Immunotherapy for Gastric Cancer: Time for a Personalized Approach?




Abstract
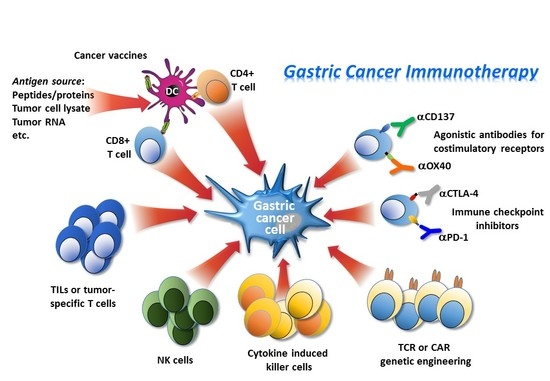
:
1. Introduction
2. Immunosurveillance and Immunoescape
3. Immune-Based Therapies
3.1. Adoptive Cell Immunotherapy
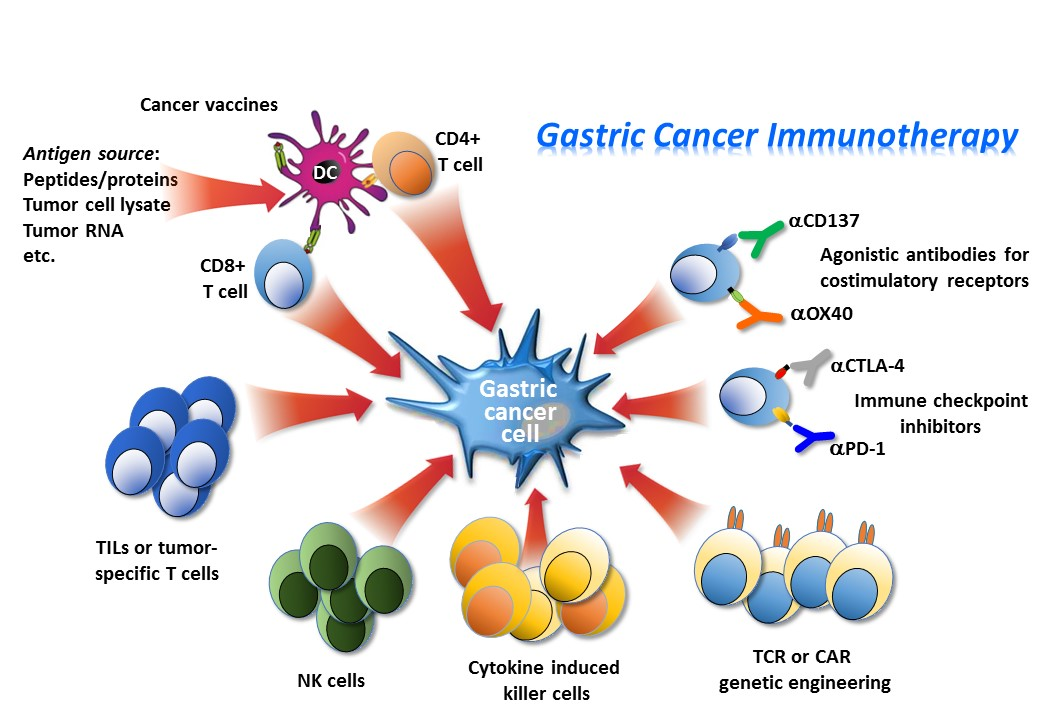
3.2. Engineered Cells for Adoptive Immunotherapy
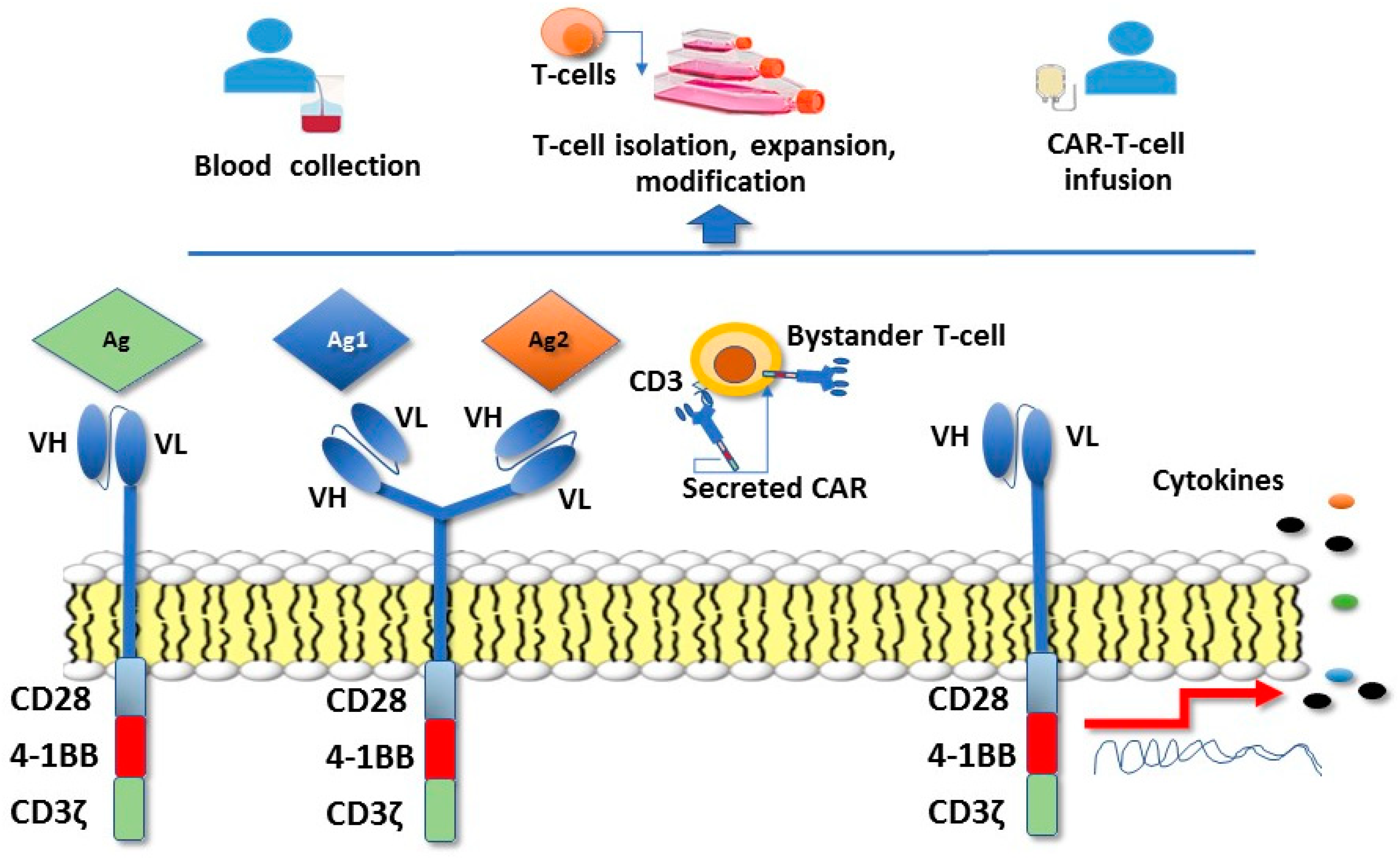
3.3. Immune Checkpoint Inhibitors/Immune Modulatory Pathways
3.4. Agonistic Antibodies for Costimulatory Receptors
3.5. Safety Issues Related to the Use of Checkpoint Inhibitors
3.6. Cancer Vaccines
4. Concluding Remarks and Future Perspectives
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bilici, A. Treatment options in patients with metastatic gastric cancer: Current status and future perspectives. World J. Gastroenterol. 2014, 20, 3905–3915. [Google Scholar] [CrossRef] [PubMed]
- Brierley, J.D.; Gospodarowicz, M.K.; Wittekind, C. TNM Classification of Malignant Tumors, 8th ed.; John Wiley & Sons, Inc.: Chichester, UK; Hoboken, NJ, USA, 2017. [Google Scholar]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Bonotto, M.; Garattini, S.K.; Basile, D.; Ongaro, E.; Fanotto, V.; Cattaneo, M.; Cortiula, F.; Iacono, D.; Cardellino, G.G.; Pella, N.; et al. Immunotherapy for gastric cancers: Emerging role and future perspectives. Expert Rev. Clin. Pharmacol. 2017, 10, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Procaccio, L.; Schirripa, M.; Fassan, M.; Vecchione, L.; Bergamo, F.; Prete, A.A.; Intini, R.; Manai, C.; Dadduzio, V.; Boscolo, A.; et al. Immunotherapy in gastrointestinal cancers. Biomed. Res. Int. 2017, 3, 4346576. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.P.; et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health. Study of Pembrolizumab (MK-3475) as First-Line Monotherapy and Combination Therapy for Treatment of Advanced Gastric or Gastroesophageal Junction Adenocarcinoma (MK-3475-062/KEYNOTE-062). Available online: https://clinicaltrials.gov/ct2/show/NCT02494583 (accessed on 10 July 2015).
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Derks, S.; Liao, X.; Chiaravalli, A.M.; Xu, X.; Camargo, M.C.; Solcia, E.; Sessa, F.; Fleitas, T.; Freeman, G.J.; Rodig, S.J.; et al. Abundant PD-L1 expression in Epstein–Barr Virus-infected gastric cancers. Oncotarget 2016, 7, 32925–32932. [Google Scholar] [CrossRef] [PubMed]
- Finn, O.J. A believer’s overview of cancer immunosurveillance and immunotherapy. J. Immunol. 2018, 200, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Davoodzadeh, G.M.; Kardar, G.A.; Saeedi, Y.; Heydari, S.; Garssen, J.; Falak, R. Exhaustion of T lymphocytes in the tumor microenvironment: Significance and effective mechanisms. Cell. Immunol. 2017, 322, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, S.; Natsugoe, S.; Tokuda, K.; Nakajo, A.; Okumura, H.; Matsumoto, M.; Miyazono, F.; Hokita, S.; Aikou, T. Tumor-associated macrophage (TAM) infiltration in gastric cancer. Anticancer Res. 2003, 23, 4079–4083. [Google Scholar] [PubMed]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Sung, J.Y.; Lee, J.; Park, Y.K.; Kim, Y.W.; Kim, G.Y.; Won, K.Y.; Lim, S.J. Polarized CD163+ tumor-associated macrophages are associated with increased angiogenesis and CXCL12 expression in gastric cancer. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.H.; Lee, W.J.; Hua, K.T.; Kuo, M.L.; Lin, M.T. Macrophage infiltration induces gastric cancer invasiveness by activating the β-catenin pathway. PLoS ONE 2015, 10, e0134122. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [PubMed]
- Ben-Meir, K.; Twaik, N.; Baniyash, M. Plasticity and biological diversity of myeloid derived suppressor cells. Curr. Opin. Immunol. 2018, 51, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Gedeon, P.C.; Herndon, J.E.; Archer, G.E.; Reap, E.A.; Sanchez-Perez, L.; Mitchell, D.A.; Bigner, D.D.; Sampson, J.H. Human regulatory T cells kill tumor cells through granzyme-dependent cytotoxicity upon retargeting with a bispecific antibody. Cancer Immunol. Res. 2013, 1, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Kandulski, A.; Malfertheiner, P.; Wex, T. Role of regulatory T-cells in H. pylori-induced gastritis and gastric cancer. Anticancer Res. 2010, 30, 1093–1103. [Google Scholar] [PubMed]
- Kang, B.W.; Seo, A.N.; Yoon, S.; Bae, H.I.; Jeon, S.W.; Kwon, O.K.; Chung, H.Y.; Yu, W.; Kang, H.; Kim, J.G. Prognostic value of tumor-infiltrating lymphocytes in Epstein–Barr virus-associated gastric cancer. Ann. Oncol. 2016, 27, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Takeoka, T.; Urakawa, S.; Morimoto-Okazawa, A.; Kawashima, A.; Iwahori, K.; Takiguchi, S.; Nishikawa, H.; Sato, E.; Sakaguchi, S.; et al. ICOS+ Foxp3+ TILs in gastric cancer are prognostic markers and effector regulatory T cells associated with Helicobacter pylori. Int. J. Cancer 2017, 140, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Sackstein, R.; Schatton, T.; Barthel, S.R. T-lymphocyte homing: An underappreciated yet critical hurdle for successful cancer immunotherapy. Lab. Investig. 2017, 97, 669–697. [Google Scholar] [CrossRef] [PubMed]
- Badalamenti, G.; Fanale, D.; Incorvaia, L.; Barraco, N.; Listi, A.; Maragliano, R.; Vincenzi, B.; Calo, V.; Iovanna, J.L.; Bazan, V.; et al. Role of tumor-infiltrating lymphocytes in patients with solid tumors: Can a drop dig a stone? Cell. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Lee, K.S.; Cho, H.J.; Kim, Y.H.; Yang, H.K.; Kim, W.H.; Kang, G.H. Prognostic implications of tumor-infiltrating FoxP3+ regulatory T cells and CD8+ cytotoxic T cells in microsatellite-unstable gastric cancers. Hum. Pathol. 2014, 45, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural killer cells for immunotherapy—Advantages of the NK-92 cell line over blood NK cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Sarvaria, A.; Madrigal, J.A.; Saudemont, A. B cell regulation in cancer and anti-tumor immunity. Cell. Mol. Immunol. 2017, 14, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Teitz-Tennenbaum, S.; Donald, E.J.; Li, M.; Chang, A.E. In vivo sensitized and in vitro activated B cells mediate tumor regression in cancer adoptive immunotherapy. J. Immunol. 2009, 183, 3195–3203. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lao, X.; Pan, Q.; Ning, N.; Yet, J.; Xu, Y.; Li, S.; Chang, A.E. Adoptive transfer of tumor reactive B cells confers host T-cell immunity and tumor regression. Clin. Cancer Res. 2011, 17, 4987–4995. [Google Scholar] [CrossRef] [PubMed]
- Perricone, M.A.; Smith, K.A.; Claussen, K.A.; Plog, M.S.; Hempel, D.M.; Roberts, B.L.; St George, J.A.; Kaplan, J.M. Enhanced efficacy of melanoma vaccines in the absence of B lymphocytes. J. Immunother. 2004, 27, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Divekar, A.A.; Hilchey, S.P.; Cho, H.M.; Newman, C.L.; Shin, S.U.; Nechustan, H.; Challita-Eid, P.M.; Segal, B.M.; Yi, K.H.; et al. Increased rejection of primary tumors in mice lacking B cells: Inhibition of anti-tumor CTL and TH1 cytokine responses by B cells. Int. J. Cancer 2005, 117, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.W.; Kim, J.G.; Lee, I.H.; Bae, H.I.; Seo, A.N. Clinical significance of tumor-infiltrating lymphocytes for gastric cancer in the era of immunology. World J. Gastrointest. Oncol. 2017, 9, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, S.; Natsugoe, S.; Tokuda, K.; Nakajo, A.; Xiangming, C.; Iwashige, H.; Aridome, K.; Hokita, S.; Aikou, T. Clinical impact of intratumoral natural killer cell and dendritic cell infiltration in gastric cancer. Cancer Lett. 2000, 159, 103–108. [Google Scholar] [CrossRef]
- Malmberg, K.J.; Carlsten, M.; Bjorklund, A.; Sohlberg, E.; Bryceson, Y.T.; Ljunggren, H.G. Natural killer cell-mediated immunosurveillance of human cancer. Semin. Immunol. 2017, 31, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Rosso, D.; Rigueiro, M.P.; Kassab, P.; Ilias, E.J.; Castro, O.A.; Novo, N.F.; Lourenco, L.G. Correlation of natural killer cells with the prognosis of gastric adenocarcinoma. Arq. Bras. Cir. Dig. 2012, 25, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Takaya, S.; Osaki, T.; Ikeguchi, M. Increased apoptosis and elevated FAS expression in circulating natural killer cells in gastric cancer patients. Gastric. Cancer 2013, 16, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Rosenberg, S.A. Adoptive T-Cell Therapy for Cancer. Adv. Immunol. 2016, 130, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Kono, K.; Ichihara, F.; Iizuka, H.; Sekikawa, T.; Matsumoto, Y. Differences in the recognition of tumor-specific CD8+ T cells derived from solid tumor, metastatic lymph nodes and ascites in patients with gastric cancer. Int. J. Cancer 1997, 71, 978–981. [Google Scholar] [CrossRef]
- Fujie, T.; Tanaka, F.; Tahara, K.; Li, J.; Tanaka, S.; Mori, M.; Ueo, H.; Takesako, K.; Akiyoshi, T. Generation of specific antitumor reactivity by the stimulation of spleen cells from gastric cancer patients with MAGE-3 synthetic peptide. Cancer Immunol. Immunother. 1999, 48, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Voskens, C.J.; Watanabe, R.; Rollins, S.; Campana, D.; Hasumi, K.; Mann, D.L. Ex-vivo expanded human NK cells express activating receptors that mediate cytotoxicity of allogeneic and autologous cancer cell lines by direct recognition and antibody directed cellular cytotoxicity. J. Exp. Clin. Cancer Res. 2010, 29, 134. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Okayama, T.; Sakamoto, N.; Ideno, M.; Oka, K.; Enoki, T.; Mineno, J.; Yoshida, N.; Katada, K.; Kamada, K.; et al. Phase I clinical trial of adoptive transfer of expanded natural killer cells in combination with IgG1 antibody in patients with gastric or colorectal cancer. Int. J. Cancer 2018, 142, 2599–2609. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, N.; Ishikawa, T.; Kokura, S.; Okayama, T.; Oka, K.; Ideno, M.; Sakai, F.; Kato, A.; Tanabe, M.; Enoki, T.; et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J. Transl. Med. 2015, 13, 277. [Google Scholar] [CrossRef] [PubMed]
- Kloss, S.; Oberschmidt, O.; Morgan, M.; Dahlke, J.; Arseniev, L.; Huppert, V.; Granzin, M.; Gardlowski, T.; Matthies, N.; Soltenborn, S.; et al. Optimization of human NK cell manufacturing: Fully automated separation, improved ex vivo expansion using IL-21 with autologous feeder cells, and generation of anti-CD123-CAR-expressing effector cells. Hum. Gene Ther. 2017, 28, 897–913. [Google Scholar] [CrossRef] [PubMed]
- Uherek, C.; Tonn, T.; Uherek, B.; Becker, S.; Schnierle, B.; Klingemann, H.G.; Wels, W. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood 2002, 100, 1265–1273. [Google Scholar] [PubMed]
- Schonfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bonig, H.; Kohl, U.; Kloess, S.; et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genssler, S.; Schonfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J. Natl. Cancer Inst. 2015, 108, 375. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Han, W. Cytokine-induced killer (CIK) cells: From basic research to clinical translation. Chin. J. Cancer 2015, 34, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Song, G.; Hu, X.; Zhou, Y.; Li, Y.; Chen, Q.; Feng, G. A positive role of cytokine-induced killer cell therapy on gastric cancer therapy in a Chinese population: A systematic meta-analysis. Med. Sci. Monit. 2015, 21, 3363–3370. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Zhou, C.H.; Chen, S.F.; Ding, J.; Zhang, Y.X.; Yang, Y.P.; Wang, W.H. Effectiveness and safety of chemotherapy combined with cytokine-induced killer cell/dendritic cell-cytokine-induced killer cell therapy for treatment of gastric cancer in China: A systematic review and meta-analysis. Cytotherapy 2016, 18, 1162–1177. [Google Scholar] [CrossRef] [PubMed]
- Introna, M.; Correnti, F. Innovative clinical perspectives for CIK cells in cancer patients. Int. J. Mol. Sci. 2018, 19, 358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, G.; Hou, Y.; Zhang, J.; Hu, J.; Zhang, K. The experimental study on the treatment of cytokine-induced killer cells combined with EGFR monoclonal antibody against gastric cancer. Cancer Biother. Radiopharm. 2014, 29, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Del Zotto, G.; Marcenaro, E.; Vacca, P.; Sivori, S.; Pende, D.; Della, C.M.; Moretta, F.; Ingegnere, T.; Mingari, M.C.; Moretta, A.; et al. Markers and function of human NK cells in normal and pathological conditions. Cytometry B Clin. Cytom. 2017, 92, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xu, L.; Ding, S.; Wu, M.; Tang, Z.; Fu, W.; Ni, Q. Treatment of 23 patients with advanced gastric cancer by intravenously transfer of autologous tumor-infiltrating lymphocytes combined with rIL-2. Chin. Med. Sci. J. 1995, 10, 185–187. [Google Scholar] [PubMed]
- Zhang, G.Q.; Zhao, H.; Wu, J.Y.; Li, J.Y.; Yan, X.; Wang, G.; Wu, L.L.; Zhang, X.G.; Shao, Y.; Wang, Y.; et al. Prolonged overall survival in gastric cancer patients after adoptive immunotherapy. World J. Gastroenterol. 2015, 21, 2777–2785. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zhou, Q.; Wu, J.; Ji, M.; Li, G.; Jiang, J.; Wu, C. Efficacy of adjuvant immunotherapy with cytokine-induced killer cells in patients with locally advanced gastric cancer. Cancer Immunol. Immunother. 2012, 61, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.T.; Shen, Y.P.; Wu, C.P.; Zhu, Y.B.; Wei, W.X.; Chen, L.J.; Zheng, X.; Sun, J.; Lu, B.F.; Zhang, X.G. Increasing the frequency of CIK cells adoptive immunotherapy may decrease risk of death in gastric cancer patients. World J. Gastroenterol. 2010, 16, 6155–6162. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guo, Z.Q.; Shi, C.M.; Zhou, Z.F.; Ye, Y.B.; Chen, Q. Efficacy of adjuvant chemotherapy combined with immunotherapy with cytokine-induced killer cells for gastric cancer after d2 gastrectomy. Int. J. Clin. Exp. Med. 2015, 8, 7728–7736. [Google Scholar] [PubMed]
- Zhao, H.; Fan, Y.; Li, H.; Yu, J.; Liu, L.; Cao, S.; Ren, B.; Yan, F.; Ren, X. Immunotherapy with cytokine-induced killer cells as an adjuvant treatment for advanced gastric carcinoma: A retrospective study of 165 patients. Cancer Biother. Radiopharm. 2013, 28, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Song, J.; Yang, Z.; Zhang, X. Effects of cytokine-induced killer cell treatment combined with FolFox4 on the recurrence and survival rates for gastric cancer following surgery. Exp. Ther. Med. 2013, 6, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.R.; Rodriguez, A.; Shepphird, J.; Brown, C.E.; Badie, B. Chimeric antigen receptors T cell therapy in solid tumor: Challenges and clinical APPLICATIONS. Front. Immunol. 2017, 8, 1850. [Google Scholar] [CrossRef] [PubMed]
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T cells: The promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Liu, Y.; Guo, Y.; Qiu, J.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Han, W. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell 2017, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, C.; Li, G.; Li, J.; Lv, X.; Shi, H.; Liu, J.; Liu, S.; Yan, P.; Wang, S.; et al. Antitumor effects and persistence of a novel HER2 CAR T cells directed to gastric cancer in preclinical models. Am. J. Cancer Res. 2018, 8, 106–119. [Google Scholar] [PubMed]
- Luo, F.; Qian, J.; Yang, J.; Deng, Y.; Zheng, X.; Liu, J.; Chu, Y. Bifunctional αHER2/CD3 RNA-engineered CART-like human T cells specifically eliminate HER2+ gastric cancer. Cell Res. 2016, 26, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Tong, C.; Wang, Y.; Gao, Y.; Dai, H.; Guo, Y.; Zhao, X.; Wang, Y.; Wang, Z.; Han, W.; et al. Effective and persistent antitumor activity of HER2-directed CAR-T cells against gastric cancer cells in vitro and xenotransplanted tumors in vivo. Protein Cell 2017, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shibaguchi, H.; Luo, N.; Shirasu, N.; Kuroki, M.; Kuroki, M. Enhancement of antitumor activity by using a fully human gene encoding a single-chain fragmented antibody specific for carcinoembryonic antigen. Onco Targets Ther. 2017, 10, 3979–3990. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.N.; Sarkissian, S.; Chao, J.; Klempner, S.J. PD-1 and PD-L1 as emerging therapeutic targets in gastric cancer: Current evidence. Gastrointest. Cancer 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Boger, C.; Behrens, H.M.; Mathiak, M.; Kruger, S.; Kalthoff, H.; Rocken, C. PD-L1 is an independent prognostic predictor in gastric cancer of Western patients. Oncotarget 2016, 7, 24269–24283. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Nam, K.H.; Ahn, S.H.; Park, D.J.; Kim, H.H.; Kim, S.H.; Chang, H.; Lee, J.O.; Kim, Y.J.; Lee, H.S.; et al. Prognostic implications of immunosuppressive protein expression in tumors as well as immune cell infiltration within the tumor microenvironment in gastric cancer. Gastric Cancer 2016, 19, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Schlosser, H.A.; Drebber, U.; Kloth, M.; Thelen, M.; Rothschild, S.I.; Haase, S.; Garcia-Marquez, M.; Wennhold, K.; Berlth, F.; Urbanski, A.; et al. Immune checkpoints programmed death 1 ligand 1 and cytotoxic T lymphocyte associated molecule 4 in gastric adenocarcinoma. Oncoimmunology 2015, 5, e1100789. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Chen, M.; Guo, D.; Zhu, H.; Zhang, W.; Pan, J.; Zhong, X.; Li, X.; Qian, H.; Wang, X. PD-L1 and gastric cancer prognosis: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0182692. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Abe, H.; Kunita, A.; Yamashita, H.; Seto, Y.; Fukayama, M. Overexpression and gene amplification of PD-L1 in cancer cells and PD-L1+ immune cells in Epstein–Barr virus-associated gastric cancer: The prognostic implications. Mod. Pathol. 2017, 30, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cheng, Y.; Xu, Y.; Wang, Z.; Du, X.; Li, C.; Peng, J.; Gao, L.; Liang, X.; Ma, C. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene 2017, 36, 6143–6153. [Google Scholar] [CrossRef] [PubMed]
- Nowak, E.C.; Lines, J.L.; Varn, F.S.; Deng, J.; Sarde, A.; Mabaera, R.; Kuta, A.; Le, M.I.; Cheng, C.; Noelle, R.J. Immunoregulatory functions of VISTA. Immunol. Rev. 2017, 276, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Boger, C.; Behrens, H.M.; Kruger, S.; Rocken, C. The novel negative checkpoint regulator VISTA is expressed in gastric carcinoma and associated with PD-L1/PD-1: A future perspective for a combined gastric cancer therapy? Oncoimmunology 2017, 6, e1293215. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Yang, M.; Turner, A.; Xu, C.; Ferris, R.L.; Huang, J.; Kane, L.P.; Lu, B. TIM-3 as a target for cancer immunotherapy and mechanisms of action. Int. J. Mol. Sci. 2017, 18, 645. [Google Scholar] [CrossRef] [PubMed]
- Takano, S.; Saito, H.; Ikeguchi, M. An increased number of PD-1+ and Tim-3+ CD8+ T cells is involved in immune evasion in gastric cancer. Surg. Today 2016, 46, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yang, L.; Yao, D.; Wu, X.; Li, J.; Liu, X.; Deng, L.; Huang, C.; Wang, Y.; Li, D.; et al. Tumor antigen-specific CD8+ T cells are negatively regulated by PD-1 and Tim-3 in human gastric cancer. Cell. Immunol. 2017, 313, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Ralph, C.; Elkord, E.; Burt, D.J.; O’Dwyer, J.F.; Austin, E.B.; Stern, P.L.; Hawkins, R.E.; T histlethwaite, F.C. Modulation of lymphocyte regulation for cancer therapy: A phase II trial of tremelimumab in advanced gastric and esophageal adenocarcinoma. Clin. Cancer Res. 2010, 16, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.; Kim, S.S.; Gold, P.J.; Philip, P.A.; Enzinger, P.C.; Coffie, J.; Schmidt, E.V.; Baldwin, M.; Nordstrom, J.L.; Bonvini, E.; et al. A phase 1b/2, open label, dose-escalation study of margetuximab (M) in combination with pembrolizumab (P) in patients with relapsed/refractory advanced HER2+ gastroesophageal (GEJ) junction or gastric (G) cancer. J. Clin. Oncol. 2018, 35, TPS219. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Chen, Y.; Frankel, P.H.; Chung, V.M.; Lim, D.; Li, D.; Fakih, M.; Lee, P.P. Combining pembrolizumab and palliative radiotherapy in gastroesophageal cancer to enhance antitumor T-cell response and augment the abscopal effect. J. Clin. Oncol. 2018, 35, TPS220. [Google Scholar] [CrossRef]
- Bang, Y.J.; Cho, J.Y.; Kim, Y.H.; Kim, J.W.; Di, B.M.; Ajani, J.A.; Yamaguchi, K.; Balogh, A.; Sanchez, T.; Moehler, M. Efficacy of sequential ipilimumab monotherapy versus best supportive care for unresectable locally advanced/metastatic gastric or gastroesophageal junction cancer. Clin. Cancer Res. 2017, 23, 5671–5678. [Google Scholar] [CrossRef] [PubMed]
- Muro, K.; Chung, H.C.; Shankaran, V.; Geva, R.; Catenacci, D.; Gupta, S.; Eder, J.P.; Golan, T.; Le, D.T.; Burtness, B.; et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): A multicentre, open-label, phase 1b trial. Lancet Oncol. 2016, 17, 717–726. [Google Scholar] [CrossRef]
- Doi, T.; Piha-Paul, S.A.; Jalal, S.I.; Saraf, S.; Lunceford, J.; Koshiji, M.; Bennouna, J. Safety and antitumor activity of the anti-programmed death-1 antibody pembrolizumab in patients with advanced esophageal carcinoma. J. Clin. Oncol. 2018, 36, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.A.; Jalal, S.; Muro, K.; Yoon, H.H.; Garrido, M.; Golan, T.; Doi, T.; Catenacci, D.V.; Geva, R.; Ku, G.; et al. Oesophageal cancer gastric cancer cancer immunology and immunotherapy. In Proceedings of the ESMO 2017 Congress, Annals of Oncology, Madrid, Spain, 8–12 September 2018; Volume 28, pp. v605–v649. [Google Scholar]
- Ohtsu, A.; Tabernero, J.; Bang, Y.; Fuchs, C.S.; Sun, L.; Wang, Z.; Csiki, I.; Koshiji, M.; Cutsem, E.V. Pembrolizumab (MK-3475) versus paclitaxel as second-line therapy for advanced gastric or gastroesophageal junction (GEJ) adenocarcinoma: Phase 3 KEYNOTE-061 study. J. Clin. Oncol. 2018, 34, TPS183. [Google Scholar] [CrossRef]
- Chau, I.; Bendell, J.C.; Calvo, E.; Santana-Davila, R.; Ahnert, J.R.; Penel, N.; Arkenau, H.; Yang, Y.; Rege, J.; Mi, G.; et al. Interim safety and clinical activity in patients (pts) with advanced gastric or gastroesophageal junction (G/GEJ) adenocarcinoma from a multicohort phase 1 study of ramucirumab (R) plus pembrolizumab (P). J. Clin. Oncol. 2018, 35, 102. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Ott, P.A.; Calvo, E.; Kim, J.W.; Ascierto, P.A.; Sharma, P.; Peltola, K.J.; Jaeger, D.; Jeffry Evans, T.R.; De Braud, F.G.; et al. Nivolumab ± ipilimumab in pts with advanced (adv)/metastatic chemotherapy-refractory (CTx-R) gastric (G), esophageal (E), or gastroesophageal junction (GEJ) cancer: CheckMate 032 study. J. Clin. Oncol. 2018, 35, 4014. [Google Scholar]
- Kang, Y.K.; Boku, N.; Satoh, T.; Ryu, M.H.; Chao, Y.; Kato, K.; Chung, H.C.; Chen, J.S.; Muro, K.; Kang, W.K.; et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 2461–2471. [Google Scholar] [CrossRef]
- Heery, C.R.; O’Sullivan-Coyne, G.; Madan, R.A.; Cordes, L.; Rajan, A.; Rauckhorst, M.; Lamping, E.; Oyelakin, I.; Marte, J.L.; Lepone, L.M.; et al. Avelumab for metastatic or locally advanced previously treated solid tumours (JAVELIN Solid Tumor): A phase 1a, multicohort, dose-escalation trial. Lancet Oncol. 2017, 18, 587–598. [Google Scholar] [CrossRef]
- Bang, Y.Y.; Wyrwicz, L.; Park, Y.L.; Ryu, M.; Muntean, A.; Gomez-Martin, C.; Guimbaud, R.; Ciardiello, F.; Boku, N.; Van Cutsem, E.; et al. Avelumab (MSB0010718C.; anti-PD-L1) + best supportive care (BSC) vs BSC ± chemotherapy as third-line treatment for patients with unresectable, recurrent, or metastatic gastric cancer: The phase 3 JAVELIN Gastric 300 trial. J. Clin. Oncol. 2018, 34, TPS4135. [Google Scholar]
- Marcus, H.; Moehler, M.H.; Taïeb, J.; Gurtler, J.S.; Xiong, H.; Zhang, J.; Cuillerot, J.; Boku, N. Maintenance therapy with avelumab (MSB0010718C.; anti-PD-L1) vs continuation of first-line chemotherapy in patients with unresectable, locally advanced or metastatic gastric cancer: The phase 3 JAVELIN Gastric 100 trial. J. Clin. Oncol. 2018, 34, TPS4134. [Google Scholar]
- Kelly, R.J.; Chung, K.; Gu, Y.; Steele, K.E.; Rebelatto, M.C.; Robbins, P.B.; Tavakkoli, F.; Karakunnel, J.J.; Lai, D.W.; Almhanna, K. Phase Ib/II study to evaluate the safety and antitumor activity of durvalumab (MEDI4736) and tremelimumab as monotherapy or in combination, in patients with recurrent or metastatic gastric/gastroesophageal junction adenocarcinoma. J. Immunother. Cancer 2018, 3, P157. [Google Scholar] [CrossRef]
- Bang, Y.J.; Golan, T.; Lin, C.; Kang, Y.; Wainberg, Z.A.; Wasserstrom, H.; Jin, J.; Mi, G.; McNeely, S.; Laing, N.; et al. Interim safety and clinical activity in patients with locally advanced and unresectable or metastatic gastric or gastroesophageal junction (G/GEJ) adenocarcinoma from a multicohort phase I study of ramucirumab plus durvalumab. J. Clin. Oncol. 2018, 36, 92. [Google Scholar]
- Linch, S.N.; McNamara, M.J.; Redmond, W.L. OX40 Agonists and combination immunotherapy: Putting the pedal to the metal. Front. Oncol. 2015, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Chester, C.; Sanmamed, M.F.; Wang, J.; Melero, I. Immunotherapy targeting 4-1BB: Mechanistic rationale, clinical results, and future strategies. Blood 2018, 131, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Burugu, S.; Dancsok, A.R.; Nielsen, T.O. Emerging targets in cancer immunotherapy. Semin. Cancer Biol. 2017, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Curti, B.D.; Kovacsovics-Bankowski, M.; Morris, N.; Walker, E.; Chisholm, L.; Floyd, K.; Walker, J.; Gonzalez, I.; Meeuwsen, T.; Fox, B.A.; et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013, 73, 7189–7198. [Google Scholar] [CrossRef] [PubMed]
- Chester, C.; Ambulkar, S.; Kohrt, H.E. 4-1BB agonism: Adding the accelerator to cancer immunotherapy. Cancer Immunol. Immunother. 2016, 65, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Kojima, Y.; Uno, T.; Hayakawa, Y.; Teng, M.W.; Yoshizawa, H.; Yagita, H.; Gejyo, F.; Okumura, K.; Smyth, M.J. Combination therapy of established tumors by antibodies targeting immune activating and suppressing molecules. J. Immunol. 2010, 184, 5493–5501. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Pastor, F.; Rodriguez, A.; Perez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Melero, I. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin. Oncol. 2015, 42, 640–655. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Quezada, S.A.; Sepulveda, M.A.; Sharma, P.; Allison, J.P. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J. Exp. Med. 2014, 211, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Taieb, J.; Moehler, M.; Boku, N.; Ajani, J.A.; Yanez, R.E.; Ryu, M.H.; Guenther, S.; Chand, V.; Bang, Y.J. Evolution of checkpoint inhibitors for the treatment of metastatic gastric cancers: Current status and future perspectives. Cancer Treat. Rev. 2018, 66, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Kottschade, L.A. Incidence and management of immune-related adverse events in patients undergoing treatment with immune checkpoint inhibitors. Curr. Oncol. Rep. 2018, 20, 24. [Google Scholar] [CrossRef] [PubMed]
- Niccolai, E.; Taddei, A.; Prisco, D.; Amedei, A. Gastric cancer and the epoch of immunotherapy approaches. World J. Gastroenterol. 2015, 21, 5778–5793. [Google Scholar] [CrossRef] [PubMed]
- Kono, K.; Takahashi, A.; Sugai, H.; Fujii, H.; Choudhury, A.R.; Kiessling, R.; Matsumoto, Y. Dendritic cells pulsed with HER-2/neu-derived peptides can induce specific T-cell responses in patients with gastric cancer. Clin. Cancer Res. 2002, 8, 3394–3400. [Google Scholar] [PubMed]
- Sadanaga, N.; Nagashima, H.; Mashino, K.; Tahara, K.; Yamaguchi, H.; Ohta, M.; Fujie, T.; Tanaka, F.; Inoue, H.; Takesako, K.; et al. Dendritic cell vaccination with MAGE peptide is a novel therapeutic approach for gastrointestinal carcinomas. Clin. Cancer Res. 2001, 7, 2277–2284. [Google Scholar] [PubMed]
- Ajani, J.A.; Hecht, J.R.; Ho, L.; Baker, J.; Oortgiesen, M.; Eduljee, A.; Michaeli, D. An open-label, multinational, multicenter study of G17DT vaccination combined with cisplatin and 5-fluorouracil in patients with untreated, advanced gastric or gastroesophageal cancer: The GC4 study. Cancer 2006, 106, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Gao, H.; Gao, M.; Qi, S.; Yang, K.; Zhang, Y.; Wang, J. Immunogenicity and safety of a novel tetanus toxoid-conjugated anti-gastrin vaccine in BALB/c mice. Vaccine 2018, 36, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Higashihara, Y.; Kato, J.; Nagahara, A.; Izumi, K.; Konishi, M.; Kodani, T.; Serizawa, N.; Osada, T.; Watanabe, S. Phase I clinical trial of peptide vaccination with URLC10 and VEGFR1 epitope peptides in patients with advanced gastric cancer. Int. J. Oncol. 2014, 44, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Peng, Z.; Huang, X.; Qiao, Z.; Wang, X.; Wang, N.; Xi, H.; Cui, J.; Gao, Y.; Huang, X.; et al. Phase II trial of adjuvant immunotherapy with autologous tumor-derived Gp96 vaccination in patients with gastric CANCER. J. Cancer 2017, 8, 1826–1832. [Google Scholar] [CrossRef] [PubMed]
- Popiela, T.; Kulig, J.; Czupryna, A.; Szczepanik, A.M.; Zembala, M. Efficiency of adjuvant immunochemotherapy following curative resection in patients with locally advanced gastric cancer. Gastric Cancer 2004, 7, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Shomura, H.; Maeda, Y.; Mine, T.; Une, Y.; Akasaka, Y.; Kondo, M.; Takahashi, S.; Shinohara, T.; Katagiri, K.; et al. Immunological evaluation of peptide vaccination for patients with gastric cancer based on pre-existing cellular response to peptide. Cancer Sci. 2003, 94, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Masuzawa, T.; Fujiwara, Y.; Okada, K.; Nakamura, A.; Takiguchi, S.; Nakajima, K.; Miyata, H.; Yamasaki, M.; Kurokawa, Y.; Osawa, R.; et al. Phase I/II study of S-1 plus cisplatin combined with peptide vaccines for human vascular endothelial growth factor receptor 1 and 2 in patients with advanced gastric cancer. Int. J. Oncol. 2012, 41, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Sugimura, K.; Miyata, H.; Omori, T.; Nakano, H.; Mochizuki, C.; Shimizu, K.; Saito, H.; Ashida, K.; Honjyo, S.; et al. A pilot study of post-operative adjuvant vaccine for advanced gastric cancer. Yonago Acta Med. 2017, 60, 101–105. [Google Scholar] [PubMed]
- Sundar, R.; Rha, S.Y.; Yamaue, H.; Katsuda, M.; Kono, K.; Kim, H.S.; Kim, C.; Mimura, K.; Kua, L.F.; Yong, W.P. A phase I/Ib study of OTSGC-A24 combined peptide vaccine in advanced gastric cancer. BMC Cancer 2018, 18, 332. [Google Scholar] [CrossRef] [PubMed]
- Koido, S. Dendritic-tumor fusion cell-based cancer vaccines. Int. J. Mol. Sci. 2016, 17, 828. [Google Scholar] [CrossRef] [PubMed]
- Takakura, K.; Kajihara, M.; Ito, Z.; Ohkusa, T.; Gong, J.; Koido, S. Dendritic-tumor fusion cells in cancer immunotherapy. Discov. Med. 2015, 19, 169–174. [Google Scholar] [PubMed]
- Lasek, W.; Zagozdzon, R.; Jakobisiak, M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014, 63, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Okamoto, M.; Namiki, Y.; Takakura, K.; Takahara, A.; Odahara, S.; Tsukinaga, S.; Yukawa, T.; Mitobe, J.; et al. Combined TLR2/4-activated dendritic/tumor cell fusions induce augmented cytotoxic T lymphocytes. PLoS ONE 2013, 8, e59280. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Tumor Stage | TNM Classification | Survival Rate (%, 5 Years) | Treatment |
|---|---|---|---|
| 1 | T1-2, N0-1, M0 | 69 | Surgical resection |
| 2 | T1-4a, N0-3a, M0 | 43 | Preoperative chemotherapy and surgery followed by post-operative adjuvant chemo/radio-therapy |
| 3 | T1-4b, N1-3b, M0 | 28 | |
| 4 | Tx, Nx, M0 | 9 | Palliative chemotherapy ± targeted therapy |
| Type of Treatment | Setting | Primary End-Point | References |
|---|---|---|---|
| Autologous tumor infiltrating lymphocytes (TILs) combined with rIL-2 | advanced GC (n = 23) | 13% CR 21.7% PR | [57] |
| Autologous peripheral blood lymphocytes activated by anti-CD3 antibody and interleukin (IL)-2 + chemotherapy | GC with a life expectancy >12 weeks (n = 84) | OS in patients that had received surgery was prolonged after EAAL immunotherapy | [58] |
| Ex vivo expanded natural killer (NK) in co-culture with K562 | [43] | ||
| NK expansion using recombinant human fibronectin fragment (FN-CH296) + target-based chemotherapy | unresectable, locally advanced, and/or metastatic GC (n = 3) | phase I trial, good tolerability | [44] |
| Expanded NK with OK432, IL-2, and modified FN-CH296 | unresectable, locally advanced and/or metastatic GC (n = 3) | phase I well tolerated with no severe adverse events | [45] |
| NK-92 cell line | advanced solid tumors | only pre-clinical studies | [29] |
| Autologous cytokine-induced killer cells (CIK) | post-operative locally advanced GC (n = 151) | 5-year OS 46.8 vs. 31.4% intestinal type (p = 0.045), 5-year DFS 28.3 versus 10.4% (p = 0.044) | [59] |
| Autologous CIK + chemotherapy | post-operative locally advanced GC (n = 95) | DFS and OS were longer in pts with higher major histocompatibility complex (MHC)-I-related gene A (MICA) | [58] |
| Autologous CIK + chemotherapy | post-operative locally advanced GC (n = 156) | longer OS | [60] |
| Autologous CIK + chemotherapy | GC stage II-III (n = 226) | longer DFS and OS | [61] |
| Autologous CIK + oxaliplatin | post-operative stage II-III GC (n = 167) | higher 5-year OS rate (56.6% vs. 26.8%, p = 0.014) and progression-free survival (PFS) rate (49.1% vs. 24.1%, p = 0.026) | [62] |
| Autologous CIK + FolFox4 | post-operative GC (n = 51) | reduced GC recurrence rates and enhanced survival rates | [63] |
| Type of Treatment | Setting | Type of Study/Trial | Reference/Trial No. |
|---|---|---|---|
| CAR T cell therapy targeting human epidermal growth factor receptor 2 (HER2) | HER2+ GC | pre-clinical studies | [67,69] |
| CAR-T-like T cells targeting HER2 | HER2+ GC | pre-clinical study | [68] |
| CAR targeting HER2+ | HER2-positive solid tumors (breast cancer, ovarian cancer, lung cancer, GC, colorectal cancer, glioma, pancreatic cancer) | ongoing phase I studies | NCT02713984 |
| CAR targeting the carcinoembryonic antigen (CEA) | GC CEA-positive | ongoing phase I studies | NCT02349724 NCT02850536 NCT02416466 |
| CAR targeting Human Mucin-1 (MUC1) | GC MUC1-positive | ongoing phase I | NCT02617134 |
| CAR targeting the epithelial cell adhesion molecule (EpCAM) | GC EpCAM-positive | ongoing phase I studies | NCT02725125 NCT03013712 |
| Type of Treatment | Setting | Primary End-Point | Reference/Trial No. |
|---|---|---|---|
| Tremelimumab (IgG2 anti B7 ligand of CTLA-4) | metastatic gastric and esophageal carcinomas (n = 18) | phase II, OS similar to conventional therapy | [86] |
| Tremelimumab + Durvalumab | GC/gastroesophageal junction (GEJ) (n = 135) | phase Ib/II, ongoing | NCT02340975 |
| Ipilimumab (IgG1κ anti CTL-4) | unresectable locally advanced/metastatic GC/ GEJ (n = 143) | phase II, OS similar to conventional therapy | [92] |
| Ipilimumab + Nivolumab (Anti-PD-1) | GC/GEJ pre-operative setting and nivolumab combined with chemo-radiation | phase Ib, ongoing | NCT03044613 |
| Pembrolizumab (IgG4 anti PD-1) | recurrent or metastatic GC/GEJ (n = 39) | phase Ib, 22% partial response, toxicity manageable | [93] |
| PD-L1+ advanced solid tumors including GC/ GEJ (n = 23) | phase Ib, 30% Overall response rate (ORR), median 15 months, better response in patients with high interferon (IFN)-γ gene signature | [94] | |
| recurrent or metastatic GC/GEJ, 2 line (n = 259) | phase II. improved ORR (12%), progression-free survival (PFS) 2 months, and OS 6 months | [95] | |
| recurrent or metastatic GC/GEJ ≥1% PD-L1+, 1 line | phase II. improved ORR (26%), PFS 3 months, and OS not reach in GC with ≥1% expression of PD-L1 | [95] | |
| Pembrolizumab + chemotherapy | recurrent or metastatic GC/GEJ | phase II. improved ORR (60%), PFS 7 months, and OS 14 months | [95] |
| recurrent or metastatic GC/GEJ | phase III ongoing | [96] | |
| Pembrolizumab + Ramucirumab (anti VEGFR2) | locally advanced and unresectable or metastatic GC and other tumors (n = 155) | phase I, study ongoing | [97] |
| Pembrolizumab + Margetuximab (anti HER2) | advanced and metastatic GC/GEJ HER2+ (n = 72) | phase I, dose escalation, safety, efficacy. Study ongoing | [88] |
| neoadjuvant Pembrolizumab + chemo/radiotherapy | resectable, locally advanced GEJ or GC of cardia (n = 68) | phase Ib/II, side effects and best way to give the treatment. Study ongoing | [91] |
| Nivolumab (IgG4 anti PD-1) | recurrent or metastatic GC/GEJ (n = 160) | phase I/II, ORR 24% Nivolumab and Ipilimumab vs 12% Nivolumab in monotherapy with lower toxicity | [98] |
| Nivolumab + Ipilumumab | unresectable advanced or recurrent gastric or GEJ cancer, refractory to, or intolerant of, two or more prior chemotherapy regimens, only patients from Asian countries | phase III, improved OS (26.6% at 1 year, median 5.32 months), PFS (1.61 months). ORR 11.2% | [99] |
| Avelumab (IgG1 anti PD-L1) | advanced or metastatic previously treated solid tumors, including GC/GEJ | phase Ia, dose escalation trial, acceptable toxicity | [100] |
| 3 line recurrent or metastatic GC/GEJ (n = 371) | phase III, Avelumab + best supportive care (BSC) vs BSC ± chemotherapy, study on going at the moment, it did not improve overall survival (OS) | [101] | |
| unresectable, locally advanced or metastatic GC | Avelumab vs continuation of first-line chemotherapy | [102] | |
| Durvalumab (IgG1κ anti PD-L1) | 2/3 line metastatic GC | phase Ib/II Durvalumab or Durvalumab + Tremelimumab vs Tremelimumab alone. study is ongoing | [103] |
| Durvalumab + Ramucirumab (anti VEGFR2) | refractory GC/GEJ (n = 114) | phase Ia/Ib. Safety and efficacy | [104] |
| Durvalumab + Indoleamine 2,3-dioxygenase (IDO) Inhibitor | selected advanced solid tumors (n = 192) | phase I/II safety, tolerability, and efficacy. study ongoing | NCT02318277 |
| Atezolizumab (IgG1κ anti PD-L1) | locally advanced or metastatic solid tumors including GC (n = 661) | phase I. Dose escalation Study of the safety and pharmacokinetics. Study is ongoing | NCT01375842 |
| Atezolizumab + IDO inhibitor | locally advanced, recurrent, or metastatic incurable solid tumors including GC (n = 158) | phase I. Dose limiting toxicity, adverse events. study is ongoing | NCT02471846 |
| Atezolizumab + FLOT (docetaxel, oxaliplatin, and fluorouracil /leucovorin) chemotherapy | locally advanced unresectable or metastatic GC/GEJ (n = 357) | phase Ib/II | NCT03281369 |
| Atezolizumab + Ramucirumab + chemotherapy | GC/GEJ (n = 295) | phase II, Atezolizumab + FLOT vs. FLOT. study is ongoing | NCT03421288 |
| Type of Vaccine | Setting | Primary End-Point | Reference |
|---|---|---|---|
| DC pulsed with melanoma-associated antigen (MAGE) A3 peptides | MAGE-3-expressing advanced GC (n = 12) | phase I, safe and exhibits antitumor effects in some patients | [117] |
| HER2(p369) peptide | advanced or recurrent GC HER2+ (n = 9) | phase I, tumor specific T-cell response | [116] |
| BCG (Bacillus Calmette–Guérin) + chemotherapy | radically resected stage III/IV GC | prolonged 10-year OS (47.1%) as compared to mono-chemotherapy (30%) or surgery alone (15.2%) | [122] |
| gastrin-17 diphtheria toxoid (G17DT) + chemotherapy | metastatic GC/GEJ (n = 94) | phase II, longer TTP and OS in responders | [118] |
| URLC10 or VEGFR1 Epitopes | chemotherapy-resistant advanced GC (n = 14) | phase I, tumor specific T cell responses | [120,124] |
| heat shock protein GP96 + oxaliplatinum | GC (n = 45) | phase II, 81.9% 2-year OS | [121] |
| OTSGC-A24 (5 HLA-A24-restricted peptides DEPDC1, FOXM1, KIF20, URLC10, and VEGFR1) | inoperable/unresectable, metastatic GC, 2 line therapy or greater (n = 23) | favourable results for safety and immune reactivity | [126] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dolcetti, R.; De Re, V.; Canzonieri, V. Immunotherapy for Gastric Cancer: Time for a Personalized Approach? Int. J. Mol. Sci. 2018, 19, 1602. https://doi.org/10.3390/ijms19061602
Dolcetti R, De Re V, Canzonieri V. Immunotherapy for Gastric Cancer: Time for a Personalized Approach? International Journal of Molecular Sciences. 2018; 19(6):1602. https://doi.org/10.3390/ijms19061602
Chicago/Turabian StyleDolcetti, Riccardo, Valli De Re, and Vincenzo Canzonieri. 2018. "Immunotherapy for Gastric Cancer: Time for a Personalized Approach?" International Journal of Molecular Sciences 19, no. 6: 1602. https://doi.org/10.3390/ijms19061602
APA StyleDolcetti, R., De Re, V., & Canzonieri, V. (2018). Immunotherapy for Gastric Cancer: Time for a Personalized Approach? International Journal of Molecular Sciences, 19(6), 1602. https://doi.org/10.3390/ijms19061602