Structural Basis for Mutations of Human Aquaporins Associated to Genetic Diseases


 and
and
Abstract
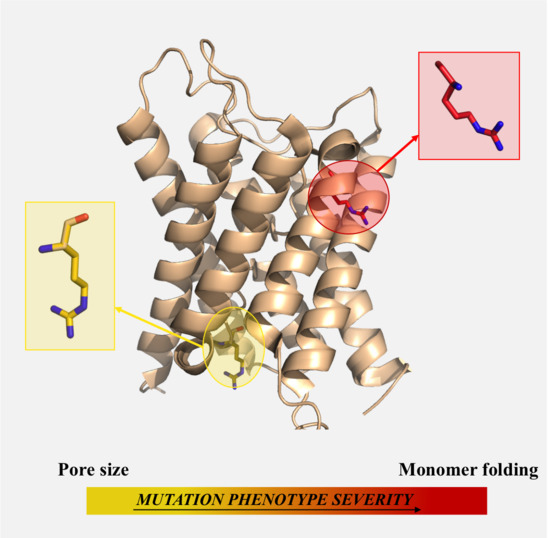
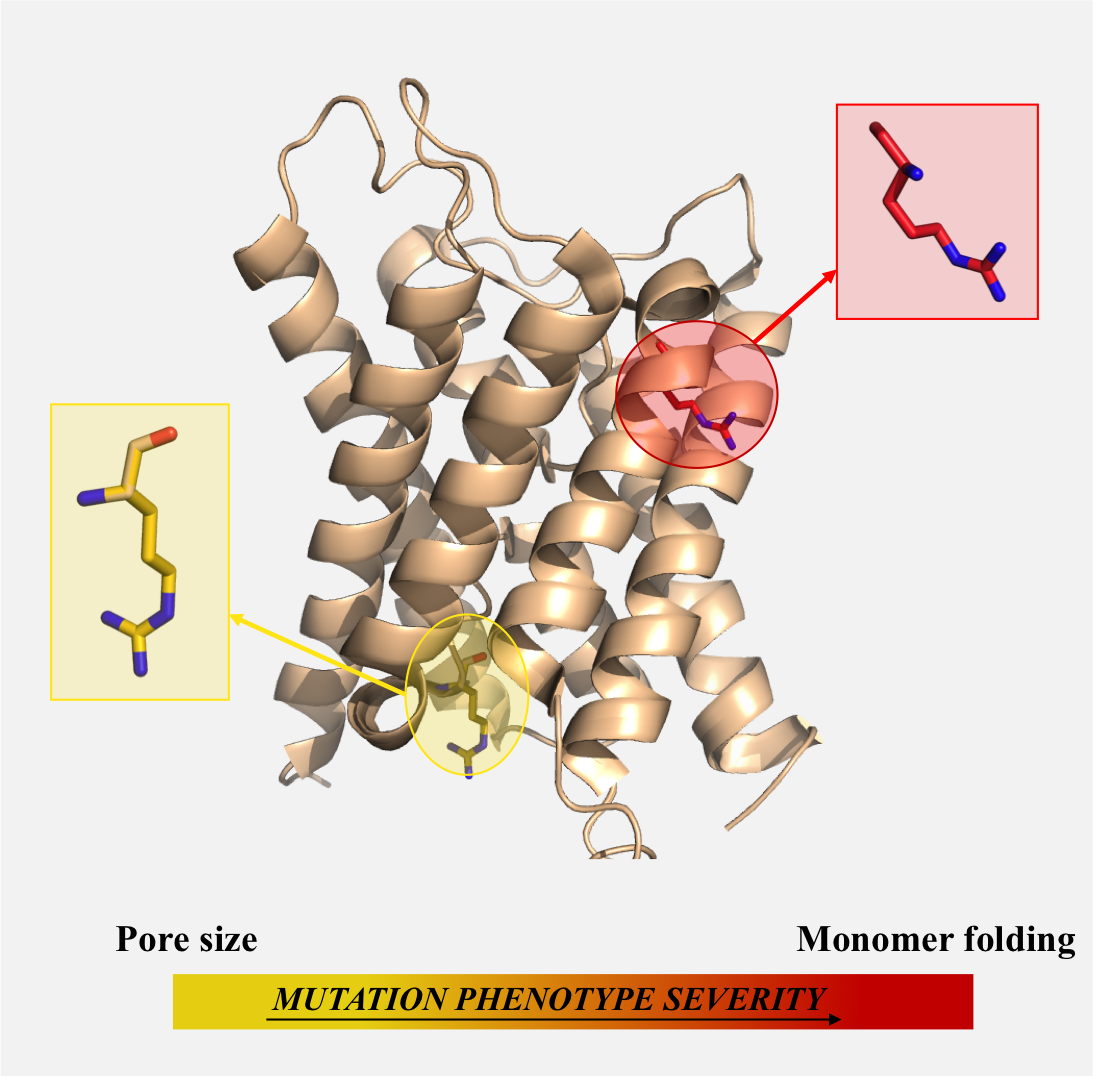
:
1. Introduction
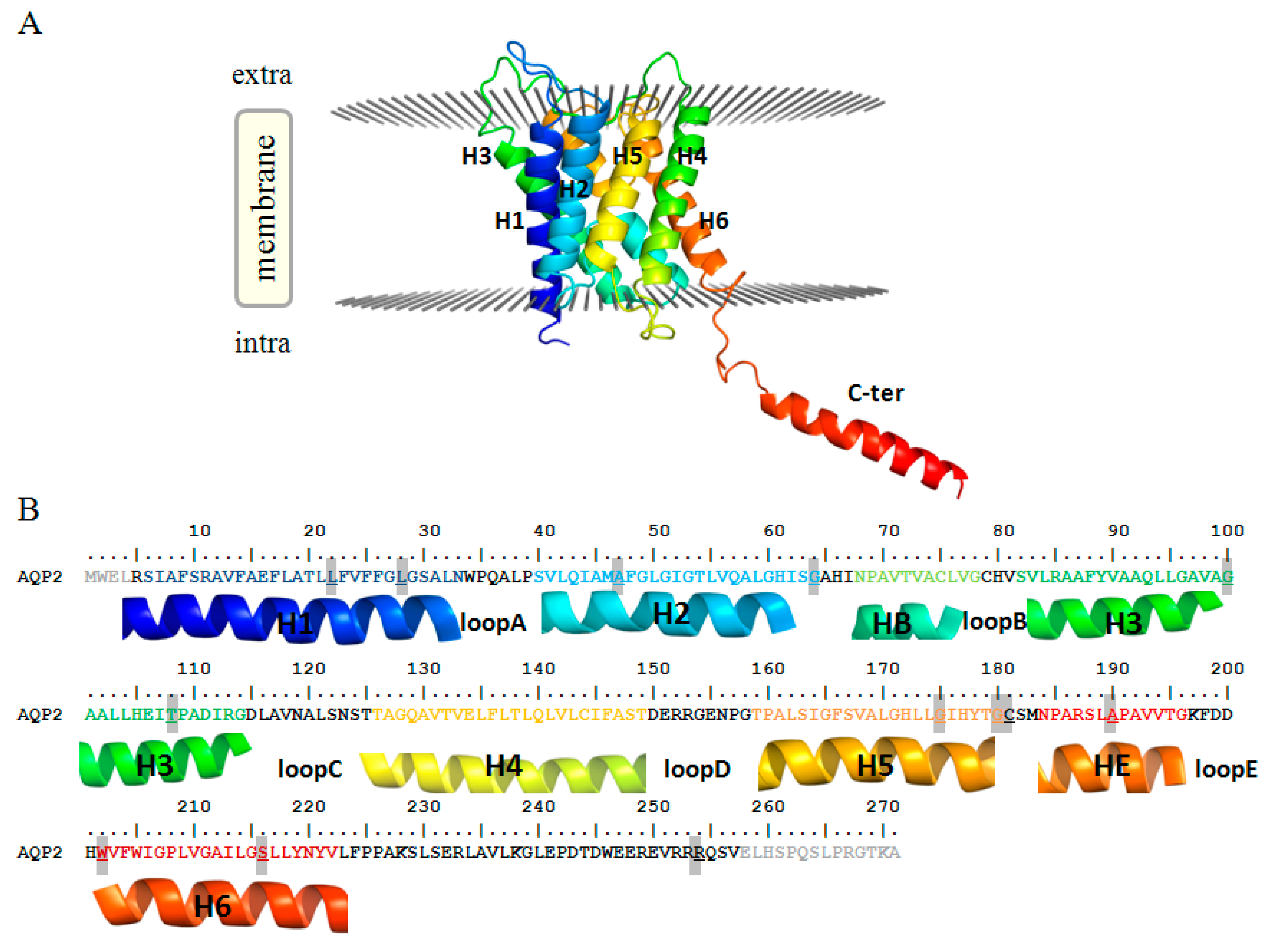
2. Results and Discussion
2.1. Mutations Affecting the Pore (AQP2-G64R, AQP2-G180S)
2.2. Mutations Affecting the Tetramer Assembly (AQP2-L22V)
2.3. Mutations Affecting the Monomer Folding (AQP2-L28P, AQP2-A47V, APQ2-G100V, AQP2-T108M, AQP2-G175R, AQP2-C181W, AQP2-A190T, AQP2-W202C, AQP2-S216P, AQP8-I229M)
2.4. Mutations Affecting the Protein Phosphorylation (AQP2-R254L, AQP2-R254Q)
2.5. Previously Described Structural Basis for Other AQP Mutants
2.5.1. AQP2 (Q57P, N68S, A70D, V71M, G100R, T125M, T126M, A147T, V168M, P185A, R187C, R187H, E258K, P262L)
2.5.2. AQP5 (A38E, I45S, N123D, I177E, R188C)
3. Materials and Methods
3.1. SAPs Collection
3.2. Model Building
3.3. Model Analysis
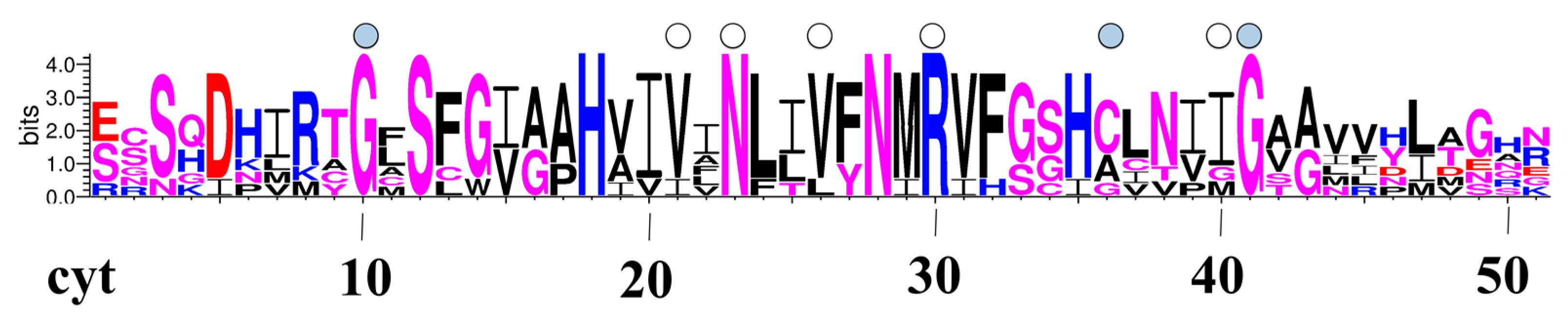
3.4. Pore-Logos
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| AQP | Aquaporin |
| CHO | Chinese Hamster Ovary |
| ER | Endoplasmic Reticulum |
| NDI | Nephrogenic Diabetes Insipidus |
| PPKB | PalmoPlantar Keratoderma of Bothnian type |
| SAP | Single Amino acid Polymorphism |
References
- Fujiyoshi, Y.; Mitsuoka, K.; de Groot, B.L.; Philippsen, A.; Grubmuller, H.; Agre, P.; Engel, A. Structure and function of water channels. Curr. Opin. Struct. Biol. 2002, 12, 509–515. [Google Scholar] [CrossRef]
- Yang, B.; Verkman, A.S. Water and glycerol permeabilities of aquaporins 1-5 and MIP determined quantitatively by expression of epitope-tagged constructs in Xenopus oocytes. J. Biol. Chem. 1997, 272, 16140–16146. [Google Scholar] [CrossRef] [PubMed]
- Gonen, T.; Walz, T. The structure of aquaporins. Q. Rev. Biophys. 2006, 39, 361–396. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Ma, S.; Shen, R.; Guo, W. Dynamic and energetic mechanisms for the distinct permeation rate in AQP1 and AQP0. Biochim. Biophys. Acta 2010, 1798, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, P.; Day, R.E.; Salman, M.M.; Conner, M.T.; Bill, R.M.; Conner, A.C. Beyond water homeostasis: Diverse functional roles of mammalian aquaporins. Biochim. Biophys. Acta 2015, 1850, 2410–2421. [Google Scholar] [CrossRef] [PubMed]
- Koyama, N.; Ishibashi, K.; Kuwahara, M.; Inase, N.; Ichioka, M.; Sasaki, S.; Marumo, F. Cloning and functional expression of human aquaporin8 cDNA and analysis of its gene. Genomics 1998, 54, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Soria, L.R.; Fanelli, E.; Altamura, N.; Svelto, M.; Marinelli, R.A.; Calamita, G. Aquaporin-8-facilitated mitochondrial ammonia transport. Biochem. Biophys. Res. Commun. 2010, 393, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Nagase, H.; Huang, C.G.; Calamita, G.; Agre, P. Purification and functional characterization of aquaporin-8. Biol. Cell 2006, 98, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Peleg, S.; Schrader, W.T.; Edwards, D.P.; McGuire, W.L.; O’Malley, B.W. Immunologic detection of a protein homologous to chicken progesterone receptor B. subunit. J. Biol. Chem. 1985, 260, 8492–8501. [Google Scholar] [PubMed]
- Morishita, Y.; Sakube, Y.; Sasaki, S.; Ishibashi, K. Molecular mechanisms and drug development in aquaporin water channel diseases: Aquaporin superfamily (superaquaporins): Expansion of aquaporins restricted to multicellular organisms. J. Pharmacol. Sci. 2004, 96, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Yakata, K.; Hiroaki, Y.; Ishibashi, K.; Sohara, E.; Sasaki, S.; Mitsuoka, K.; Fujiyoshi, Y. Aquaporin-11 containing a divergent NPA motif has normal water channel activity. Biochim. Biophys. Acta 2007, 1768, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Calvanese, L.; Pellegrini-Calace, M.; Oliva, R. In silico study of human aquaporin AQP11 and AQP12 channels. Protein Sci. 2013, 22, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Kreida, S.; Tornroth-Horsefield, S. Structural insights into aquaporin selectivity and regulation. Curr. Opin. Struct. Biol. 2015, 33, 126–134. [Google Scholar] [CrossRef] [PubMed]
- De Groot, B.L.; Grubmuller, H. Water permeation across biological membranes: Mechanism and dynamics of aquaporin-1 and GlpF. Science 2001, 294, 2353–2357. [Google Scholar] [CrossRef] [PubMed]
- Hub, J.S.; de Groot, B.L. Mechanism of selectivity in aquaporins and aquaglyceroporins. Proc. Natl. Acad. Sci. USA 2008, 105, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.V.; Tornroth-Horsefield, S. Aquaporin Protein-Protein Interactions. Int. J. Mol. Sci. 2017, 18, 2255. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S. Aquaporins at a glance. J. Cell Sci. 2011, 124, 2107–2112. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S. Aquaporins in clinical medicine. Annu. Rev. Med. 2012, 63, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Yip, Y.L.; Famiglietti, M.; Gos, A.; Duek, P.D.; David, F.P.; Gateau, A.; Bairoch, A. Annotating single amino acid polymorphisms in the UniProt/Swiss-Prot knowledgebase. Hum. Mutat. 2008, 29, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Deen, P.M.; Verdijk, M.A.; Knoers, N.V.; Wieringa, B.; Monnens, L.A.; van Os, C.H.; van Oost, B.A. Requirement of human renal water channel aquaporin-2 for vasopressin-dependent concentration of urine. Science 1994, 264, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Frick, A.; Eriksson, U.K.; de Mattia, F.; Oberg, F.; Hedfalk, K.; Neutze, R.; de Grip, W.J.; Deen, P.M.; Tornroth-Horsefield, S. X-ray structure of human aquaporin 2 and its implications for nephrogenic diabetes insipidus and trafficking. Proc. Natl. Acad. Sci. USA 2014, 111, 6305–6310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeller, H.B.; Rittig, S.; Fenton, R.A. Nephrogenic diabetes insipidus: Essential insights into the molecular background and potential therapies for treatment. Endocr. Rev. 2013, 34, 278–301. [Google Scholar] [CrossRef] [PubMed]
- Wesche, D.; Deen, P.M.; Knoers, N.V. Congenital nephrogenic diabetes insipidus: The current state of affairs. Pediatr. Nephrol. 2012, 27, 2183–2204. [Google Scholar] [CrossRef] [PubMed]
- King, L.S.; Kozono, D.; Agre, P. From structure to disease: The evolving tale of aquaporin biology. Nat. Rev. Mol. Cell Biol. 2004, 5, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Blaydon, D.C.; Lind, L.K.; Plagnol, V.; Linton, K.J.; Smith, F.J.; Wilson, N.J.; McLean, W.H.; Munro, C.S.; South, A.P.; Leigh, I.M.; et al. Mutations in AQP5, encoding a water-channel protein, cause autosomal-dominant diffuse nonepidermolytic palmoplantar keratoderma. Am. J. Hum. Genet. 2013, 93, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Kroigard, A.B.; Hetland, L.E.; Clemmensen, O.; Blaydon, D.C.; Hertz, J.M.; Bygum, A. The first Danish family reported with an AQP5 mutation presenting diffuse non-epidermolytic palmoplantar keratoderma of Bothnian type, hyperhidrosis and frequent Corynebacterium infections: A case report. BMC Dermatol. 2016, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, K.; Kuwahara, M.; Kageyama, Y.; Tohsaka, A.; Marumo, F.; Sasaki, S. Cloning and functional expression of a second new aquaporin abundantly expressed in testis. Biochem. Biophys. Res. Commun. 1997, 237, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Yamamoto, T.; Kondo, D.; Funaki, H.; Yaoita, E.; Kawasaki, K.; Sato, N.; Hatakeyama, K.; Kihara, I. Molecular cloning of a new aquaporin from rat pancreas and liver. J. Biol. Chem. 1997, 272, 30329–30333. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Yang, B.; Verkman, A.S. Cloning of a novel water and urea-permeable aquaporin from mouse expressed strongly in colon, placenta, liver, and heart. Biochem. Biophys. Res. Commun. 1997, 240, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Calamita, G.; Mazzone, A.; Bizzoca, A.; Svelto, M. Possible involvement of aquaporin-7 and -8 in rat testis development and spermatogenesis. Biochem. Biophys. Res. Commun. 2001, 288, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Day, R.E.; Kitchen, P.; Owen, D.S.; Bland, C.; Marshall, L.; Conner, A.C.; Bill, R.M.; Conner, M.T. Human aquaporins: Regulators of transcellular water flow. Biochim. Biophys. Acta 2014, 1840, 1492–1506. [Google Scholar] [CrossRef] [PubMed]
- Wellner, R.B.; Redman, R.S.; Swaim, W.D.; Baum, B.J. Further evidence for AQP8 expression in the myoepithelium of rat submandibular and parotid glands. Pflugers Arch. 2006, 451, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Stenling, R.; Rubio, C.; Lindblom, A. Differential expression of aquaporin 8 in human colonic epithelial cells and colorectal tumors. BMC Physiol. 2001, 1, 1. [Google Scholar] [CrossRef]
- Murata, K.; Mitsuoka, K.; Hirai, T.; Walz, T.; Agre, P.; Heymann, J.B.; Engel, A.; Fujiyoshi, Y. Structural determinants of water permeation through aquaporin-1. Nature 2000, 407, 599–605. [Google Scholar] [CrossRef] [PubMed]
- De Groot, B.L.; Engel, A.; Grubmuller, H. A refined structure of human aquaporin-1. FEBS Lett. 2001, 504, 206–211. [Google Scholar] [CrossRef]
- Ren, G.; Reddy, V.S.; Cheng, A.; Melnyk, P.; Mitra, A.K. Visualization of a water-selective pore by electron crystallography in vitreous ice. Proc. Natl. Acad. Sci. USA 2001, 98, 1398–1403. [Google Scholar] [CrossRef] [PubMed]
- Ruiz Carrillo, D.; To Yiu Ying, J.; Darwis, D.; Soon, C.H.; Cornvik, T.; Torres, J.; Lescar, J. Crystallization and preliminary crystallographic analysis of human aquaporin 1 at a resolution of 3.28 angstrom. Acta Cryst. F Struct. Biol. Commun. 2014, 70, 1657–1663. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.D.; Yeh, R.; Sandstrom, A.; Chorny, I.; Harries, W.E.; Robbins, R.A.; Miercke, L.J.; Stroud, R.M. Crystal structure of human aquaporin 4 at 1.8 A and its mechanism of conductance. Proc. Natl. Acad. Sci. USA 2009, 106, 7437–7442. [Google Scholar] [CrossRef] [PubMed]
- Horsefield, R.; Norden, K.; Fellert, M.; Backmark, A.; Tornroth-Horsefield, S.; Terwisscha van Scheltinga, A.C.; Kvassman, J.; Kjellbom, P.; Johanson, U.; Neutze, R. High-resolution X-ray structure of human aquaporin 5. Proc. Natl. Acad. Sci. USA 2008, 105, 13327–13332. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, P.; Oberg, F.; Sjohamn, J.; Hedfalk, K.; Bill, R.M.; Conner, A.C.; Conner, M.T.; Tornroth-Horsefield, S. Plasma Membrane Abundance of Human Aquaporin 5 Is Dynamically Regulated by Multiple Pathways. PLoS ONE 2015, 10, e0143027. [Google Scholar] [CrossRef] [PubMed]
- De Mattia, F.; Savelkoul, P.J.; Kamsteeg, E.J.; Konings, I.B.; van der Sluijs, P.; Mallmann, R.; Oksche, A.; Deen, P.M. Lack of arginine vasopressin-induced phosphorylation of aquaporin-2 mutant AQP2-R254L explains dominant nephrogenic diabetes insipidus. J. Am. Soc. Nephrol. 2005, 16, 2872–2880. [Google Scholar] [CrossRef] [PubMed]
- Savelkoul, P.J.; De Mattia, F.; Li, Y.; Kamsteeg, E.J.; Konings, I.B.; van der Sluijs, P.; Deen, P.M. p.R254Q mutation in the aquaporin-2 water channel causing dominant nephrogenic diabetes insipidus is due to a lack of arginine vasopressin-induced phosphorylation. Hum. Mutat. 2009, 30, E891–E903. [Google Scholar] [CrossRef] [PubMed]
- Kamsteeg, E.J.; Savelkoul, P.J.; Hendriks, G.; Konings, I.B.; Nivillac, N.M.; Lagendijk, A.K.; van der Sluijs, P.; Deen, P.M. Missorting of the Aquaporin-2 mutant E258K to multivesicular bodies/lysosomes in dominant NDI is associated with its monoubiquitination and increased phosphorylation by PKC but is due to the loss of E258. Pflugers Arch. Eur. J. Physiol. 2008, 455, 1041–1054. [Google Scholar] [CrossRef] [PubMed]
- Trimpert, C.; van den Berg, D.T.; Fenton, R.A.; Klussmann, E.; Deen, P.M. Vasopressin increases S261 phosphorylation in AQP2-P262L, a mutant in recessive nephrogenic diabetes insipidus. Nephrol. Dial. Transplant. 2012, 27, 4389–4397. [Google Scholar] [CrossRef] [PubMed]
- Calvanese, L.; Pellegrini-Calace, M.; Oliva, R. Mutations at key pore-lining positions differentiate the water permeability of fish lens aquaporin from other vertebrates. Febs Lett. 2010, 584, 4797–4801. [Google Scholar] [CrossRef] [PubMed]
- Oliva, R.; Calamita, G.; Thornton, J.M.; Pellegrini-Calace, M. Electrostatics of aquaporin and aquaglyceroporin channels correlates with their transport selectivity. Proc. Natl. Acad. Sci. USA 2010, 107, 4135–4140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, R.; Thornton, J.M.; Pellegrini-Calace, M. PoreLogo: A new tool to analyse, visualize and compare channels in transmembrane proteins. Bioinformatics 2009, 25, 3183–3184. [Google Scholar] [CrossRef] [PubMed]
- Marr, N.; Kamsteeg, E.J.; van Raak, M.; van Os, C.H.; Deen, P.M. Functionality of aquaporin-2 missense mutants in recessive nephrogenic diabetes insipidus. Pflugers Arch. Eur. J. Physiol. 2001, 442, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Smart, O.S.; Neduvelil, J.G.; Wang, X.; Wallace, B.A.; Sansom, M.S. HOLE: A program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 1996, 14, 354–360, 376. [Google Scholar] [CrossRef]
- Canfield, M.C.; Tamarappoo, B.K.; Moses, A.M.; Verkman, A.S.; Holtzman, E.J. Identification and characterization of aquaporin-2 water channel mutations causing nephrogenic diabetes insipidus with partial vasopressin response. Hum. Mol. Genet. 1997, 6, 1865–1871. [Google Scholar] [CrossRef] [PubMed]
- Tamarappoo, B.K.; Verkman, A.S. Defective aquaporin-2 trafficking in nephrogenic diabetes insipidus and correction by chemical chaperones. J. Clin. Invest. 1998, 101, 2257–2267. [Google Scholar] [CrossRef] [PubMed]
- Marr, N.; Bichet, D.G.; Hoefs, S.; Savelkoul, P.J.; Konings, I.B.; De Mattia, F.; Graat, M.P.; Arthus, M.F.; Lonergan, M.; Fujiwara, T.M.; et al. Cell-biologic and functional analyses of five new Aquaporin-2 missense mutations that cause recessive nephrogenic diabetes insipidus. J. Am. Soc. Nephrol. 2002, 13, 2267–2277. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Fushimi, K.; Sasaki, S.; Marumo, F. Structure of aquaporin-2 vasopressin water channel. J. Biol. Chem. 1996, 271, 5171–5176. [Google Scholar] [PubMed]
- Preston, G.M.; Jung, J.S.; Guggino, W.B.; Agre, P. The mercury-sensitive residue at cysteine 189 in the CHIP28 water channel. J. Biol. Chem. 1993, 268, 17–20. [Google Scholar] [PubMed]
- Goji, K.; Kuwahara, M.; Gu, Y.; Matsuo, M.; Marumo, F.; Sasaki, S. Novel mutations in aquaporin-2 gene in female siblings with nephrogenic diabetes insipidus: Evidence of disrupted water channel function. J. Clin. Endocrinol. Metabol. 1998, 83, 3205–3209. [Google Scholar] [CrossRef]
- De Mattia, F.; Savelkoul, P.J.; Bichet, D.G.; Kamsteeg, E.J.; Konings, I.B.; Marr, N.; Arthus, M.F.; Lonergan, M.; van Os, C.H.; van der Sluijs, P.; et al. A novel mechanism in recessive nephrogenic diabetes insipidus: Wild-type aquaporin-2 rescues the apical membrane expression of intracellularly retained AQP2-P262L. Hum. Mol. Genet. 2004, 13, 3045–3056. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.H.; Bichet, D.G.; Sasaki, S.; Kuwahara, M.; Arthus, M.F.; Lonergan, M.; Lin, Y.F. Two novel aquaporin-2 mutations responsible for congenital nephrogenic diabetes insipidus in Chinese families. J. Clin. Endocrinol. Metabol. 2002, 87, 2694–2700. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, M. Aquaporin-2, a vasopressin-sensitive water channel, and nephrogenic diabetes insipidus. Intern. Med. 1998, 37, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Shinbo, I.; Fushimi, K.; Kasahara, M.; Yamauchi, K.; Sasaki, S.; Marumo, F. Functional analysis of aquaporin-2 mutants associated with nephrogenic diabetes insipidus by yeast expression. Am. J. Physiol. 1999, 277, F734–F741. [Google Scholar] [CrossRef] [PubMed]
- Buck, T.M.; Eledge, J.; Skach, W.R. Evidence for stabilization of aquaporin-2 folding mutants by N-linked glycosylation in endoplasmic reticulum. Am. J. Physiol. Cell Physiol. 2004, 287, C1292–C1299. [Google Scholar] [CrossRef] [PubMed]
- Klein, N.; Neumann, J.; O’Neil, J.D.; Schneider, D. Folding and stability of the aquaglyceroporin GlpF: Implications for human aqua(glycero)porin diseases. Biochim. Biophys. Acta 2015, 1848, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Van Balkom, B.W.; Savelkoul, P.J.; Markovich, D.; Hofman, E.; Nielsen, S.; van der Sluijs, P.; Deen, P.M. The role of putative phosphorylation sites in the targeting and shuttling of the aquaporin-2 water channel. J. Biol. Chem. 2002, 277, 41473–41479. [Google Scholar] [CrossRef] [PubMed]
- Kamsteeg, E.J.; Wormhoudt, T.A.; Rijss, J.P.; van Os, C.H.; Deen, P.M. An impaired routing of wild-type aquaporin-2 after tetramerization with an aquaporin-2 mutant explains dominant nephrogenic diabetes insipidus. EMBO J. 1999, 18, 2394–2400. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, M.; Iwai, K.; Ooeda, T.; Igarashi, T.; Ogawa, E.; Katsushima, Y.; Shinbo, I.; Uchida, S.; Terada, Y.; Arthus, M.F.; et al. Three families with autosomal dominant nephrogenic diabetes insipidus caused by aquaporin-2 mutations in the C-terminus. Am. J. Hum. Genet. 2001, 69, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Mulders, S.M.; Bichet, D.G.; Rijss, J.P.; Kamsteeg, E.J.; Arthus, M.F.; Lonergan, M.; Fujiwara, M.; Morgan, K.; Leijendekker, R.; van der Sluijs, P.; et al. An aquaporin-2 water channel mutant which causes autosomal dominant nephrogenic diabetes insipidus is retained in the Golgi complex. J. Clin. Investig. 1998, 102, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, G.; Koudijs, M.; van Balkom, B.W.; Oorschot, V.; Klumperman, J.; Deen, P.M.; van der Sluijs, P. Glycosylation is important for cell surface expression of the water channel aquaporin-2 but is not essential for tetramerization in the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 2975–2983. [Google Scholar] [CrossRef] [PubMed]
- Kamsteeg, E.J.; Stoffels, M.; Tamma, G.; Konings, I.B.; Deen, P.M. Repulsion between Lys258 and upstream arginines explains the missorting of the AQP2 mutant p.Glu258Lys in nephrogenic diabetes insipidus. Hum. Mutat. 2009, 30, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- McKusick, V.A. Mendelian Inheritance in Man and its online version, OMIM. Am. J. Hum. Genet. 2007, 80, 588–604. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Soding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [PubMed]
- Wallner, B.; Elofsson, A. Identification of correct regions in protein models using structural, alignment, and consensus information. Protein Sci. 2006, 15, 900–913. [Google Scholar] [CrossRef] [PubMed]
- Feyfant, E.; Sali, A.; Fiser, A. Modeling mutations in protein structures. Protein Sci. 2007, 16, 2030–2041. [Google Scholar] [CrossRef] [PubMed]
- DeLano WL (2002) The PyMOL Molecular Graphics System. Available online: http://www.pymol.org (accessed on 20 May 2018).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Vangone, A.; Spinelli, R.; Scarano, V.; Cavallo, L.; Oliva, R. COCOMAPS: A web application to analyze and visualize contacts at the interface of biomolecular complexes. Bioinformatics 2011, 27, 2915–2916. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Vargyas, M.; Vasko-Szedlar, J.; Roux, B.; Im, W. PBEQ-Solver for online visualization of electrostatic potential of biomolecules. Nucleic Acids Res. 2008, 36, W270–W275. [Google Scholar] [CrossRef] [PubMed]
- The UniProt, C. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X. version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Milano, S.; Carmosino, M.; Gerbino, A.; Svelto, M.; Procino, G. Hereditary Nephrogenic Diabetes Insipidus: Pathophysiology and Possible Treatment. An Update. Int. J. Mol. Sci. 2017, 18, 2385. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhao, D.; Verkman, A.S. Hsp90 inhibitor partially corrects nephrogenic diabetes insipidus in a conditional knock-in mouse model of aquaporin-2 mutation. FASEB J. 2009, 23, 503–512. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| hAQP | Ress | PDB ID | Mutation | Asymmetric Unit | Pore 1 | Method 2 | Resolution (Å) | Deposition |
|---|---|---|---|---|---|---|---|---|
| AQP1 | 8-233 | 1FQY [34] | -- | monomer | -- | EC | 3.80 | 2000 |
| 9-233 | 1H6I [35] | -- | monomer | -- | EC | 3.80 | 2001 | |
| 9-232 | 1IH5 [36] | -- | monomer | -- | EC | 3.70 | 2001 | |
| 3-235 | 4CSK [37] | -- | monomer | -- | XRD | 3.28 | 2014 | |
| AQP2 | 2-240 | 4NEF [21] | -- | tetramer | -- | XRD | 2.75 | 2013 |
| 5-257 | 4OJ2 3 | S256A | monomer | water | XRD | 3.05 | 2014 | |
| AQP4 | 32-254 | 3GD8 [38] | -- | monomer | water | XRD | 1.80 | 2009 |
| AQP5 | 1-245 | 3D9S [39] | -- | tetramer | water | XRD | 2.00 | 2008 |
| 2-245 | 5C5X [40] | S156E | octamer | water | XRD | 2.60 | 2015 | |
| 2-244 | 5DYE [40] | S156E | tetramer | -- | XRD | 3.50 | 2015 |
| hAQP | Mutation | Disease | Location | Hypothesized Structural Defect |
|---|---|---|---|---|
| AQP2 | L22V | NDI | H1 | tetramer assembly |
| AQP2 | L28P | NDI | H1 | monomer folding |
| AQP2 | A47V | NDI | H2 | monomer folding |
| AQP2 | Q57P | NDI | H2 | impaired metal binding [21] |
| AQP2 | G64R | NDI | H2 | pore features |
| AQP2 | N68S | NDI | HB | pore features (NPA) [21] |
| AQP2 | A70D | NDI | HB | pore features (NPA) [21] |
| AQP2 | V71M | NDI | HB | pore features [21] |
| AQP2 | G100R | NDI | H3 | monomer folding [21] |
| AQP2 | G100V | NDI | H3 | monomer folding |
| AQP2 | T108M | NDI | H3 | monomer folding |
| AQP2 | T125M | NDI | loop C | tetramer assembly [21] |
| AQP2 | T126M | NDI | H4 | tetramer assembly [21] |
| AQP2 | A147T | NDI | H4 | impaired metal binding [21] |
| AQP2 | V168M | NDI | H5 | pore features [21] |
| AQP2 | G175R | NDI | H5 | monomer folding |
| AQP2 | G180S | NDI | H5 | pore features |
| AQP2 | C181W | NDI | loop E | monomer folding |
| AQP2 | P185A | NDI | HE | pore features (NPA) [21] |
| AQP2 | R187C | NDI | HE | pore features (ar/R) [21] |
| AQP2 | R187H | NDI | HE | pore features (ar/R) [21] |
| AQP2 | A190T | NDI | HE | monomer folding |
| AQP2 | W202C | NDI | H6 | monomer folding |
| AQP2 | S216P | NDI | H6 | monomer folding |
| AQP2 | R254L | NDI | C-ter | signal loss [41] |
| AQP2 | R254Q | NDI | C-ter | signal loss [42] |
| AQP2 | E258K | NDI | C-ter | signal loss [43] |
| AQP2 | P262L | NDI | C-ter | signal loss [44] |
| AQP5 | A38E | PPKB | loop A | tetramer assembly [25] |
| AQP5 | I45S | PPKB | H2 | pore features [25] |
| AQP5 | N123D | PPKB | loop C | tetramer assembly [25] |
| AQP5 | I177F | PPKB | H5 | pore features [25] |
| AQP5 | R188C | PPKB | HE | pore features [25] |
| AQP8 | I229M | colorectal tumor | H6 | monomer folding |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calvanese, L.; D’Auria, G.; Vangone, A.; Falcigno, L.; Oliva, R. Structural Basis for Mutations of Human Aquaporins Associated to Genetic Diseases. Int. J. Mol. Sci. 2018, 19, 1577. https://doi.org/10.3390/ijms19061577
Calvanese L, D’Auria G, Vangone A, Falcigno L, Oliva R. Structural Basis for Mutations of Human Aquaporins Associated to Genetic Diseases. International Journal of Molecular Sciences. 2018; 19(6):1577. https://doi.org/10.3390/ijms19061577
Chicago/Turabian StyleCalvanese, Luisa, Gabriella D’Auria, Anna Vangone, Lucia Falcigno, and Romina Oliva. 2018. "Structural Basis for Mutations of Human Aquaporins Associated to Genetic Diseases" International Journal of Molecular Sciences 19, no. 6: 1577. https://doi.org/10.3390/ijms19061577
APA StyleCalvanese, L., D’Auria, G., Vangone, A., Falcigno, L., & Oliva, R. (2018). Structural Basis for Mutations of Human Aquaporins Associated to Genetic Diseases. International Journal of Molecular Sciences, 19(6), 1577. https://doi.org/10.3390/ijms19061577