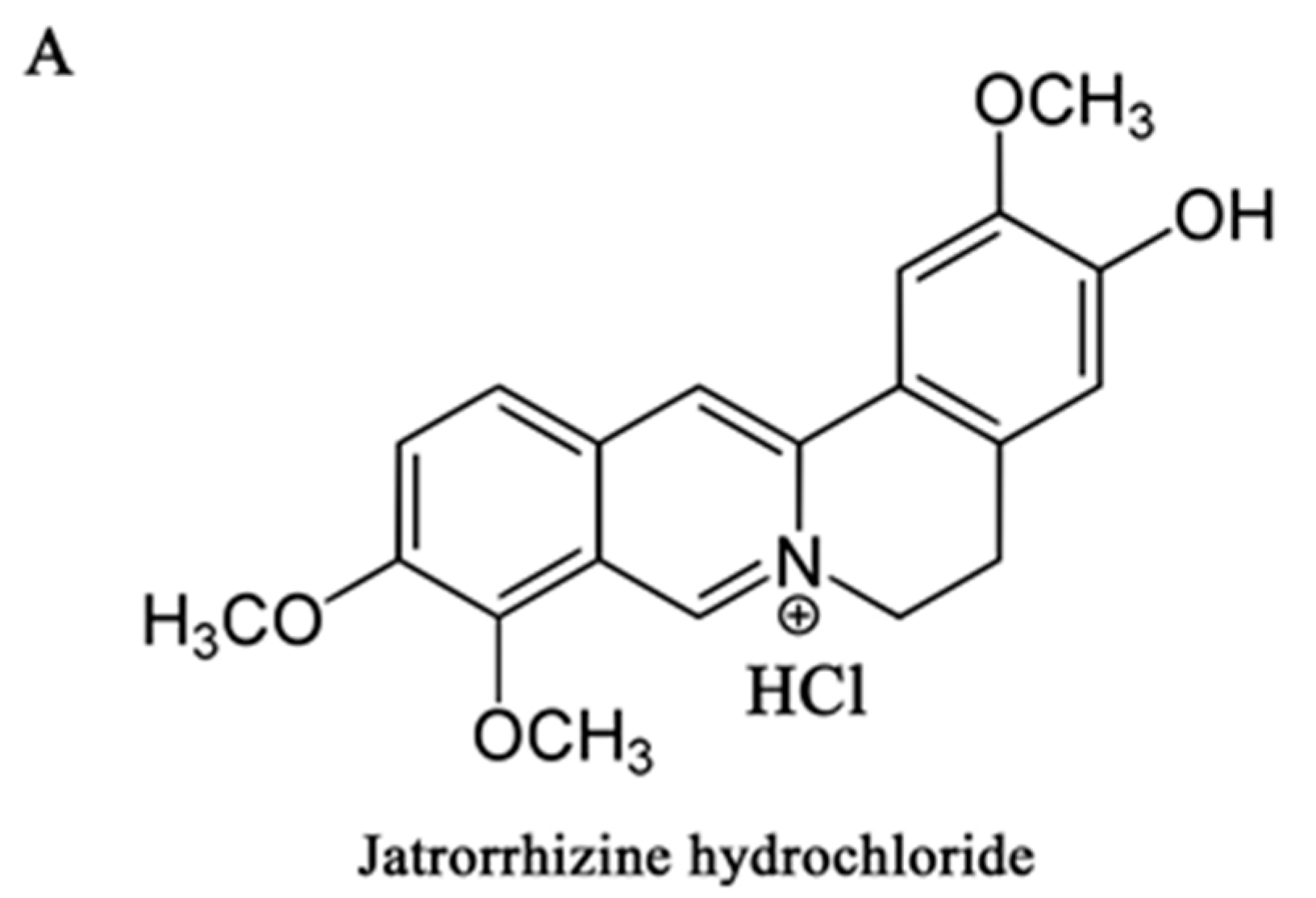
Jatrorrhizine Hydrochloride Suppresses Proliferation, Migration, and Secretion of Synoviocytes In Vitro and Ameliorates Rat Models of Rheumatoid Arthritis In Vivo

Abstract
:1. Introduction
2. Results
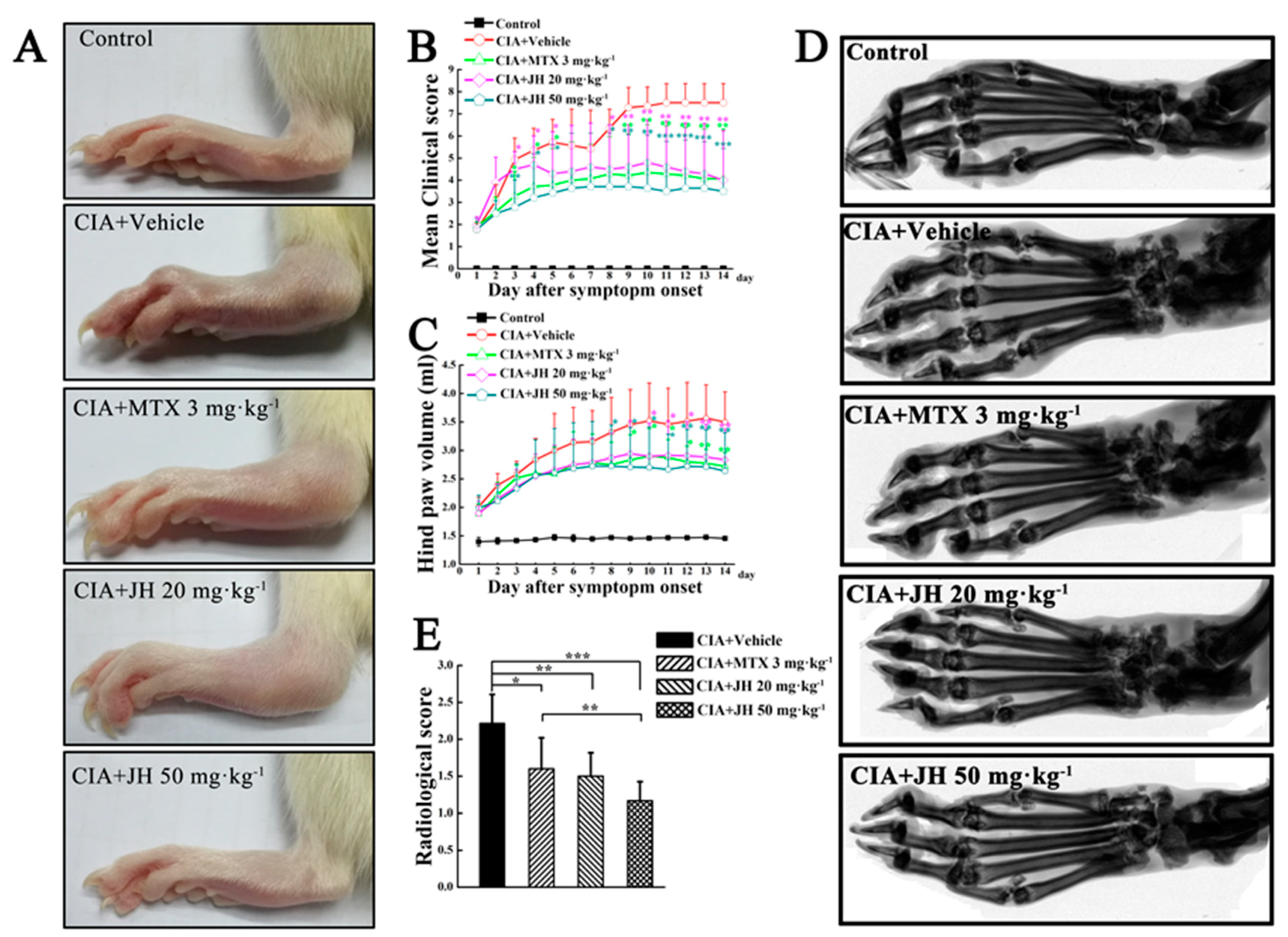
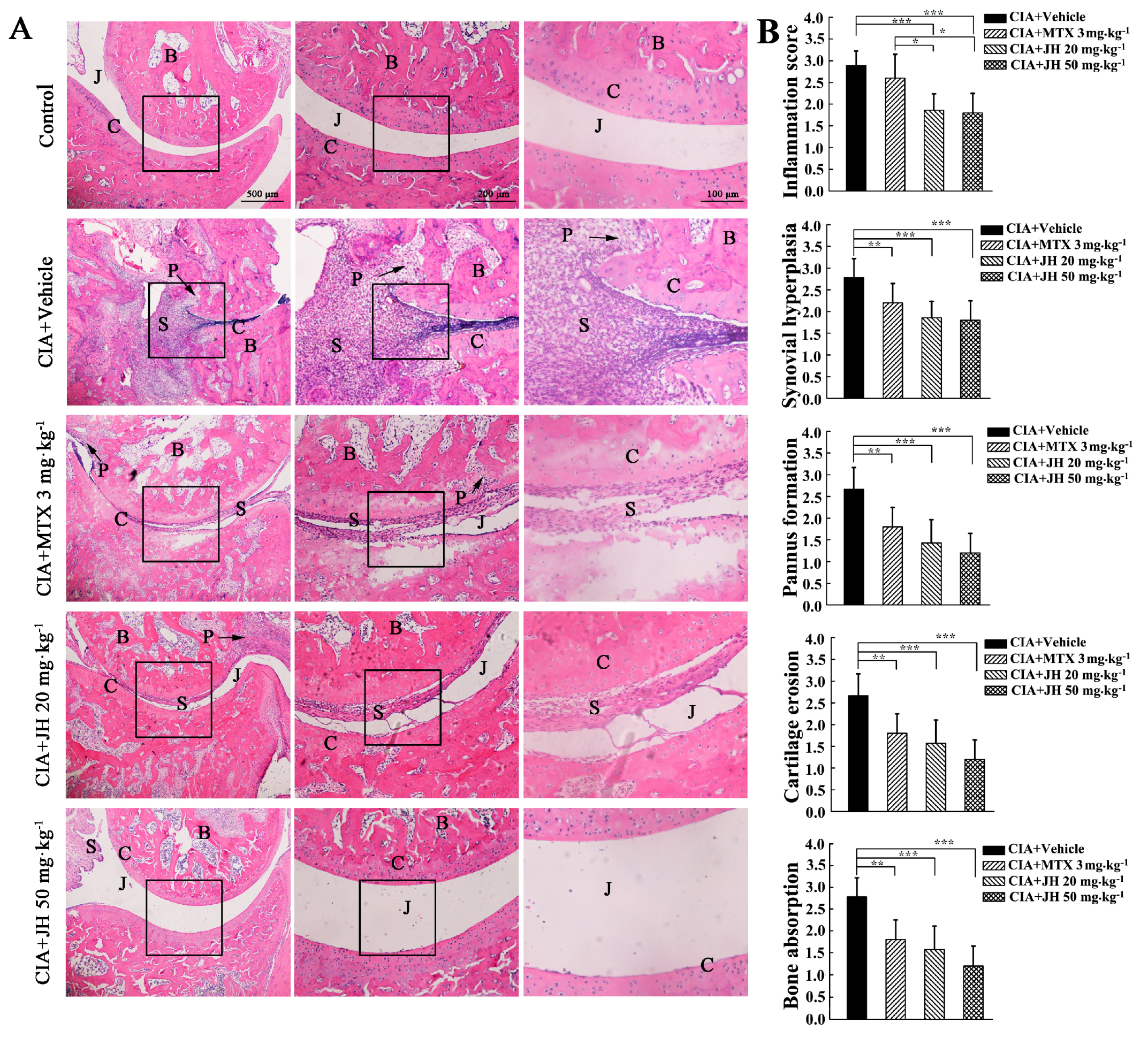
2.1. JH Treatment Suppressed Synovial Inflammation and Joint Damage
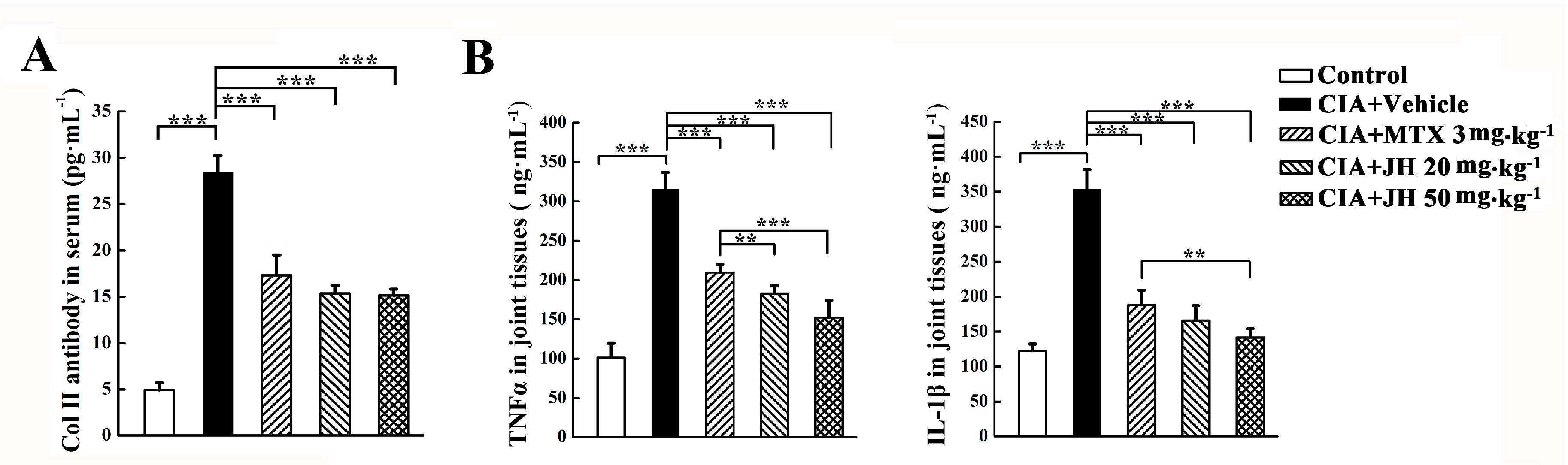
2.2. JH Treatment Significantly Suppressed the Production of Anti-CII Antibody and Proinflammatory Cytokines in CIA Rats
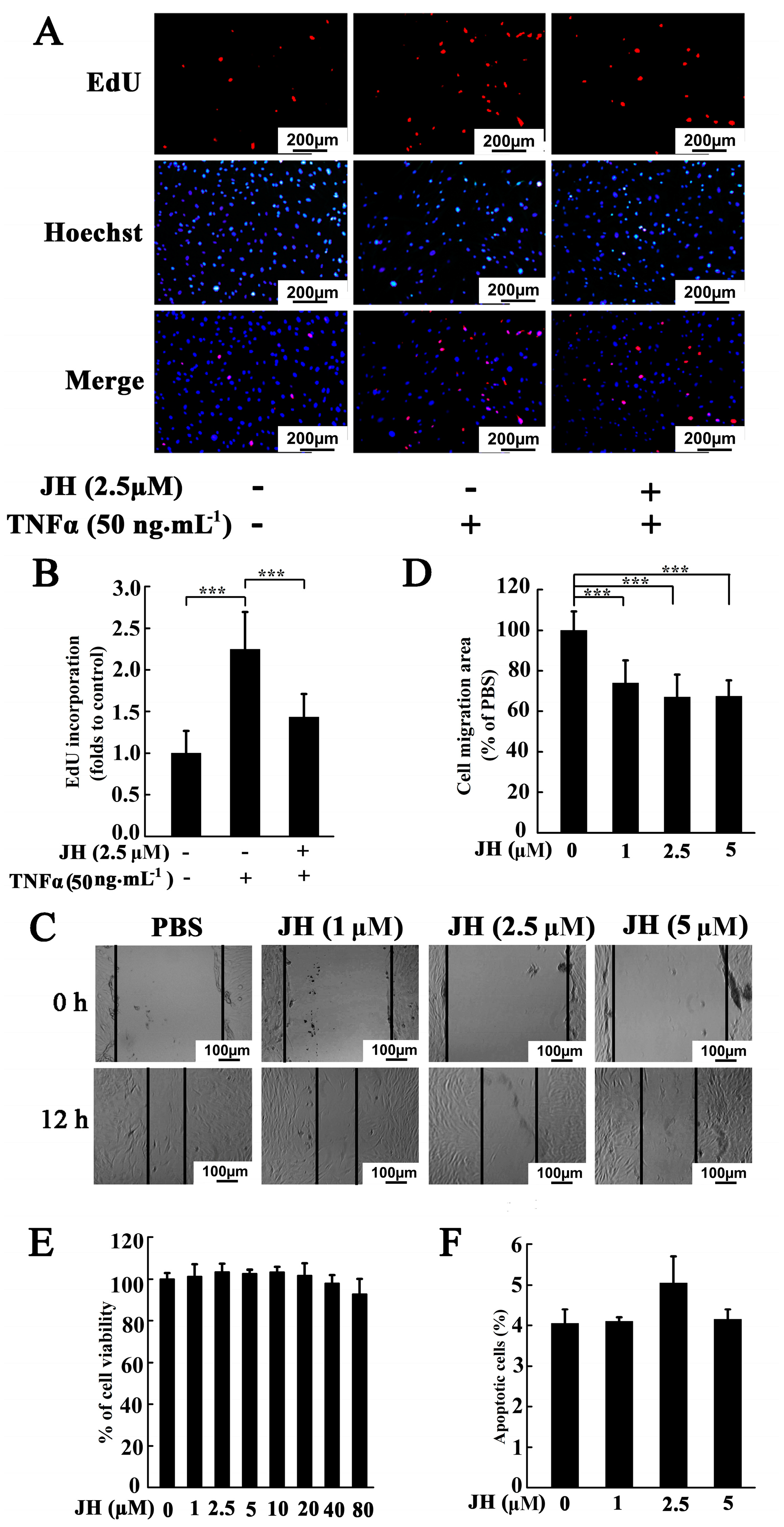
2.3. JH Decreased TNFα-Stimulated Proliferation and Migration in Cultured MH7A Cells
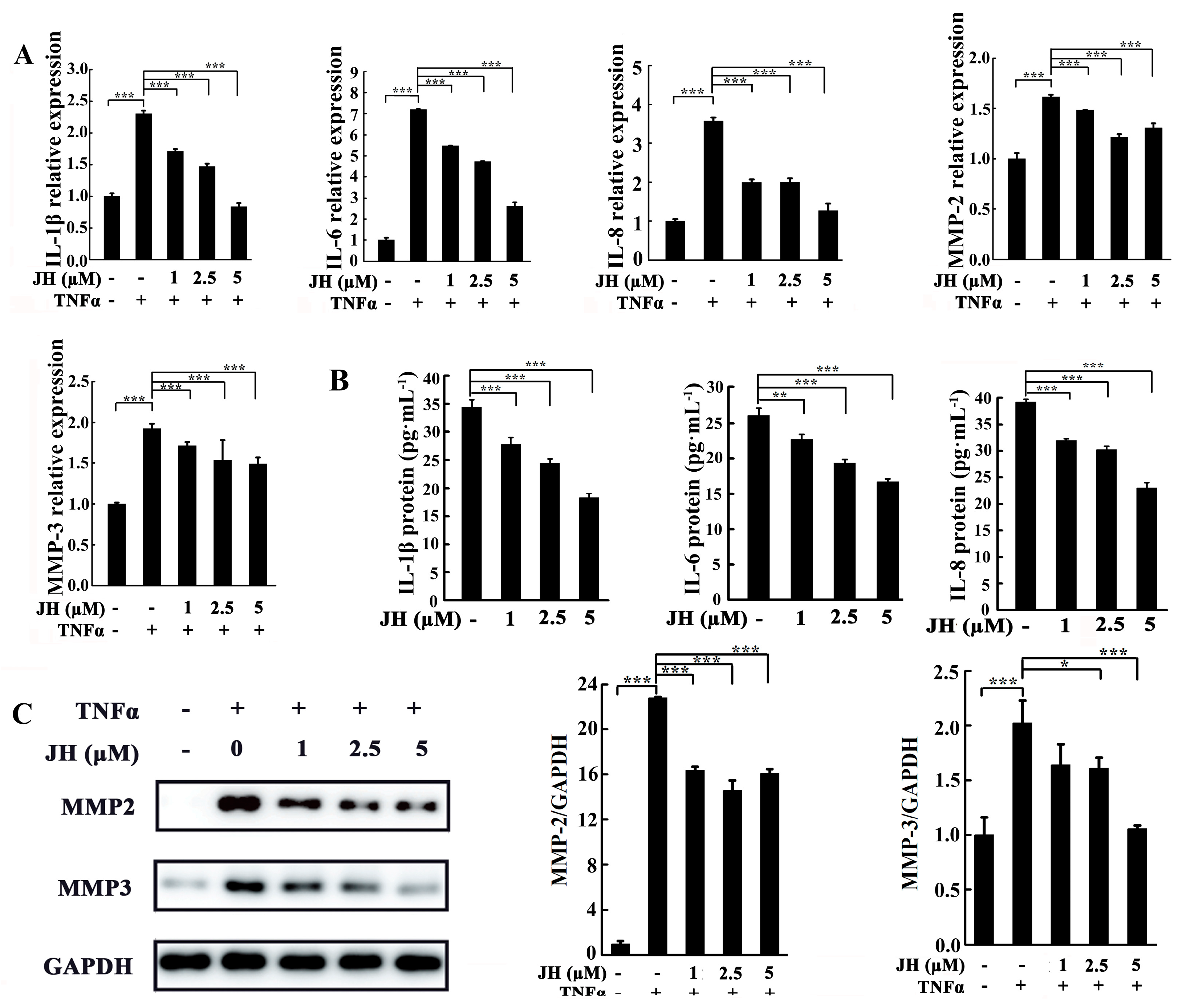
2.4. JH Treatment Inhibited TNFα-Stimulated Inflammatory Cytokine Production in Cultured MH7A Cells
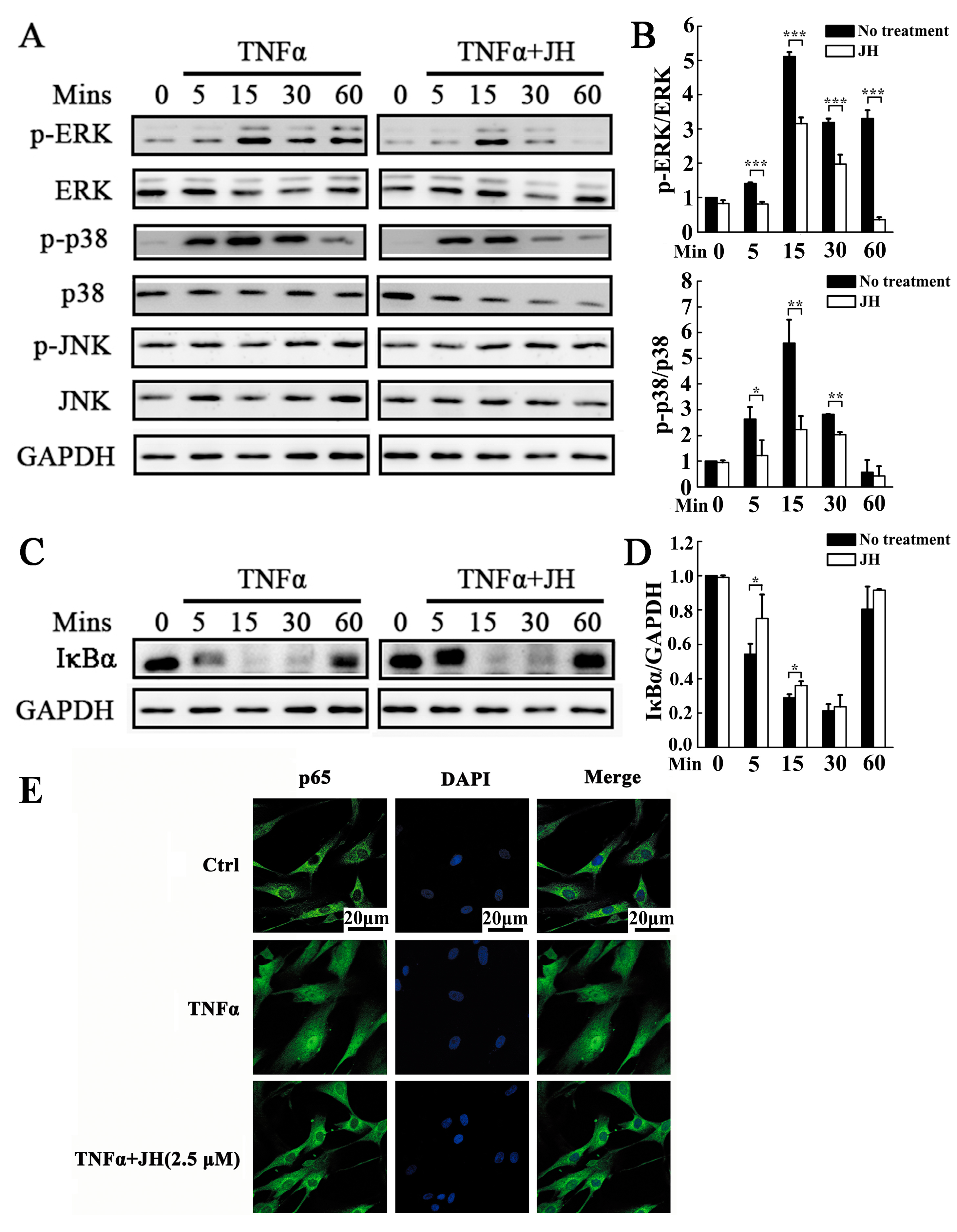
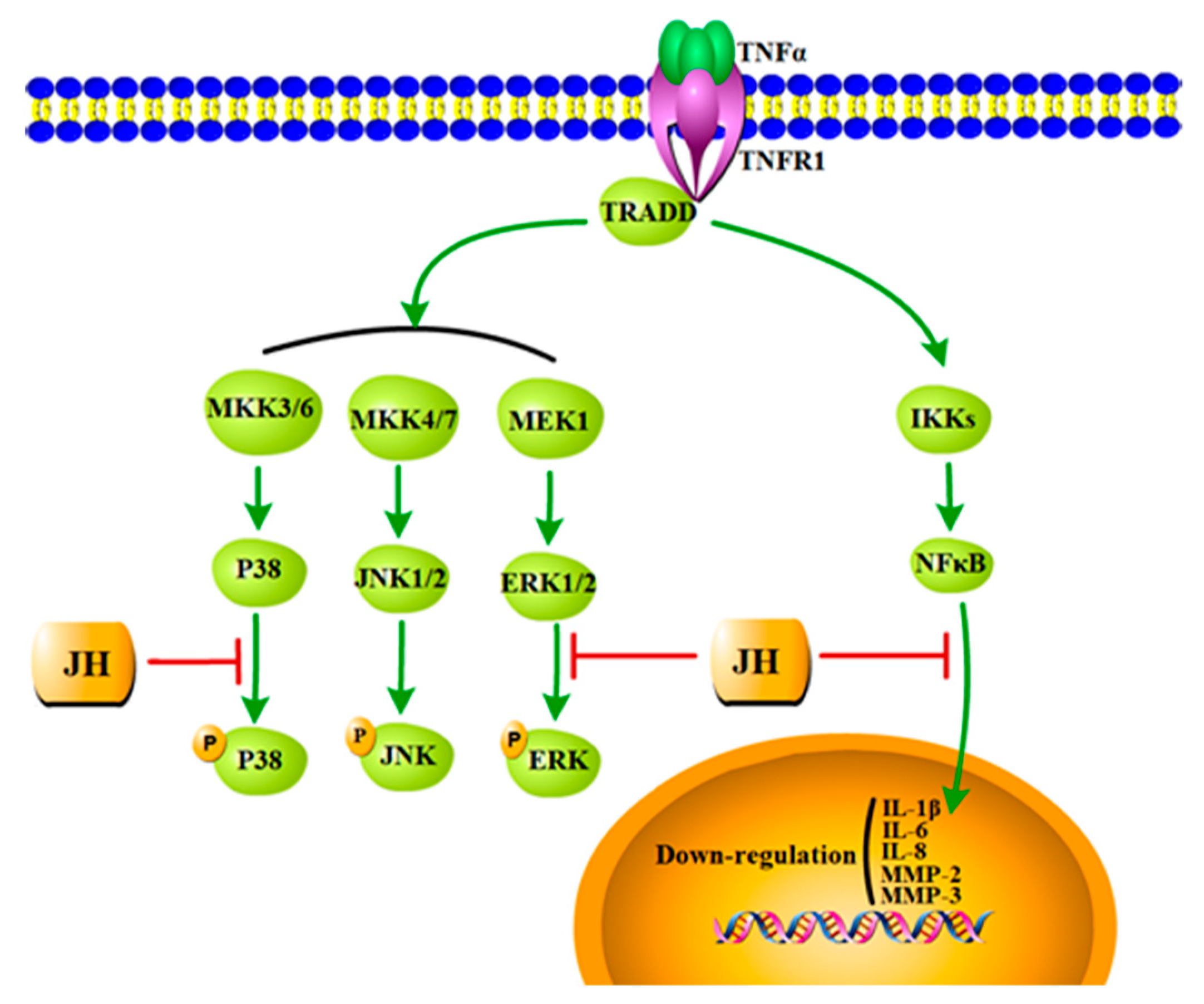
2.5. JH Treatment Inhibited TNFα-Stimulated Activations of MAPKs and NF-κB
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Reagents
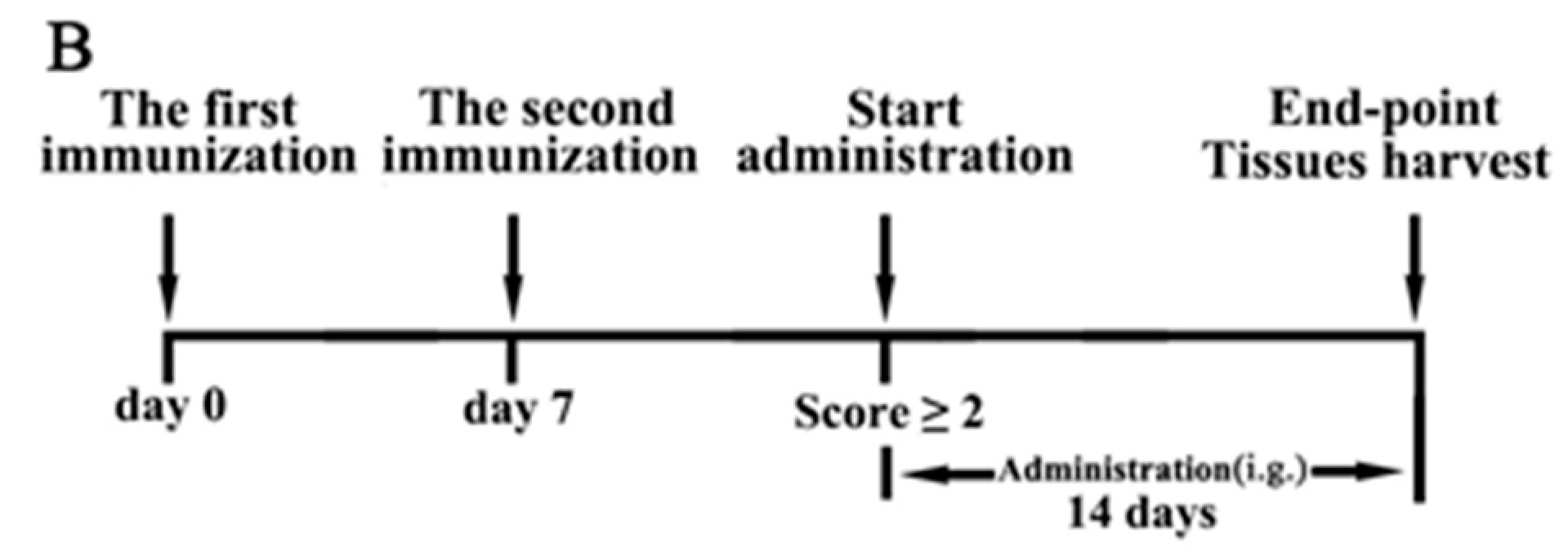
4.3. Induction of CIA and Treatment with JH
4.4. Radiographic Evaluation
4.5. Histopathological Assessment
4.6. Serum ALT and AST Measurement
4.7. Serum Anti-CII Antibody Measurement
4.8. Cytokine Levels in Rats with CIA
4.9. Cell Culture
4.10. Cell Viability Assay
4.11. TNFα-Induced Cell Proliferation
4.12. Wound Healing Assay
4.13. Analysis of Apoptosis by Flow Cytometry
4.14. Cytokine Levels in Cultured MH7A Cells
4.15. Western Blot Analysis
4.16. Immunofluorescent Staining for NF-κB Localization
4.17. Statistical Analysis
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Gravallese, E.M. Bone destruction in arthritis. Ann. Rheum. Dis. 2002, 61 (Suppl. 2), ii84–ii86. [Google Scholar] [CrossRef] [PubMed]
- Skapenko, A.; Leipe, J.; Lipsky, P.E.; Schulzekoops, H. The role of the t cell in autoimmune inflammation. Arthritis Res. Ther. 2005, 7 (Suppl. 2), S4–S14. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Pope, R.M. The role of macrophages in rheumatoid arthritis. Curr. Pharm. Des. 2005, 11, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Huber, L.C.; Distler, O.; Tarner, I.; Gay, R.E.; Gay, S.; Pap, T. Synovial fibroblasts: Key players in rheumatoid arthritis. Rheumatology 2006, 45, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Saegusa, J.; Sendo, S.; Okano, T.; Akashi, K.; Irino, Y.; Morinobu, A. Glutaminase 1 plays a key role in the cell growth of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 76. [Google Scholar] [CrossRef] [PubMed]
- Lefèvre, S.; Knedla, A.; Tennie, C.; Kampmann, A.; Wunrau, C.; Dinser, R.; Korb, A.; Schnäker, E.M.; Tarner, I.H.; Robbins, P.D. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat. Med. 2009, 15, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- Laragione, T.; Brenner, M.; Mello, A.; Symons, M.; Gulko, P.S. The arthritis severity locus cia5d is a novel genetic regulator of the invasive properties of synovial fibroblasts. Arthritis Rheum. 2008, 58, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Tolboom, T.; van der Helm-Van Mil, A.; Nelissen, R.; Breedveld, F.; Toes, R.; Huizinga, T. Invasiveness of fibroblast-like synoviocytes is an individual patient characteristic associated with the rate of joint destruction in patients with rheumatoid arthritis. Arthritis Rheum. 2005, 52, 1999–2002. [Google Scholar] [CrossRef] [PubMed]
- Péntek, M.; Poór, G.; Wiland, P.; Olejárová, M.; Brzosko, M.; Codreanu, C.; Brodszky, N.; Gulácsi, L. Biological therapy in inflammatory rheumatic diseases: Issues in central and eastern european countries. Eur. J. Health Econ. 2014, 15, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.J. New drugs for rheumatoid arthritis. Med. Lett. Drugs Ther. 2005, 351, 110–112. [Google Scholar]
- Smolen, J.S.; Beaulieu, A.; Rubbert-Roth, A.; Ramos-Remus, C.; Rovensky, J.; Alecock, E.; Woodworth, T.; Alten, R.; Investigators, O. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (option study): A double-blind, placebo-controlled, randomised trial. Lancet 2008, 371, 987–997. [Google Scholar] [CrossRef]
- Tarp, S.; Furst, D.E.; Boers, M.; Luta, G.; Bliddal, H.; Tarp, U.; Asmussen, K.H.; Brock, B.; Dossing, A.; Jørgensen, T.S. Risk of serious adverse effects of biological and targeted drugs in patients with rheumatoid arthritis: A systematic review meta-analysis. Rheumatology 2017, 56, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Varela, H.; Romero, J.; Villamañán, E.; Rueda, C.; Baumann, T.; Balsa, A.; Herrero, A. PS-008 The effect of disease-modifying antirheumatic drugs on the suspension of biological treatment due to adverse effects. Eur. J. Hosp. Pharm. 2015, 22, A145. [Google Scholar] [CrossRef]
- Slobodníková, L.; Kost'Álová, D.; Labudová, D.; Kotulová, D.; Kettmann, V. Antimicrobial activity of Mahonia aquifolium crude extract and its major isolated alkaloids. Phytother. Res. 2004, 18, 674–676. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.B.; Mishra, S. Hypoglycemic activity of alkaloidal fraction of tinospora cordifolia. Phytomed. Int. J. Phytother. Phytopharmacol. 2011, 18, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Zhang, H.; Zhang, W.W.; Huang, J.T.; Song, E.L.; Chen, S.G.; He, F.; Xu, J.; Wang, H.Q. Neuroprotective effect of jatrorrhizine on hydrogen peroxide-induced cell injury and its potential mechanisms in PC12 cells. Neurosci. Lett. 2011, 498, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Rackova, L.; Oblozinsky, M.; Kostalova, D.; Kettmann, V.; Bezakova, L. Free radical scavenging activity and lipoxygenase inhibition of Mahonia aquifolium extract and isoquinoline alkaloids. J. Inflamm. 2007, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, Z.; Yang, S.; Wang, Y.; Huang, Z.; Gao, J. Berberine ameliorates collagen-induced arthritis in rats associated with anti-inflammatory and anti-angiogenic effects. Inflammation 2014, 37, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Pietrosimone, K.M.; Jin, M.; Poston, B.; Liu, P. Collagen-induced arthritis: A model for murine autoimmune arthritis. Bio-Protocol 2015, 5, e1626. [Google Scholar] [CrossRef] [PubMed]
- Myers, L.K.; Rosloniec, E.F.; Cremer, M.A.; Kang, A.H. Collagen-induced arthritis, an animal model of autoimmunity. Life Sci. 1997, 61, 1861–1878. [Google Scholar] [CrossRef]
- Smolen, J.S.; Redlich, K.; Zwerina, J.; Aletaha, D.; Steiner, G.; Schett, G. Pro-inflammatory cytokines in rheumatoid arthritis. Clin. Rev. Allergy Immunol. 2005, 28, 239–248. [Google Scholar] [CrossRef]
- Chang, W.N. Regulation of a Cytokine Pathway in Inflammation and Arthritis. Doctoral Dissertation, The University of Melbourne, Melbourne, Australia, 2015. [Google Scholar]
- Brzustewicz, E.; Bryl, E. The role of cytokines in the pathogenesis of rheumatoid arthritis—Practical and potential application of cytokines as biomarkers and targets of personalized therapy. Cytokine 2015, 76, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Makarov, S.S. Nf-κb in rheumatoid arthritis: A pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthritis Res. 2001, 3, 200–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, J.; Liao, J.W.; Peng, W.H.; Lee, M.S.; Pao, L.H.; Cheng, H.Y. Antioxidant, analgesic, anti-inflammatory, and hepatoprotective effects of the ethanol extract of mahonia oiwakensis stem. Int. J. Mol. Sci. 2013, 14, 2928–2945. [Google Scholar] [CrossRef] [PubMed]
- Volleková, A.; Košt'Álová, D.; Kettmann, V.; Tóth, J. Antifungal activity of Mahonia aquifolium extract and its major protoberberine alkaloids. Phytother. Res. 2003, 17, 834–837. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Cao, Z.; Pan, Y.; Zhang, G.; Yang, P.; Guo, P.; Zhou, Q. Jatrorrhizine hydrochloride inhibits the proliferation and neovascularization of c8161 metastatic melanoma cells. Anti-Cancer Drugs 2013, 24, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Ng, L.T.; Hsu, F.F.; Shieh, D.E.; Chiang, L.C. Cytotoxic effects of coptis chinensis and epimedium sagittatum extracts and their major constituents (berberine, coptisine and icariin) on hepatoma and leukaemia cell growth. Clin. Exp. Pharmacol. Physiol. 2004, 31, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.U.; Ben-Rong, H.U.; Tang, Q.; Qin, F.U.; Zhang, Q.Y.; Xiang, J.Z. Effect of jatrorrhizine, berberine, huanglian decoction and compound-mimic prescription on blood glucose in mice. Chin. Tradit. Herbal Drugs 2005, 36, 548–551. [Google Scholar]
- Yang, W.; She, L.; Yu, K.; Yan, S.; Zhang, X.; Tian, X.; Ma, S.; Zhang, X. Jatrorrhizine hydrochloride attenuates hyperlipidemia in a high-fat diet-induced obesity mouse model. Mol. Med. Rep. 2016, 14, 3277–3284. [Google Scholar] [CrossRef] [PubMed]
- Mcinnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.M.; Mcinnes, I.B. Evidence that cytokines play a role in rheumatoid arthritis. J. Clin. Investig. 2008, 118, 3537–3545. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.X.; Zhu, S.L.; Zhang, B.Y.; Shi, Y.M.; Feng, X.X.; Liu, F.; Huang, J.L.; Zheng, S.G. Smoothened regulates migration of fibroblast-like synoviocytes in rheumatoid arthritisviaactivation of rho GTPase signaling. Front. Immunol. 2017, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Tao, S.S.; Zhao, M.Q.; Li, J.; Wang, X.; Pan, H.F.; Ye, D.Q. Association study of matrix metalloproteinases gene polymorphisms with susceptibility to rheumatoid arthritis: A meta-analysis. Immunol. Investig. 2015, 44, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Burrage, P.S.; Mix, K.S.; Brinckerhoff, C.E. Matrix metalloproteinases: Role in arthritis. Front. Biosci. A J. Virtual Libr. 2006, 11, 529–543. [Google Scholar] [CrossRef]
- Klein, T.; Bischoff, R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids 2011, 41, 271–290. [Google Scholar] [CrossRef] [PubMed]
- Tchetverikov, I.; Ronday, H.K.; Van, E.B.; Kiers, G.H.; Verzijl, N.; Tekoppele, J.M.; Huizinga, T.W.; Degroot, J.; Hanemaaijer, R. Mmp profile in paired serum and synovial fluid samples of patients with rheumatoid arthritis. Ann. Rheum. Dis. 2004, 63, 881–883. [Google Scholar] [CrossRef] [PubMed]
- Uemura, Y.; Hayashi, H.; Takahashi, T.; Saitho, T.; Umeda, R.; Ichise, Y.; Sendo, S.; Tsuji, G.; Kumagai, S. [MMP-3 as a biomarker of disease activity of rheumatoid arthritis]. Rinsho Byori 2015, 63, 1357–1364. [Google Scholar] [PubMed]
- Šenolt, L.; Vencovský, J.; Pavelka, K.; Ospelt, C.; Gay, S. Prospective new biological therapies for rheumatoid arthritis. Autoimmun. Rev. 2009, 9, 102–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogoyevitch, M.A.; Ngoei, K.R.W.; Zhao, T.T.; Yeap, Y.Y.C.; Ng, D.C.H. c-Jun N-terminal kinase (JNK) signaling: Recent advances and challenges. Biochim. Biophys. Acta 2010, 1804, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.Y.; Lee, S.J.; Park, S.H.; Kim, S.K. Tacrolimus (FK506) inhibits interleukin-1β-induced angiopoietin-1, Tie-2 receptor, and vascular endothelial growth factor through down-regulation of JNK and p38 pathway in human rheumatoid fibroblast-like synoviocytes. Joint Bone Spine 2011, 79, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kweon, S.; Kwon, K.B.; Park, J.; Yoon, T.; Park, B. Inhibition of IL-1beta-mediated inflammatory responses by the IkappaB alpha super-repressor in human fibroblast-like synoviocytes. Biochem. Biophys. Res. Commun. 2009, 378, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Scheinman, R. NF-κb and rheumatoid arthritis: Will understanding genetic risk lead to a therapeutic reward? For. Immunopathol. Dis. Therap. 2013, 4, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, Y.; Kong, X.; Zhu, L.; Pang, J.; Xu, Y.; Chen, W.; Zhan, H.; Lu, A.; Lin, N. Triptolide prevents bone destruction in the collagen-induced arthritis model of rheumatoid arthritis by targeting RANKL/RANK/OPG signal pathway. Evid.-Based Complement. Alternat. Med. 2013, 2013, 626038. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zhou, H.; Wong, Y.F.; Xie, Y.; Liu, Z.Q.; Jiang, Z.H.; Bian, Z.X.; Xu, H.X.; Liu, L. Suppression of the onset and progression of collagen-induced arthritis in rats by QFGJS, a preparation from an anti-arthritic Chinese herbal formula. J. Ethnopharmacol. 2007, 110, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.A.; Green, J.R.; Glatt, M.; Schlict, S.; Martin, T.J.; Gillespie, M.T.; Romas, E. Targeting osteoclasts with zoledronic acid prevents bone destruction in collagen-induced arthritis. Arthritis Rheumatol. 2004, 50, 2338–2346. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.; Huang, C.; Zhang, X.; Xin, S.; Zhou, Y.; Ma, X.; Zhang, D.; Li, Y.; Zhou, S.; Zhang, D. Methyl salicylate lactoside inhibits inflammatory response of fibroblast-like synoviocytes and joint destruction in collagen-induced arthritis in mice. Br. J. Pharmacol. 2014, 171, 3526–3538. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Control | Vehicle | MTX (3 mg∙kg−1) | JH (20 mg∙kg−1) | JH (50 mg∙kg−1) |
|---|---|---|---|---|---|
| Body weight gain (%) | 12.38 ± 2.18 | −1.05 ± 1.07 ### | 0.08 ± 0.60 | 2.63 ± 0.69 * | 3.5 ± 1.55 ** |
| Liver (g) | 8.69 ± 0.29 | 7.59 ± 0.77 | 7.67 ± 0.94 | 7.27 ± 0.59 | 7.51 ± 0.72 |
| Spleen (g) | 0.67 ± 0.055 | 0.57 ± 0.059 | 0.62 ± 0.061 | 0.63 ± 0.061 | 0.65 ± 0.98 |
| AST (U/L) | 22.47 ± 3.25 | 23.54 ± 7.77 | 24.66 ± 5.93 | 24.54 ± 3.61 | 22.41 ± 4.72 |
| ALT (U/L) | 4.56 ± 2.78 | 3.27 ± 1.87 | 2.90 ± 1.52 | 3.39 ± 2.88 | 3.24 ± 2.11 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, H.; Sun, S.; Ma, X.; Cui, C.; Chen, G.; Liu, Z.; Li, H.; Liu, M. Jatrorrhizine Hydrochloride Suppresses Proliferation, Migration, and Secretion of Synoviocytes In Vitro and Ameliorates Rat Models of Rheumatoid Arthritis In Vivo. Int. J. Mol. Sci. 2018, 19, 1514. https://doi.org/10.3390/ijms19051514
Qiu H, Sun S, Ma X, Cui C, Chen G, Liu Z, Li H, Liu M. Jatrorrhizine Hydrochloride Suppresses Proliferation, Migration, and Secretion of Synoviocytes In Vitro and Ameliorates Rat Models of Rheumatoid Arthritis In Vivo. International Journal of Molecular Sciences. 2018; 19(5):1514. https://doi.org/10.3390/ijms19051514
Chicago/Turabian StyleQiu, Haiwen, Shengnan Sun, Xuemei Ma, Congcong Cui, Gang Chen, Zhenzhou Liu, Hui Li, and Mei Liu. 2018. "Jatrorrhizine Hydrochloride Suppresses Proliferation, Migration, and Secretion of Synoviocytes In Vitro and Ameliorates Rat Models of Rheumatoid Arthritis In Vivo" International Journal of Molecular Sciences 19, no. 5: 1514. https://doi.org/10.3390/ijms19051514
APA StyleQiu, H., Sun, S., Ma, X., Cui, C., Chen, G., Liu, Z., Li, H., & Liu, M. (2018). Jatrorrhizine Hydrochloride Suppresses Proliferation, Migration, and Secretion of Synoviocytes In Vitro and Ameliorates Rat Models of Rheumatoid Arthritis In Vivo. International Journal of Molecular Sciences, 19(5), 1514. https://doi.org/10.3390/ijms19051514