The Neuromuscular Junction and Wide Heterogeneity of Congenital Myasthenic Syndromes


Abstract
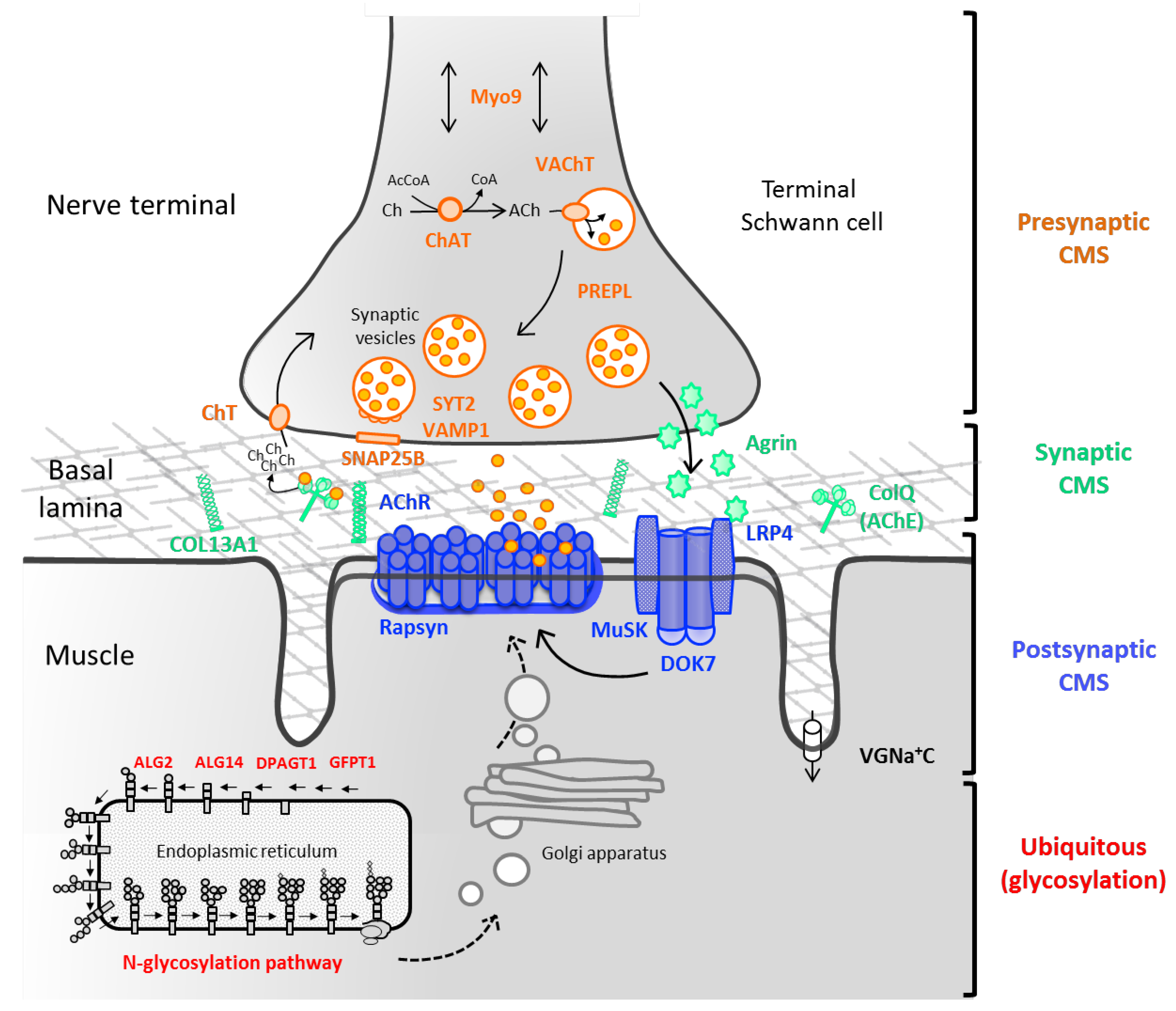
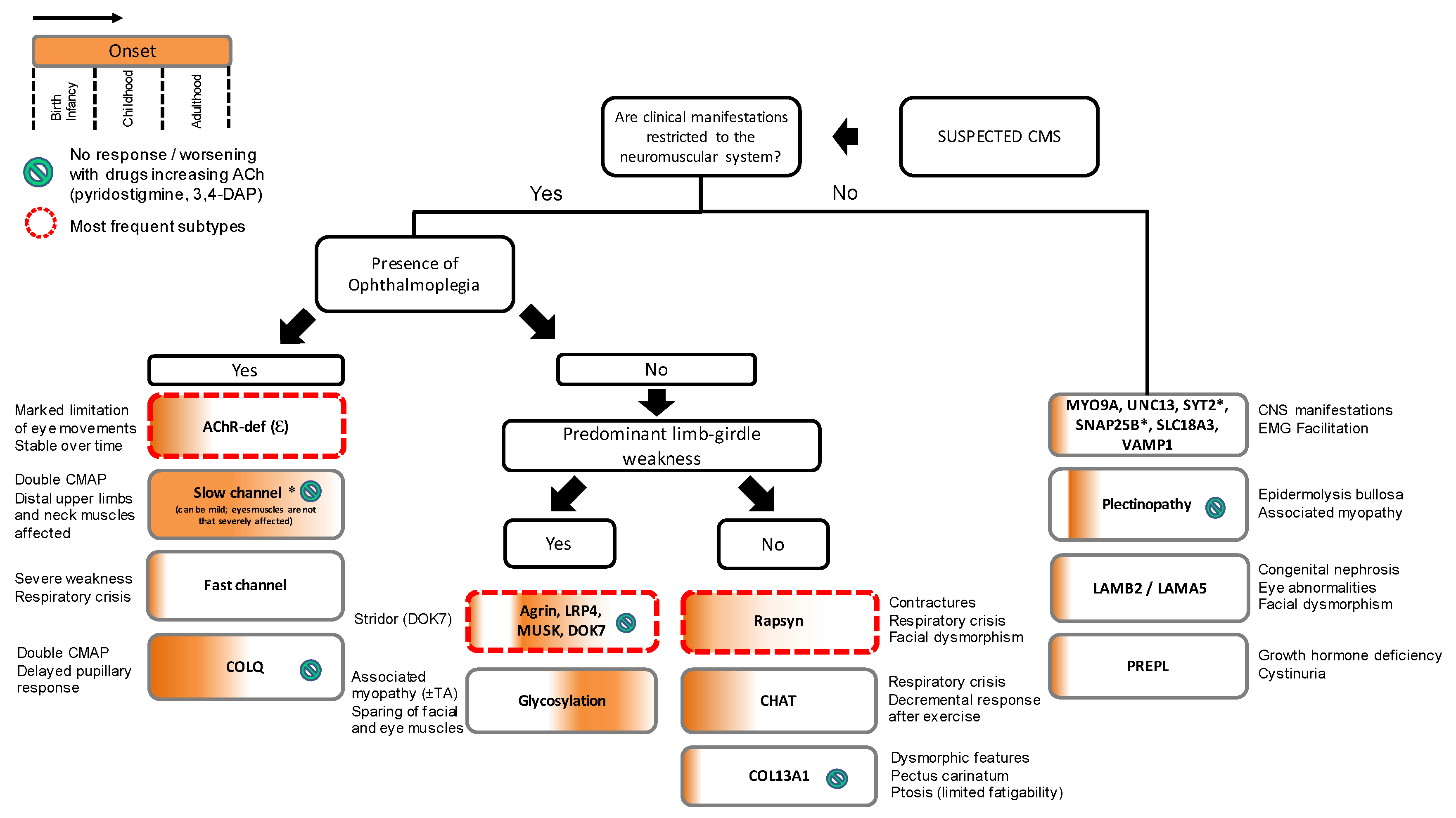
:1. Introduction
2. Presynaptic Syndromes
2.1. Axonal Transport
MYO9A
2.2. Synthesis and Recycling of Acetylcholine
2.2.1. ChAT
2.2.2. PREPL Deficiency
2.2.3. SLC5A7
2.2.4. SLC18A3
2.3. Synaptic Vesicles Exocytosis
2.3.1. SNAP25
2.3.2. SYT2
2.3.3. VAMP1
2.3.4. UNC13A1
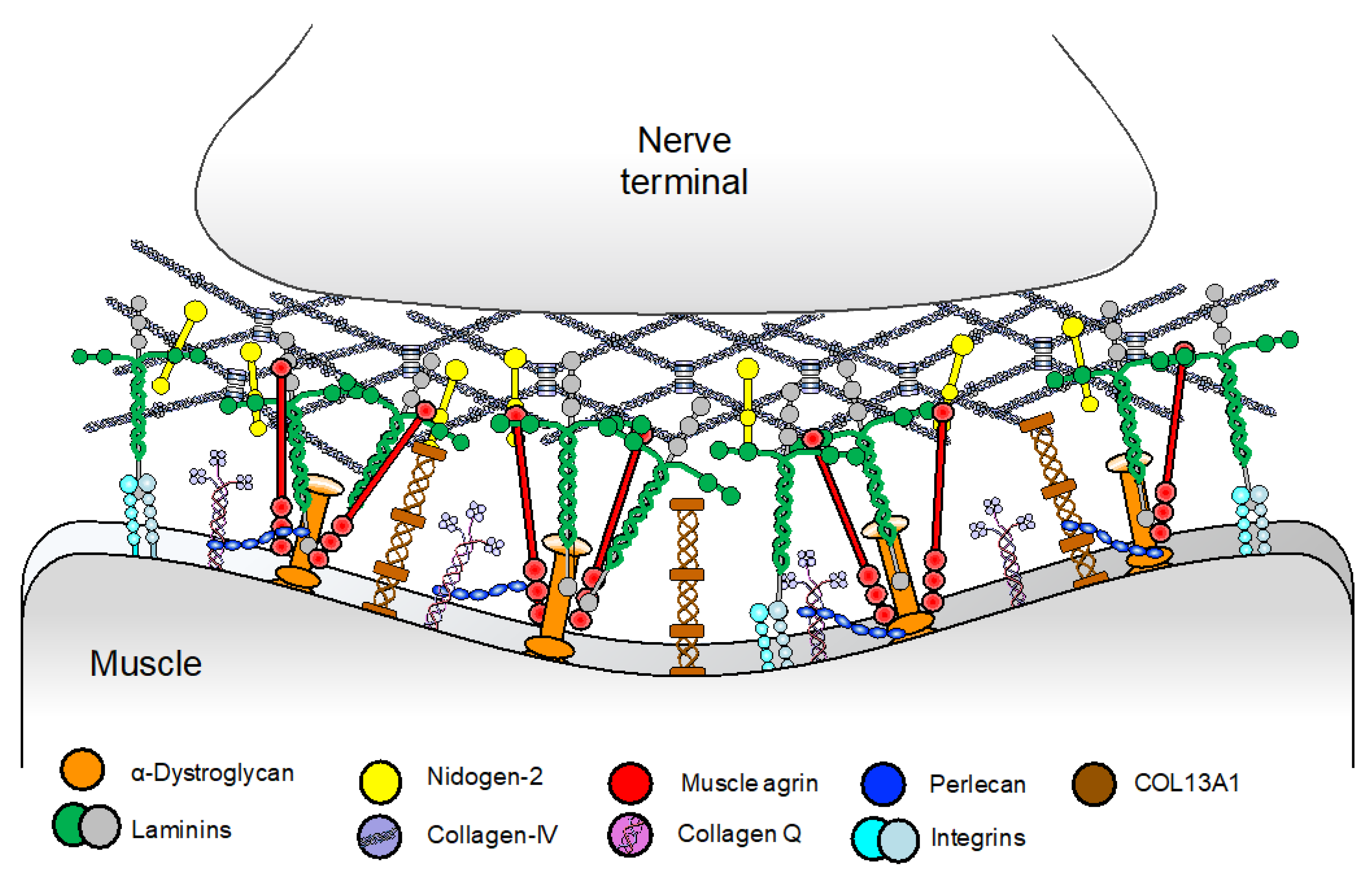
3. Synaptic and Basal-Lamina Associated Syndromes
3.1. COLQ
3.2. COL13A1
3.3. Laminin β2 and Laminin α5 Deficiencies
4. Postsynapatic Syndromes
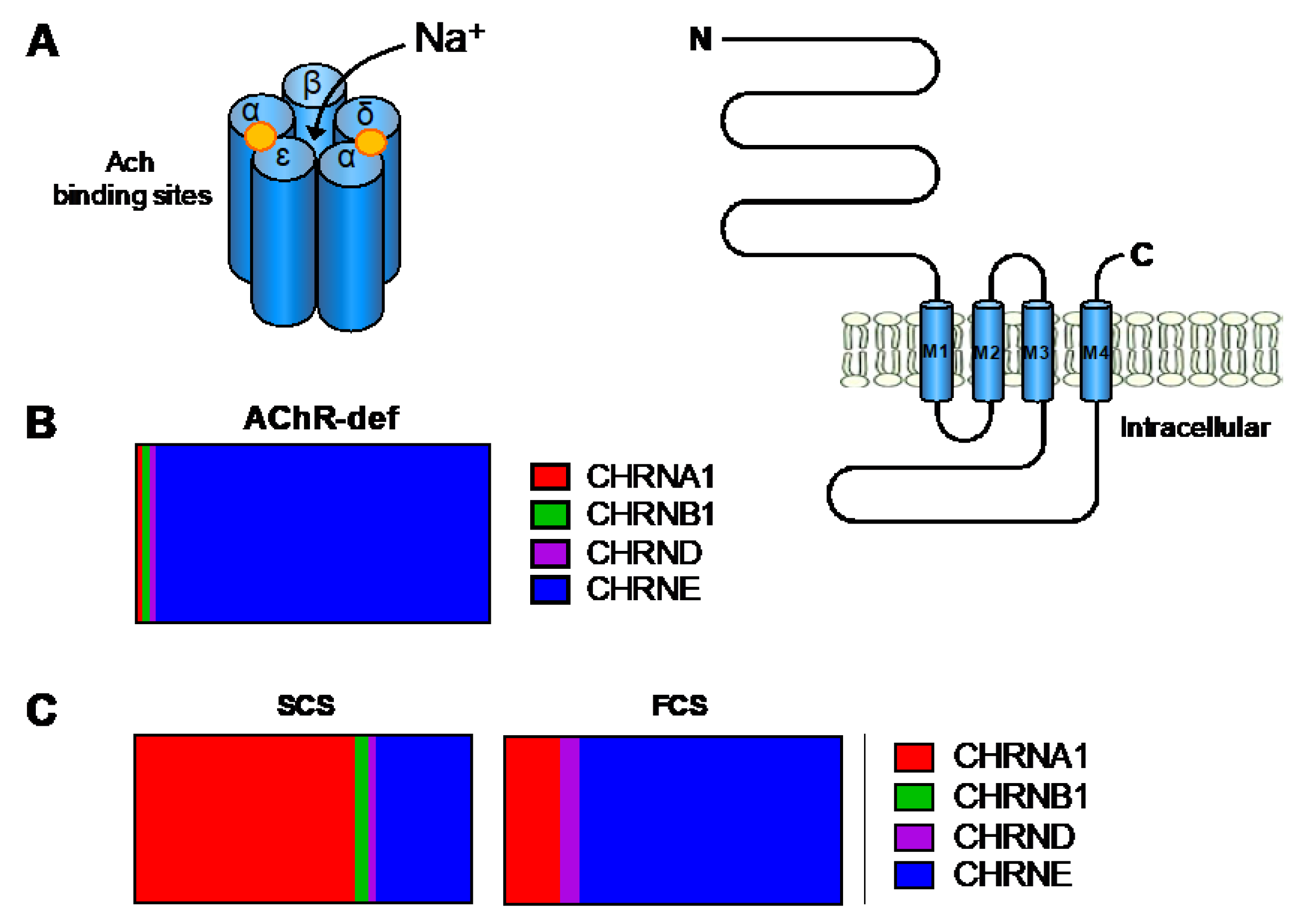
4.1. Primary AChR Deficiency
4.2. Kinetic Abnormalities of the AChR (with or without AChR Deficiency)
4.3. Defects within the AChR-Clustering Pathway
4.3.1. AGRN
4.3.2. LRP4
4.3.3. MuSK
4.3.4. DOK7
4.4. Rapsyn Deficiency
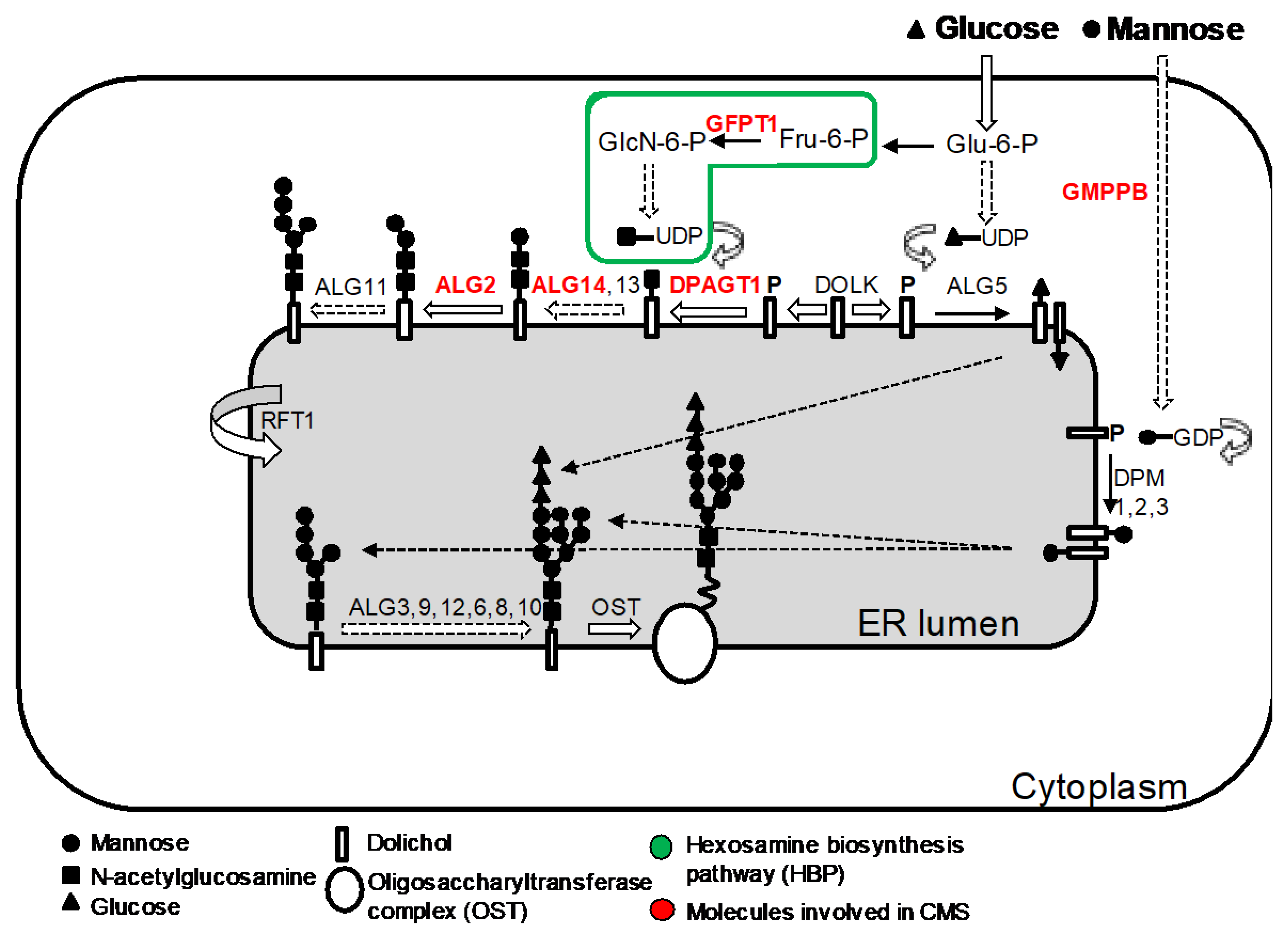
5. CMS Due to Abnormal Glycosylation
5.1. GFPT1
5.2. DPAGT1
5.3. ALG2 and ALG14
5.4. GMPPB
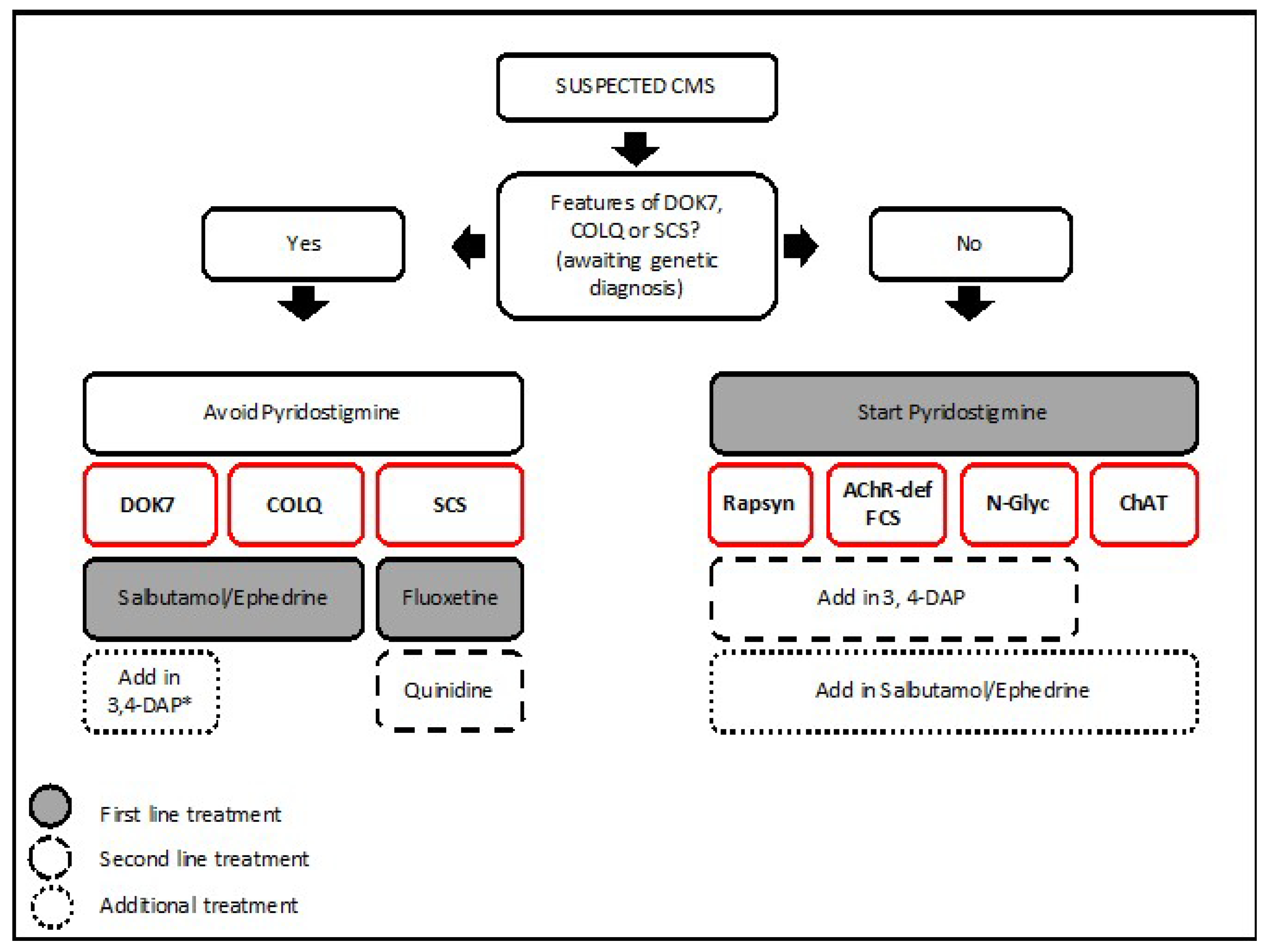
6. Treatment
Acknowledgments
Conflicts of Interest
References
- Palace, J.; Beeson, D. The congenital myasthenic syndromes. J. Neuroimmunol. 2008, 201–202, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Parr, J.R.; Andrew, M.J.; Finnis, M.; Beeson, D.; Vincent, A.; Jayawant, S. How common is childhood myasthenia? The UK incidence and prevalence of autoimmune and congenital myasthenia. Arch. Dis. Child. 2014, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Shen, C.; Bealmear, B.; Ragheb, S.; Xiong, W.-C.; Lewis, R.A.; Lisak, R.P.; Mei, L. Autoantibodies to agrin in myasthenia gravis patients. PLoS ONE 2014, 9, e91816. [Google Scholar] [CrossRef] [PubMed]
- Pevzner, A.; Schoser, B.; Peters, K.; Cosma, N.-C.; Karakatsani, A.; Schalke, B.; Melms, A.; Kröger, S. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J. Neurol. 2012, 259, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Leite, M.I.; Jacob, S.; Viegas, S.; Cossins, J.; Clover, L.; Morgan, B.P.; Beeson, D.; Willcox, N.; Vincent, A. IgG1 antibodies to acetylcholine receptors in “seronegative” myasthenia gravis. Brain 2008, 1940–1952. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Cruz, P.M.; Sewry, C.; Beeson, D.; Jayawant, S.; Squier, W.; McWilliam, R.; Palace, J. Congenital myopathies with secondary neuromuscular transmission defects; A case report and review of the literature. Neuromuscul. Disord. 2014, 24, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; Martens, K.; Francois, I.; Eyskens, F.; Lecointre, C.; Derua, R.; Meulemans, S.; Slootstra, J.W.; Waelkens, E.; de Zegher, F.; et al. Deletion of PREPL, a gene encoding a putative serine oligopeptidase, in patients with hypotonia-cystinuria syndrome. Am. J. Hum. Genet. 2006, 78, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Choi, U.B.; Leitz, J.; Brose, N.; Rhee, J.; Brunger, A.T.; Lai, Y.; Choi, U.B.; Leitz, J.; Rhee, H.J.; et al. Molecular Mechanisms of Synaptic Vesicle Priming Article Molecular Mechanisms of Synaptic Vesicle Priming by Munc13 and Munc18. Neuron 2017, 95, 591–607. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Su, L.; Seven, A.B.; Xu, Y.; Rizo, J. Reconstitution of the Vital Functions of Munc18 and Munc13 in Neurotransmitter Release. Science 2013, 339, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.A.; Finan, D.; Sivaramakrishnan, S.; Spudich, J.A. Principles of Unconventional Myosin Function and Targeting. Annu. Rev. Cell Dev. Biol. 2011, 27, 133–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgman, P.C. Myosin Motor Proteins in the Cell Biology of Axons and Other Neuronal Compartments. In Cell Biology of the Axon; Koenig, E., Ed.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 91–105. [Google Scholar]
- O’Connor, E.; Töpf, A.; Müller, J.S.; Cox, D.; Evangelista, T.; Colomer, J.; Abicht, A.; Senderek, J.; Hasselmann, O.; Yaramis, A.; et al. Identification of mutations in the MYO9A gene in patients with congenital myasthenic syndrome. Brain 2016, 1–11. [Google Scholar] [CrossRef]
- Shen, X.-M.; Crawford, T.O.; Brengman, J.; Acsadi, G.; Iannaconne, S.; Karaca, E.; Khoury, C.; Mah, J.K.; Edvardson, S.; Bajzer, Z.; et al. Functional consequences and structural interpretation of mutations of human choline acetyltransferase. Hum. Mutat. 2011, 32, 1259–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maselli, R.A.; Chen, D.; Delores, M.; Bowe, C.; Fenton, G.; Wollmann, R.L. Choline acetyltransferase mutations in myasthenic syndrome due to deficient acetylcholine resynthesis. Muscle Nerve 2003, 27, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Tsujino, A.; Brengman, J.M.; Harper, C.M.; Bajzer, Z.; Udd, B.; Beyring, R.; Robb, S.; Kirkham, F.J.; Engel, A.G. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc. Natl. Acad. Sci. USA 2001, 98, 2017–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schara, U.; Christen, H.-J.; Durmus, H.; Hietala, M.; Krabetz, K.; Rodolico, C.; Schreiber, G.; Topaloglu, H.; Talim, B.; Voss, W.; et al. Long-term follow-up in patients with congenital myasthenic syndrome due to CHAT mutations. Eur. J. Paediatr. Neurol. 2010, 14, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Lone, A.M.; Nolte, W.M.; Tinoco, A.D.; Saghatelian, A. Peptidomics of the prolyl peptidases. AAPS J. 2010, 12, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Martens, K.; Derua, R.; Meulemans, S.; Waelkens, E.; Jaeken, J.; Matthijs, G.; Creemers, J.W.M. PREPL: A putative novel oligopeptidase propelled into the limelight. Biol. Chem. 2006, 387, 879–883. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-H.; Hersh, L.B. The vesicular acetylcholine transporter interacts with clathrin-associated adaptor complexes AP-1 and AP-2. J. Biol. Chem. 2004, 279, 12580–12587. [Google Scholar] [CrossRef] [PubMed]
- Régal, L.; Shen, X.-M.; Selcen, D.; Verhille, C.; Meulemans, S.; Creemers, J.W.M.; Engel, A.G. PREPL deficiency with or without cystinuria causes a novel myasthenic syndrome. Neurology 2014. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; Haga, T.; Kanai, Y.; Endou, H.; Ishihara, T. Identification and characterization of the high-affinity choline transporter. Nat. Neurosci. 2000, 3, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Bauché, S.; O’Regan, S.; Azuma, Y.; Laffargue, F.; McMacken, G.; Sternberg, D.; Brochier, G.; Buon, C.; Bouzidi, N.; Topf, A.; et al. Impaired Presynaptic High-Affinity Choline Transporter Causes a Congenital Myasthenic Syndrome with Episodic Apnea. Am. J. Hum. Genet. 2016, 99, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Arvidsson, U.L.F.; Riedl, M.; Elde, R.; Meister, B. Vesicular acetylcholine transporter (VAChT) protein: A novel and unique marker for cholinergic neurons in the central and peripheral nervous systems. J. Comp. Neurol. 1997, 467, 454–467. [Google Scholar] [CrossRef]
- O’Grady, G.L.; Verschuuren, C.; Yuen, M.; Webster, R.; Menezes, M.; Fock, J.M.; Pride, N.; Best, H.A.; Benavides Damm, T.; Turner, C.; et al. Variants in SLC18A3, vesicular acetylcholine transporter, cause congenital myasthenic syndrome. Neurology 2016, 87, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Aran, A.; Renbaum, P.; Oliphant, S.; Weinberg, A.; Zeligson, S.; Lee, M.K.; Samson, A.O.; Parsons, S.M.; King, M. Vesicular acetylcholine transporter defect underlies devastating congenital myasthenia syndrome. Neurology 2017, 88, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.A.; Scheller, R.H.; Medical, H.H. Snare-mediated membrane fusion. Nature 2001, 2, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Eaton, L.M.; Lambert, E.H. Electromyography and electric stimulation of nerves in diseases of motor unit; observations on myasthenic syndrome associated with malignant tumors. J. Am. Med. Assoc. 1957, 163, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Oyler, G.A.; Higgins, G.A.; Hart, R.A.; Battenberg, E.; Billingsley, M.; Bloom, F.E.; Wilson, M.C. The Identification of a Novel Synaptosomal-associated Protein, SNAP-25, Differentially Expressed by Neuronal Subpopulations. J. Cell Biol. 1989, 109, 3039–3052. [Google Scholar] [CrossRef] [PubMed]
- Chapman, E.R.; An, S.; Barton, N.; Jahn, R. SNAP-25, a t-SNARE Which Binds to Both Syntaxin and Synaptobrevin via Domains That May Form Coiled Coils. J. Biol. Chem. 1994, 269, 27427–27432. [Google Scholar] [PubMed]
- Shen, X.; Brengman, J.; Engel, A.G. Mutant SNAP25B causes myasthenia, cortical hyperexcitability, ataxia, and intellectual disability. Neurology 2014, 83, 2247–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Z.P.; Melicoff, E.; Padgett, D.; Liu, Y.; Teich, A.F.; Dickey, B.F.; Lin, W.; Adachi, R.; Su, T.C. Synaptotagmin-2 Is Essential for Survival and Contributes to Ca2+ Triggering of Neurotransmitter Release in Central and Neuromuscular Synapses. J. Neurosci. 2006, 26, 13493–13504. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, D.N.; Horvath, R.; Sowden, J.E.; Gonzalez, M.; Gonzales, M.; Sanchez-Mejias, A.; Guan, Z.; Whittaker, R.G.; Almodovar, J.L.; Lane, M.; et al. Synaptotagmin 2 mutations cause an autosomal-dominant form of lambert-eaton myasthenic syndrome and nonprogressive motor neuropathy. Am. J. Hum. Genet. 2014, 95, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, R.G.; Herrmann, D.N.; Bansagi, B.; Hasan, B.A.S.; Lofra, R.M.; Logigian, E.L.; Sowden, J.E.; Almodovar, J.L.; Littleton, J.T.; Zuchner, S.; et al. Electrophysiologic features of SYT2 mutations causing a treatable neuromuscular syndrome. Neurology 2015, 85, 1964–1971. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sugiura, Y.; Lin, W. The role of Synaptobrevin1/VAMP1 in Ca2+-triggered neurotransmitter release at the mouse neuromuscular junction. J. Physiol. 2011, 7, 1603–1618. [Google Scholar] [CrossRef] [PubMed]
- Elferink, L.A.; Trimbles, W.S. Two Vesicle-associated Membrane Protein Genes Are Differentially Expressed in the Rat Central Nervous System. J. Biol. Chem. 1989, 264, 11061–11064. [Google Scholar] [PubMed]
- Salpietro, V.; Lin, W.; Vedove, A.D.; Storbeck, M.; Liu, Y.; Efthymiou, S.; Manole, A.; Ye, Q.; Saggar, A.; Mcelreavey, K.; et al. Homozygous Mutations in VAMP1 cause a presynaptic congenital myasthenic syndrome. Ann. Neurol. 2017, 81, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Brose, N.; Hofmann, K.; Hata, Y.; Su, T.C. Mammalian Homologues of Caenorhabditis elegans unc-13 Gene Define Novel Family of C2-domain Proteins. J. Biol. Chem. 1995, 270, 25273–25280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, A.G.; Selcen, D.; Shen, X.-M.; Milone, M.; Harper, C.M. Loss of MUNC13-1 function causes microcephaly, cortical hyperexcitability, and fatal myasthenia. Neurol. Genet. 2016, 5, e105. [Google Scholar] [CrossRef] [PubMed]
- Sanes, J.R. The Basement Membrane/Basal Lamina of Skeletal Muscle. J. Biol. Chem. 2003, 287, 12601–12604. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Fu, A.K.Y.; Ip, N.Y. Molecular mechanisms underlying maturation and maintenance of the vertebrate neuromuscular junction. Trends Neurosci. 2012, 35, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Mouw, J.K.; Ou, G.; Weaver, V.M.; Regeneration, T.; Francisco, S.; Francisco, S.; Francisco, S.; Sciences, T.; Francisco, S.; Francisco, S.; et al. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Mokkapati, S.; Nischt, R.; Smyth, N.; Ho, M.S.P.; Bo, K. Nidogens—Extracellular Matrix Linker Molecules. Microsc. Res. Tech. 2008, 395, 387–395. [Google Scholar] [CrossRef]
- Fox, M.A.; Ho, M.S.P.; Smyth, N.; Sanes, J.R. A synaptic nidogen: Developmental regulation and role of nidogen-2 at the neuromuscular junction. Neural Dev. 2008, 17, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latvanlehto, A.; Fox, M.A.; Sormunen, R.; Tu, H.; Oikarainen, T.; Koski, A.; Naumenko, N.; Shakirzyanova, A.; Kallio, M.; Ilves, M.; Giniatullin, R.; et al. Muscle-derived collagen XIII regulates maturation of the skeletal neuromuscular junction. J. Neurosci. 2010, 30, 12230–12241. [Google Scholar] [CrossRef] [PubMed]
- Denzer, A.J.; Brandenberger, R.; Gesemann, M.; Chiquet, M.; Ruegg, M.A. Agrin Binds to the Nerve-Muscle Basal Lamina via Laminin. J. Cell Biol. 1997, 137, 671–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, J.; Bowen, D.C.; Hall, Z.W. Dystroglycan Binds Nerve and Muscle Agrin. Neuron 1994, 13, 103–115. [Google Scholar] [CrossRef]
- Samuel, M.A.; Valdez, G.; Tapia, J.C.; Lichtman, J.W.; Sanes, J.R. Agrin and Synaptic Laminin Are Required to Maintain Adult Neuromuscular Junctions. PLos ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferns, M.; Deiner, M.; Hall, Z. Agrin-induced Acetylcholine Receptor Clustering in Mammalian Muscle Rexluires Tyrosine Phosphorylation. J. Cell Biol. 1996, 132, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Arikawa-hirasawa, E.; Rossi, S.G.; Rotundo, R.L.; Yamada, Y. Absence of acetylcholinesterase at the neuromuscular junctions of perlecan-null mice. Nat. Neurosci. 2002. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.B.; Ali, A.; Dagget, D.; Rauvala, H.; Hassell, J.; Smalheiser, N. The relationship between perlecan and dystroglycan and its implication in the formation of the neuromuscular junction. Cell Adhes. Commun. 1998, 5, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Brengman, J.; Tsujino, A.; Engel, A.G. Human endplate acetylcholinesterase deficiency caused by mutations in the collagen-like tail subunit (ColQ) of the asymmetric enzyme. Proc. Natl. Acad. Sci. USA 1998, 95, 9654–9659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legay, C. Congenital myasthenic syndromes with acetylcholinesterase deficiency, the pathophysiological mechanisms. Ann. N. Y. Acad. Sci. 2018, 1413, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Cartaud, A.; Strochlic, L.; Guerra, M.; Blanchard, B.; Lambergeon, M.; Krejci, E.; Cartaud, J.; Legay, C. MuSK is required for anchoring acetylcholinesterase at the neuromuscular junction. J. Cell Biol. 2004, 165, 505–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaylova, V.; Müller, J.S.; Vilchez, J.J.; Salih, M.A.; Kabiraj, M.M.; D’Amico, A.; Bertini, E.; Wölfle, J.; Schreiner, F.; Kurlemann, G.; Rasic, V.M.; et al. Clinical and molecular genetic findings in COLQ-mutant congenital myasthenic syndromes. Brain 2008, 131, 747–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchinson, D.O.; Walls, T.J.; Nakano, S.; Camp, S.; Taylor, P.; Harper, C.M.; Groover, R.V.; Peterson, H.A.; Jamieson, D.G.; Engel, A.G. Congenital endplate acetylcholinesterase deficiency. Brain 1993, 116, 633–653. [Google Scholar] [CrossRef] [PubMed]
- Bestue-Cardiel, M.; Sáenz de Cabezón-Alvarez, A.; Capablo-Liesa, J.L.; López-Pisón, J.; Peña-Segura, J.L.; Martin-Martinez, J.; Engel, A.G. Congenital endplate acetylcholinesterase deficiency responsive to ephedrine. Neurology 2005, 65, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Nykvist, P.; Tu, H.; Ivaska, J.; Ka, J.; Pihlajaniemi, T. Distinct Recognition of Collagen Subtypes by α1β1 and α2β1 Integrins. J. Biol. Chem. 2000, 275, 8255–8261. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Sasaki, T.; Snellman, A.; Göhring, W.; Pirilä, P.I.; Timpl, R.; Pihlajaniemi, T. The type XIII collagen ectodomain is a 150-nm rod and capable of binding to fibronectin, nidogen-2, perlecan, and heparin. J. Biol. Chem. 2002, 277, 23092–23099. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.V.; Cossins, J.; Rodríguez Cruz, P.M.; Parry, D.A.; Maxwell, S.; Martínez-Martínez, P.; Riepsaame, J.; Abdelhamed, Z.A.; Lake, A.V.R.; Moran, M.; et al. Congenital Myasthenic Syndrome Type 19 Is Caused by Mutations in COL13A1, Encoding the Atypical Non-fibrillar Collagen Type XIII α1 Chain. Am. J. Hum. Genet. 2015, 97, 878–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maselli, R.A.; Ng, J.J.; Anderson, J.A.; Cagney, O.; Arredondo, J.; Williams, C.; Wessel, H.B.; Abdel-Hamid, H.; Wollmann, R.L. Mutations in LAMB2 causing a severe form of synaptic congenital myasthenic syndrome. J. Med. Genet. 2009, 46, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Zenker, M.; Aigner, T.; Wendler, O.; Tralau, T.; Mu, H.; Royer-pokora, B.; Wu, E.; Fenski, R.; Pitz, S.; Cochat, P.; et al. Human laminin b 2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum. Mol. Genet. 2004, 13, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Maselli, R.A.; Chong, J.X.; Arredondo, J.; Vázquez, J.; Bamshad, M.J.; Nickerson, D.A.; Lara, M.; Ng, F.; Lo, V.L.; Pytel, P.; Mcdonald, C.M. Presynaptic congenital myasthenic syndrome with a homozygous sequence variant in LAMA5 combines myopia, facial tics, and failure of neuromuscular transmission. Am. J. Med. Genet. Part A 2017, 2240–2245. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Newson-Davis, J.; Wray, D.; Shillito, P.; Harrison, J.; Betty, B.; Beeson, D.; Mills, K.; Palace, J.; Molenaar, P.; et al. Clinical and Experimental Observations in Patients with Congenital Myasthenic Syndromes. Ann. N. Y. Acad. Sci. 1993, 681, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Burke, G.; Cossins, J.; Maxwell, S.; Robb, S.; Nicolle, M.; Vincent, A.; Newson-Davis, J.; Palace, J.; Beeson, D. Distinct phenotypes of congenital acetylcholine receptor deficiency. Neuromuscul. Disord. 2004, 14, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Abicht, A.; Müller, J.S.; Lochmüller, H. Congenital Myasthenic Syndromes; Pagon, R., Adam, M., Ardinger, H., Eds.; University of Washington: Seattle, WA, USA, 2016. [Google Scholar]
- Cossins, J.; Webster, R.; Maxwell, S.; Burke, G.; Vincent, A.; Beeson, D. A mouse model of AChR deficiency syndrome with a phenotype reflecting the human condition. Hum. Mol. Genet. 2004, 13, 2947–2957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, A.G. The Therapy of Congenital Myasthenic Syndromes. Neurotherapeutics 2007, 4, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Cruz, P.M.; Palace, J.; Ramjattan, H.; Robb, S.A.; Beeson, D. Salbutamol and ephedrine in the treatment of severe AChR-deficiency syndromes. Neurology 2015, 85, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Hutchinson, D.; Milone, M.; Brengman, J.M.; Bouzatt, C.; Sinet, S.M.; Engel, A.G. Congenital myasthenic syndrome caused by prolonged acetylcholine receptor channel openings due to a mutation in the M2 domain of the E subunit. Proc. Natl. Acad. Sci. USA 1995, 92, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Pytel, P.; Gomez, C.M. Selective inhibition of caspases in skeletal muscle reverses the apoptotic synaptic degeneration in slow-channel myasthenic syndrome. Hum. Mol. Genet. 2014, 23, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Chaouch, A.; Müller, J.S.; Guergueltcheva, V.; Dusl, M.; Schara, U.; Rakocević-Stojanović, V.; Lindberg, C.; Scola, R.H.; Werneck, L.C.; Colomer, J.; et al. A retrospective clinical study of the treatment of slow-channel congenital myasthenic syndrome. J. Neurol. 2012, 259, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Wang, H.; Milone, M.; Bren, N.; Brengman, J.M.; Nakano, S.; Quiram, P.; Pruitt, J.N.; Sine, S.M.; Engel, A.G. Congenital Myasthenic Syndrome Caused by Decreased Agonist Binding Affinity Due to a Mutation in the Acetylcholine Receptor epsilon Subunit. Neuron 1996, 17, 157–170. [Google Scholar] [CrossRef]
- Palace, J.; Lashley, D.; Bailey, S.; Jayawant, S.; Carr, A.; McConville, J.; Robb, S.; Beeson, D. Clinical features in a series of fast channel congenital myasthenia syndrome. Neuromuscul. Disord. 2012, 22, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Sine, S.M.; Wang, H.; Shen, X.-M.; Lee, W.Y.; Engel, A.G. Mechanistic diversity underlying fast channel congenital myasthenic syndrome. Ann. N. Y. Acad. Sci. 2003, 998, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xiong, W.C.; Mei, L. To build a synapse: Signaling pathways in neuromuscular junction assembly. Development 2010, 1033, 1017–1033. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Stiegler, A.L.; Cameron, T.O.; Hallock, P.T.; Gomez, A.M.; Huang, J.H.; Hubbard, S.R.; Dustin, M.L.; Burden, S.J. Lrp4 Is a Receptor for Agrin and Forms a Complex with MuSK. Cell 2008, 135, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burden, S.J.; Yumoto, N.; Zhang, W. The role of MuSK in synapse formation and neuromuscular disease. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hallock, P.T.; Xu, C.; Park, T.; Neubert, T.A.; Curran, T.; Burden, S.J. Dok-7 regulates neuromuscular synapse formation by recruiting Crk and Crk-L. Genes Dev. 2010, 24, 2451–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Z.G.; Wang, Q.; Zhou, J.Z.; Wang, J.; Luo, Z.; Liu, M.; He, X.; Wynshaw-boris, A.; Xiong, W.C.; Lu, B.; Mei, L. Regulation of AChR Clustering by Dishevelled Interacting with MuSK and PAK1. Neuron 2002, 35, 489–505. [Google Scholar] [CrossRef] [Green Version]
- Henriquez, J.P.; Webb, A.; Bence, M.; Bildsoe, H.; Sahores, M.; Hughes, S.M.; Salinas, P.C. Wnt signaling promotes AChR aggregation at the neuromuscular synapse in collaboration with agrin. Proc. Natl. Acad. Sci. USA 2008, 105, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Dominguez, B.; Yang, J.; Aryal, P.; Brandon, E.P.; Gage, F.H.; Lee, K.F. Neurotransmitter acetylcholine negatively regulates neuromuscular synapse formation by a Cdk5-dependent mechanism. Neuron 2005, 46, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Qian, L.; Yang, Z.; Huang, Y.; Ngo, S.T.; Ruan, N.; Wang, J.; Schneider, C.; Noakes, P.G.; Ding, Y.; et al. Rapsyn Interaction with Calpain Stabilizes AChR Clusters at the Neuromuscular Junction. Neuron 2007, 55, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de la Monte, S.; Dikkes, P.; Tsai, L.-H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Maselli, R.A.; Fernandez, J.M.; Arredondo, J.; Navarro, C.; Ngo, M.; Beeson, D.; Cagney, Ó.; Williams, D.C.; Wollmann, R.L.; Ferns, V.Y.M.J. LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural (z-) agrin. Hum. Genet. 2012, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Goillot, E.; Huze, C.; Gaudon, K.; Ammar, A.B.; Chaboud, A.; Grosjean, I.; Lecuyer, H.; Koenig, J.; Fournier, E.; et al. Identification of an Agrin Mutation that Causes Congenital Myasthenia and Affects Synapse Function. Am. J. Hum. Genet. 2009, 155–167. [Google Scholar] [CrossRef]
- Xi, J.; Yan, C.; Liu, W.W.; Qiao, K.; Lin, J.; Tian, X.; Wu, H.; Lu, J.; Wong, L.J.; Beeson, D.; et al. Novel SEA and LG2 Agrin mutations causing congenital Myasthenic syndrome. Orphanet J. Rare Dis. 2017, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Karakaya, M.; Ceyhan-Birsoy, O.; Beggs, A.H.; Topaloglu, H. A novel missense variant in the AGRN gene; congenital myasthenic syndrome presenting with head drop. J. Clin. Neuromuscul. Dis. 2017, 18, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Nicole, S.; Chaouch, A.; Torbergsen, T.; Bauché, S.; de Bruyckere, E.; Fontenille, M.-J.; Horn, M.A.; van Ghelue, M.; Løseth, S.; Issop, Y.; Cox, D.; et al. Agrin mutations lead to a congenital myasthenic syndrome with distal muscle weakness and atrophy. Brain 2014, 137, 2429–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Coldefy, A.-S.; Hubbard, S.R.; Burden, S.J. Agrin binds to the N-terminal region of Lrp4 protein and stimulates association between Lrp4 and the first immunoglobulin-like domain in muscle-specific kinase (MuSK). J. Biol. Chem. 2011, 286, 40624–40630. [Google Scholar] [CrossRef] [PubMed]
- Ohkawara, B.; Cabrera-Serrano, M.; Nakata, T.; Milone, M.; Asai, N.; Ito, K.; Ito, M.; Masuda, A.; Ito, Y.; Engel, A.G.; et al. LRP4 third β-propeller domain mutations cause novel congenital myasthenia by compromising agrin-mediated MuSK signaling in a position-specific manner. Hum. Mol. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Selcen, D.; Ohkawara, B.; Shen, X.-M.; McEvoy, K.; Ohno, K.; Engel, A.G. Impaired Synaptic Development, Maintenance, and Neuromuscular Transmission in LRP4-Related Myasthenia. JAMA Neurol. 2015, 72, 889–896. [Google Scholar] [CrossRef] [PubMed]
- DeChiara, T.M.; Bowen, D.C.; Valenzuela, D.M.; Simmons, M.V.; Poueymirou, W.T.; Thomas, S.; Kinetz, E.; Compton, D.L.; Rojas, E.; Park, J.S.; et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell 1996, 85, 501–512. [Google Scholar] [CrossRef]
- Chevessier, F.; Faraut, B.; Ravel-Chapuis, A.; Richard, P.; Gaudon, K.; Bauché, S.; Prioleau, C.; Herbst, R.; Goillot, E.; Ioos, C.; et al. MUSK, a new target for mutations causing congenital myasthenic syndrome. Hum. Mol. Genet. 2004, 13, 3229–3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maselli, R.A.; Arredondo, J.; Cagney, O.; Ng, J.J.; Anderson, J.A.; Williams, C.; Gerke, B.J.; Soliven, B.; Wollmann, R.L. Mutations in MUSK causing congenital myasthenic syndrome impair MuSK–Dok-7 interaction. Hum. Mol. Genet. 2010, 19, 2370–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammar, A.B.; Soltanzadeh, P.; Bauché, S.; Richard, P.; Goillot, E.; Herbst, R.; Gaudon, K.; Huze, C.; Schaeffer, L.; Yamanashi, Y.; et al. A Mutation Causes MuSK Reduced Sensitivity to Agrin and Congenital Myasthenia. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Owen, D.; Töpf, A.; Preethish-Kumar, V.; Lorenzoni, P.J.; Vroling, B.; Scola, R.H.; Dias-Tosta, E.; Geraldo, A.; Polavarapu, K.; Nashi, S.; et al. Recessive variants of MuSK are associated with late onset CMS and predominant limb girdle weakness. Am. J. Med. Genet. Part A 2018, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Inoue, A.; Okada, M.; Murata, Y.; Kakuta, S.; Jigami, T.; Kubo, S.; Shiraishi, H.; Eguchi, K.; Motomura, M.; et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science 2006, 312, 1802–1805. [Google Scholar] [CrossRef] [PubMed]
- Beeson, D.; Higuchi, O.; Palace, J.; Cossins, J.; Spearman, H.; Maxwell, S.; Newsom-Davis, J.; Burke, G.; Fawcett, P.; Motomura, M.; et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science 2006, 313, 1975–1978. [Google Scholar] [CrossRef] [PubMed]
- Cossins, J.; Liu, W.W.; Belaya, K.; Maxwell, S.; Oldridge, M.; Lester, T.; Robb, S.; Beeson, D. The spectrum of mutations that underlie the neuromuscular junction synaptopathy in DOK7 congenital myasthenic syndrome. Hum. Mol. Genet. 2012, 21, 3765–3775. [Google Scholar] [CrossRef] [PubMed]
- Palace, J.; Lashley, D.; Newsom-davis, J.; Cossins, J.; Maxwell, S.; Kennett, R.; Jayawant, S.; Yamanashi, Y.; Beeson, D. Clinical features of the DOK7 neuromuscular junction synaptopathy. Brain 2007, 130, 1507–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lashley, D.; Palace, J.; Jayawant, S.; Robb, S.; Beeson, D. Ephedrine treatment in congenital myasthenic syndrome due to mutations in DOK7. Neurology 2010, 74, 1517–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobel, A.; Weber, M.; JP, C. Large-Scale Purification of the Acetylcholine-Receptor Protein in Its Membrane-Bound and Detergent -Ext ract ed Forms from. Eur. J. Biochem. 1977, 80, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Noakes, P.G.; Mudd, J.; Nichol, M.; Chu, G.C.; Sanes, J.R.; Merlie, J.P. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature 1995, 377, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Moransard, M.; Borges, L.S.; Willmann, R.; Marangi, P.A.; Brenner, H.R.; Ferns, M.J.; Fuhrer, C. Agrin Regulates Rapsyn Interaction with Surface Acetylcholine Receptors, and This Underlies Cytoskeletal Anchoring and Clustering. J. Biol. Chem. 2003, 278, 7350–7359. [Google Scholar] [CrossRef] [PubMed]
- Apel, E.D.; Roberds, S.L.; Campbell, K.P.; Merlie, J.P. Rapsyn May Function as a Link between the Acetylcholine Receptor and the Agrin-Binding Glycoprotein Complex. Neuron 1995, 15, 115–126. [Google Scholar] [CrossRef]
- Zuber, B.; Unwin, N. Structure and superorganization of acetylcholine receptor–rapsyn complexes. Proc. Natl. Acad. Sci. USA 2013, 110, 10622–10627. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Engel, A.G.; Shen, X.-M.; Selcen, D.; Brengman, J.; Harper, C.M.; Tsujino, A.; Milone, M. Rapsyn mutations in humans cause endplate acetylcholine-receptor deficiency and myasthenic syndrome. Am. J. Hum. Genet. 2002, 70, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Cossins, J.; Burke, G.; Maxwell, S.; Spearman, H.; Man, S.; Kuks, J.; Vincent, A.; Palace, J.; Fuhrer, C.; Beeson, D. Diverse molecular mechanisms involved in AChR deficiency due to rapsyn mutations. Brain 2006, 129, 2773–2783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, G.; Cossins, J.; Maxwell, S.; Owens, G.; Vincent, A.; Robb, S.; Nicolle, M.; Jones, D.H.; Davis, J.N.; Palace, J.; et al. Rapsyn mutations in hereditary myasthenia Distinct early- and late-onset phenotypes. Neurology 2003. [Google Scholar] [CrossRef]
- Milone, M.; Shen, X.M.; Selcen, D.; Ohno, K.; Brengman, J.; Iannaccone, S.T.; Harper, C.M.; Engel, A.G. Myasthenic syndrome due to defects in rapsyn: Clinical and molecular findings in 39 patients. Neurology 2009, 73, 228–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, J.; Harrison, B.J.; Spearman, H.; Cossins, J.; Vermeer, S.; Naudin, L.; Morgan, N.V.; Beeson, D.; Maher, E.R. Mutation Analysis of CHRNA1, CHRNB1, CHRND, and RAPSN Genes in Multiple Pterygium Syndrome/Fetal Akinesia Patients. Am. J. Hum. Genet. 2008, 88, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Natera-de Benito, D.; Bestué, M.; Vilchez, J.J.; Evangelista, T.; Töpf, A.; García-ribes, A. Long-term follow-up in patients with congenital myasthenic syndrome due to RAPSN mutations. Neuromuscul. Disord. 2016, 26, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; Matthijs, G. Congenital disorders of glycosylation. Annu. Rev. Genom. Hum. Genet. 2001, 2, 129–151. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Rush, J.S.; Karaoglu, D.; Krasnewich, D.; Lubinsky, M.S.; Waechter, C.J.; Gilmore, R.; Freeze, H.H. Deficiency of UDP-GlcNAc:Dolichol Phosphate N-Acetylglucosamine-1 Phosphate Transferase (DPAGT1) causes a novel congenital disorder of Glycosylation Type Ij. Hum. Mutat. 2003, 22, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Thiel, C.; Schwarz, M.; Peng, J.; Grzmil, M.; Hasilik, M.; Braulke, T.; Kohlschütter, A.; von Figura, K.; Lehle, L.; Körner, C. A new type of congenital disorders of glycosylation (CDG-Ii) provides new insights into the early steps of dolichol-linked oligosaccharide biosynthesis. J. Biol. Chem. 2003, 278, 22498–22505. [Google Scholar] [CrossRef] [PubMed]
- Belaya, K.; Finlayson, S.; Cossins, J.; Liu, W.W.; Maxwell, S.; Palace, J.; Beeson, D. Identification of DPAGT1 as a new gene in which mutations cause a congenital myasthenic syndrome. Ann. N. Y. Acad. Sci. 2012, 1275, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Senderek, J.; Müller, J.S.; Dusl, M.; Strom, T.M.; Guergueltcheva, V.; Diepolder, I.; Laval, S.H.; Maxwell, S.; Cossins, J.; Krause, S.; et al. Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am. J. Hum. Genet. 2011, 88, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Cossins, J.; Belaya, K.; Hicks, D.; Salih, M.A.; Finlayson, S.; Carboni, N.; Liu, W.W.; Maxwell, S.; Zoltowska, K.; Farsani, G.T.; Laval, S.; et al. Congenital myasthenic syndromes due to mutations in ALG2 and ALG14. Brain 2013, 136, 944–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehle, V.M.; Walcott, E.C.; Nishizaki, T.; Sumikawa, K. N-glycosylation at the conserved sites ensures the expression of properly folded functional ACh receptors. Brain Res. Mol. Brain Res. 1997, 45, 219–229. [Google Scholar] [CrossRef]
- Zoltowska, K.; Webster, R.; Finlayson, S.; Maxwell, S.; Cossins, J.; Müller, J.; Lochmüller, H.; Beeson, D. Mutations in GFPT1 that underlie limb-girdle congenital myasthenic syndrome result in reduced cell-surface expression of muscle AChR. Hum. Mol. Genet. 2013, 22, 2905–2913. [Google Scholar] [CrossRef] [PubMed]
- Haltiwanger, R.S.; Lowe, J.B. Role of glycosylation in development. Annu. Rev. Biochem. 2004, 73, 491–537. [Google Scholar] [CrossRef] [PubMed]
- Selcen, D.; Shen, X.-M.; Milone, M.; Brengman, J.; Ohno, K.; Deymeer, F.; Finkel, R.; Rowin, J.; Engel, A.G. GFPT1-myasthenia: Clinical, structural, and electrophysiologic heterogeneity. Neurology 2013, 81, 370–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauché, S.; Vellieux, G.; Sternberg, D.; Fontenille, M.J.; Bruyckere, E. De; Sophie, C.; Guy, D.; Messéant, J.; Wolf, L.; Fardeau, M.; Lacène, E.; et al. Mutations in GFPT1-related congenital myasthenic syndromes are associated with synaptic morphological defects and underlie a tubular aggregate myopathy with synaptopathy. J. Neurol. 2017, 264, 1791–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, S.-Y.; Kim, H.-S.; Jang, H.-J.; Park, Y.-E.; Kim, D.-S. Limb-girdle myasthenia with tubular aggregates associated with novel GFPT1 mutations. Muscle Nerve 2012, 46, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Guergueltcheva, V.; Müller, J.S.; Dusl, M.; Senderek, J.; Oldfors, A.; Lindbergh, C.; Maxwell, S.; Colomer, J.; Mallebrera, C.J.; Nascimento, A.; et al. Congenital myasthenic syndrome with tubular aggregates caused by GFPT1 mutations. J. Neurol. 2012, 259, 838–850. [Google Scholar] [CrossRef] [PubMed]
- Bretthauer, R.K. Structure, expression, and regulation of UDP-GlcNAc: Dolichol phosphate GlcNAc-1-phosphate transferase (DPAGT1). Curr. Drug Targets 2009, 10, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Basiri, K.; Belaya, K.; Liu, W.W.; Maxwell, S.; Sedghi, M.; Beeson, D. Clinical features in a large Iranian family with a limb-girdle congenital myasthenic syndrome due to a mutation in DPAGT1. Neuromuscul. Disord. 2013, 23, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Selcen, D.; Shen, X.; Brengman, J.; Li, Y.; Stans, A.A.; Wieben, E.; Engel, A. DPAGT1 myasthenia and myopathy: Genetic, phenotypic, and expression studies. Neurology 2014, 82, 1822–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belaya, K.; Finlayson, S.; Slater, C.R.; Cossins, J.; Liu, W.W.; Maxwell, S.; McGowan, S.J.; Maslau, S.; Twigg, S.R.F.; Walls, T.J.; et al. Mutations in DPAGT1 cause a limb-girdle congenital myasthenic syndrome with tubular aggregates. Am. J. Hum. Genet. 2012, 91, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Finlayson, S.; Palace, J.; Belaya, K.; Walls, T.; Norwood, F.; Burke, G.; Holton, J.; Pascual-Pascual, S.; Cossins, J.; Beeson, D. Clinical features of congenital myasthenic syndrome due to mutations in DPAGT1. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Takahashi, T.; Ohoka, A.; Nakajima, K.; Hashimoto, R.; Miura, N.; Tachikawa, H.; Gao, X.-D. Alg14 organizes the formation of a multiglycosyltransferase complex involved in initiation of lipid-linked oligosaccharide biosynthesis. Glycobiology 2012, 22, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.J.; Kukuruzinska, M.A.; Robbins, P. Biosynthesis of asparagine-linked oligosaccharides in Saccharomyces cerevisiae: The alg2 mutation. Glycobiology 1993, 3, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Monies, D.M.; Al-Hindi, H.N.; Al-Muhaizea, M.A.; Jaroudi, D.J.; Al-Younes, B.; Naim, E.A.; Wakil, S.M.; Meyer, B.F.; Bohlega, S. Clinical and pathological heterogeneity of a congenital disorder of glycosylation manifesting as a myasthenic/myopathic syndrome. Neuromuscul. Disord. 2014. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Torelli, S.; Wells, D.J.; Brown, S.C. Muscular dystrophies due to glycosylation defects: Diagnosis and therapeutic strategies. Curr. Opin. Neurol. 2011, 24, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Belaya, K. Mutations in GMPPB cause congenital myasthenic syndrome and bridge myasthenic disorders with dystroglycanopathies. Brain 2015. [Google Scholar] [CrossRef] [PubMed]
- Cruz, P.M.R.; Belaya, K.; Basiri, K.; Sedghi, M.; Farrugia, M.E.; Holton, J.L.; Liu, W.W.; Maxwell, S.; Petty, R.; Walls, T.J.; et al. Clinical features of the myasthenic syndrome arising from mutations in GMPPB. BMJ 2016, 1–8. [Google Scholar] [CrossRef]
- Witting, N.; Vissing, J. Pharmacologic Treatment of Downstream of Tyrosine Kinase 7 Congenital Myasthenic Syndrome. JAMA Neurol. 2014, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.S.; Timberlake, W.H. Pyridostigmin (Mestinon) in the treatment of myasthenia gravis. N. Engl. J. Med. 1954, 251, 271–272. [Google Scholar] [CrossRef] [PubMed]
- Palace, J.; Wiles, C.M.; Newsom-Davis, J. 3,4-Diaminopyridine in the treatment of congenital (hereditary) myasthenia. J. Neurol. Neurosurg. Psychiatry 1991, 54, 1069–1072. [Google Scholar] [CrossRef] [PubMed]
- Harper, C.M.; Fukodome, T.; Engel, A.G. Treatment of slow-channel congenital myasthenic syndrome with fluoxetine. Neurology 2003, 60, 1710–1713. [Google Scholar] [CrossRef] [PubMed]
- Burke, G.; Hiscock, A.; Klein, A.; Niks, E.H.; Main, M.; Manzur, A.Y.; Ng, J.; de Vile, C.; Muntoni, F.; Beeson, D.; et al. Salbutamol benefits children with congenital myasthenic syndrome due to DOK7 mutations. Neuromuscul. Disord. 2013, 23, 170–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzoni, P.J.; Scola, R.H.; Kay, C.S.K.; Filla, L.; Miranda, A.P.P.; Pinheiro, J.M.R.; Chaouch, A.; Lochmüller, H.; Werneck, L.C. Salbutamol therapy in congenital myasthenic syndrome due to DOK7 mutation. J. Neurol. Sci. 2013, 331, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Liewluck, T.; Selcen, D.; Engel, A.G. Beneficial effects of Albuterol in congenital endplate acetylcholinesterase deficiency and DOK-7 myasthenia. Muscle Nerve 2011, 44, 789–794. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CMS Subtype | Gene |
|---|---|
| Proteins with defined NMJ function | |
| Presynaptic | |
| Choline O-Acetyltransferase | CHAT |
| Unconventional myosin 9 | MYO9A |
| PREPL | PREPL |
| Vesicular ACh transporter (VAChT) | SLC18A3 |
| High-affinity choline transporter 1 (ChT) | SLC5A7 |
| Synaptosome Associated Protein 25 | SNAP25B |
| Synaptotagmin 2 | SYT2 |
| MUNC13-1 | UNC13-1 |
| Synaptobrevin 1 | VAMP1 |
| Synaptic | |
| Collagen Type XIII α1 Chain | COL13A1 |
| Endplate AChE deficiency | COLQ |
| Laminin α5 deficiency | LAMA5 |
| Laminin β2 deficiency | LAMB2 |
| Postsynaptic | |
| Agrin (neuronal) | AGRN |
| Primary AChR deficiency | CHRNA, CHRNB, CHRND, CHRNE |
| Slow channel syndrome | CHRNA, CHRNB, CHRND, CHRNE |
| Fast channel syndrome | CHRNA, CHRNB, CHRND, CHRNE |
| Low conductance syndrome | CHRNE |
| Escobar syndrome | CHRNG |
| DOK7 | DOK7 |
| LRP4 | LRP4 |
| MuSK | MUSK |
| Plectin deficiency | PLEC1 |
| Rapsyn | RAPSN |
| Sodium channel myasthenia | SCN4A |
| Ubiquitously expressed proteins | |
| ALG2 | ALG2 |
| ALG14 | ALG14 |
| DPAGT1 | DPAGT1 |
| GFPT1 | GFPT1 |
| GMPPB | GMPPB |
| SLC25A1 | SCL25A1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez Cruz, P.M.; Palace, J.; Beeson, D. The Neuromuscular Junction and Wide Heterogeneity of Congenital Myasthenic Syndromes. Int. J. Mol. Sci. 2018, 19, 1677. https://doi.org/10.3390/ijms19061677
Rodríguez Cruz PM, Palace J, Beeson D. The Neuromuscular Junction and Wide Heterogeneity of Congenital Myasthenic Syndromes. International Journal of Molecular Sciences. 2018; 19(6):1677. https://doi.org/10.3390/ijms19061677
Chicago/Turabian StyleRodríguez Cruz, Pedro M., Jacqueline Palace, and David Beeson. 2018. "The Neuromuscular Junction and Wide Heterogeneity of Congenital Myasthenic Syndromes" International Journal of Molecular Sciences 19, no. 6: 1677. https://doi.org/10.3390/ijms19061677
APA StyleRodríguez Cruz, P. M., Palace, J., & Beeson, D. (2018). The Neuromuscular Junction and Wide Heterogeneity of Congenital Myasthenic Syndromes. International Journal of Molecular Sciences, 19(6), 1677. https://doi.org/10.3390/ijms19061677