Amyloid Beta Peptide Is Released during Thrombosis in the Skin



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
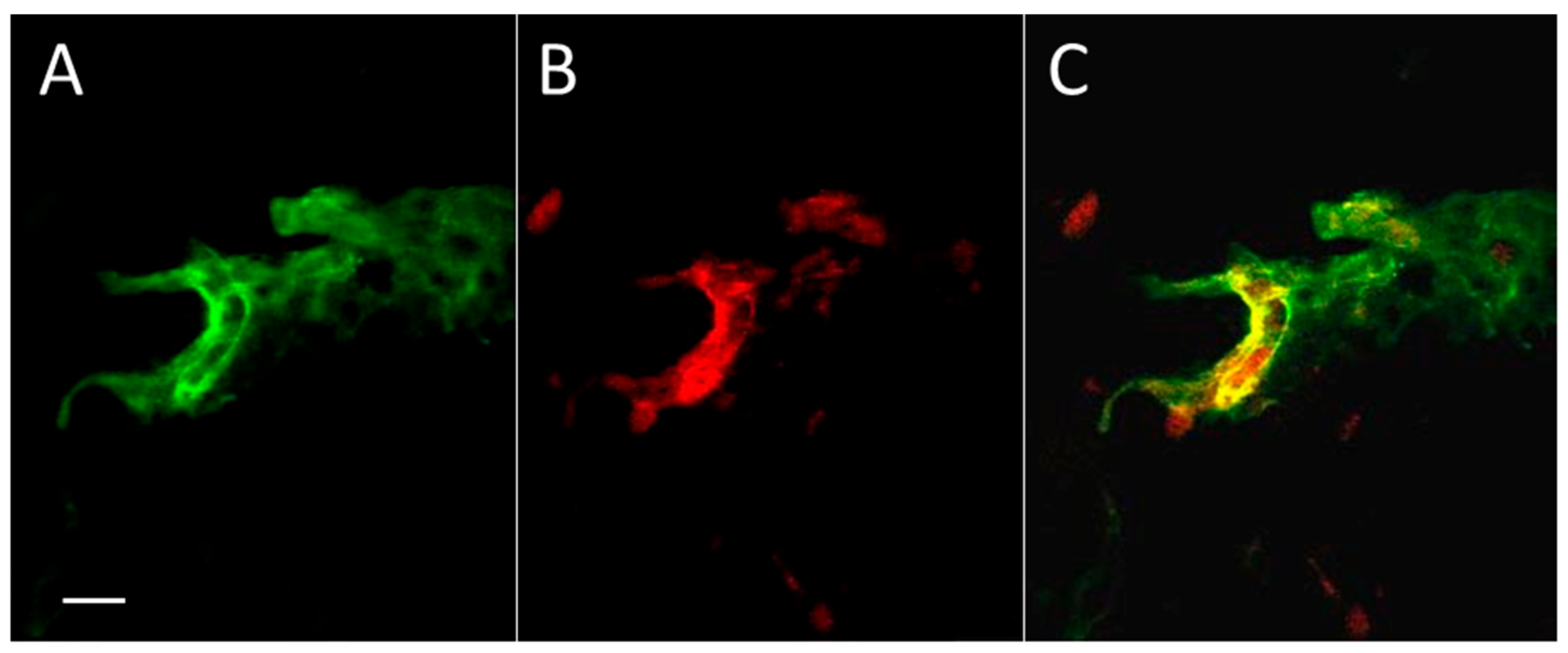
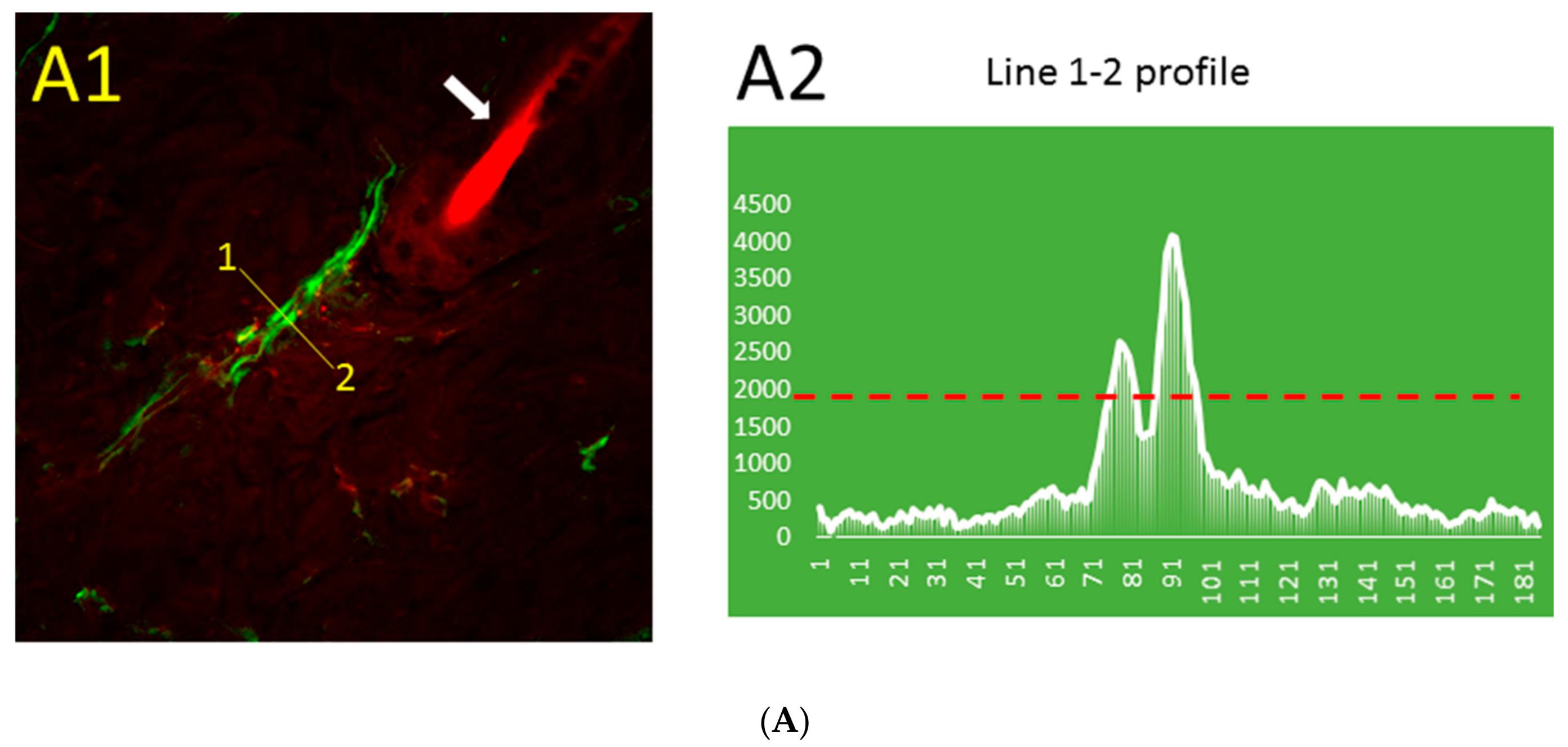
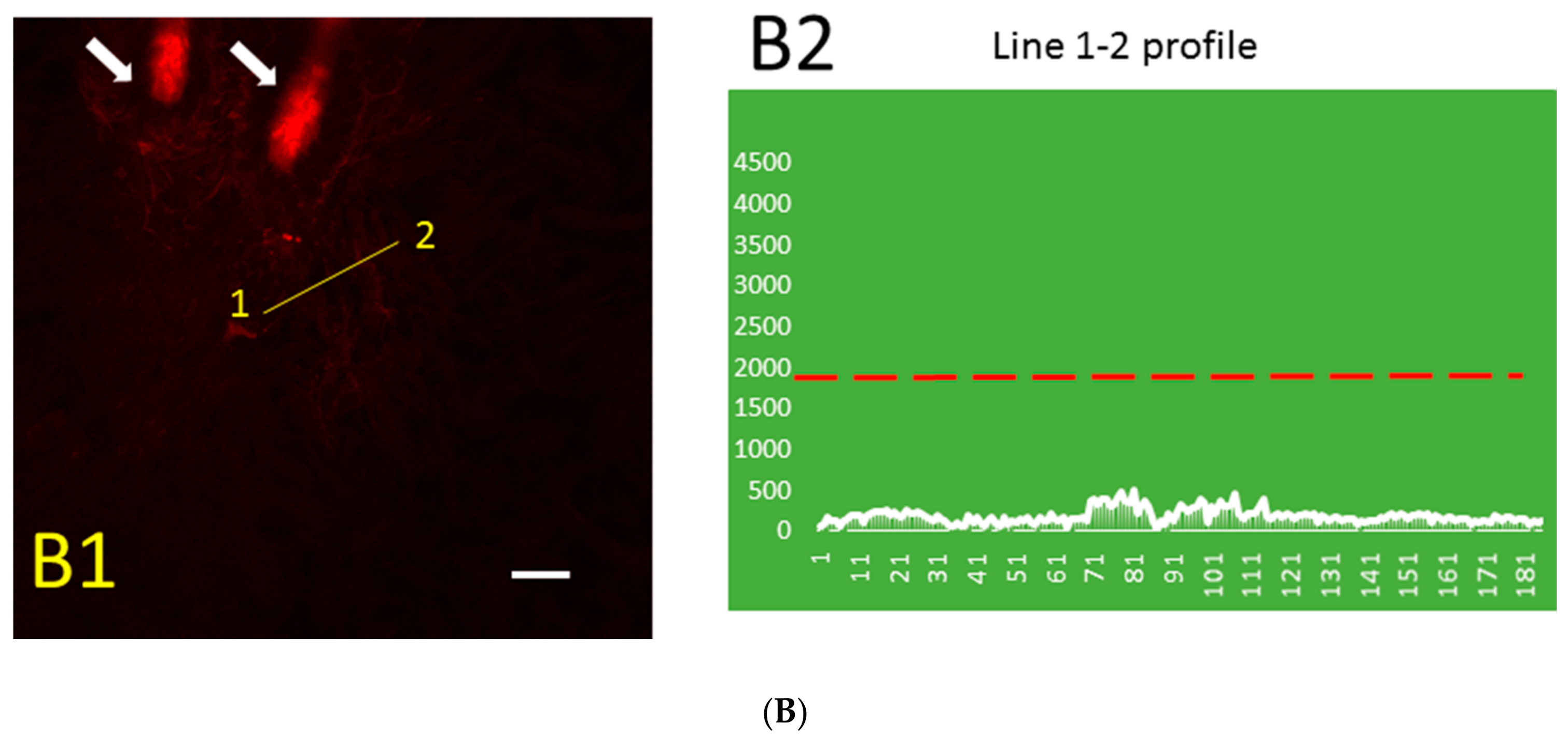
2.1. Aβ Immunoreactivity Was Concentrated in Dermal Blood Vessels a Few Minutes after Thrombosis Activation in the Skin and Became More Diffuse Later
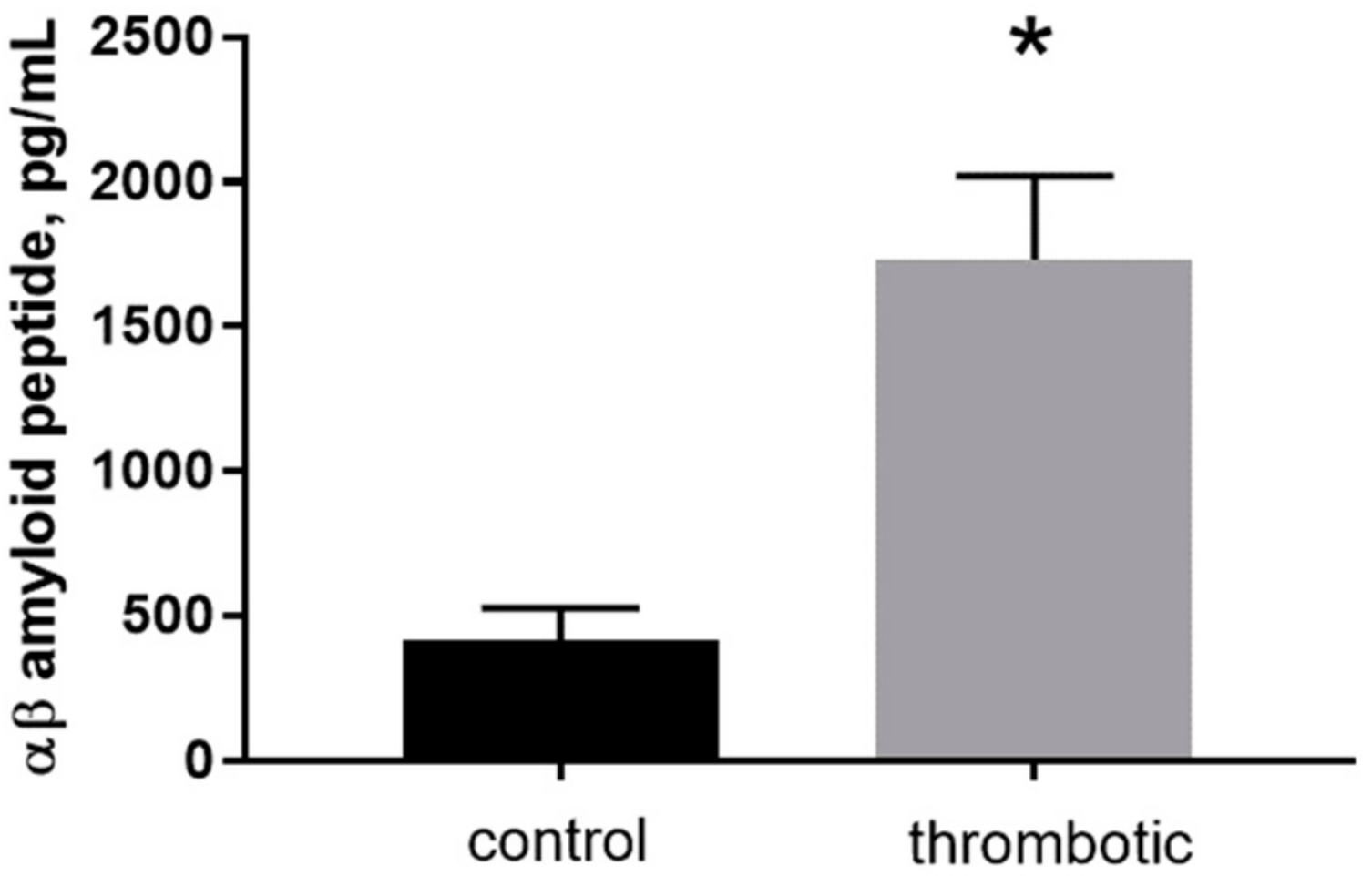
2.2. Formation of Blood Clots in the Skin Augments the Quantity of Aβ Peptide in Thrombotic Skin
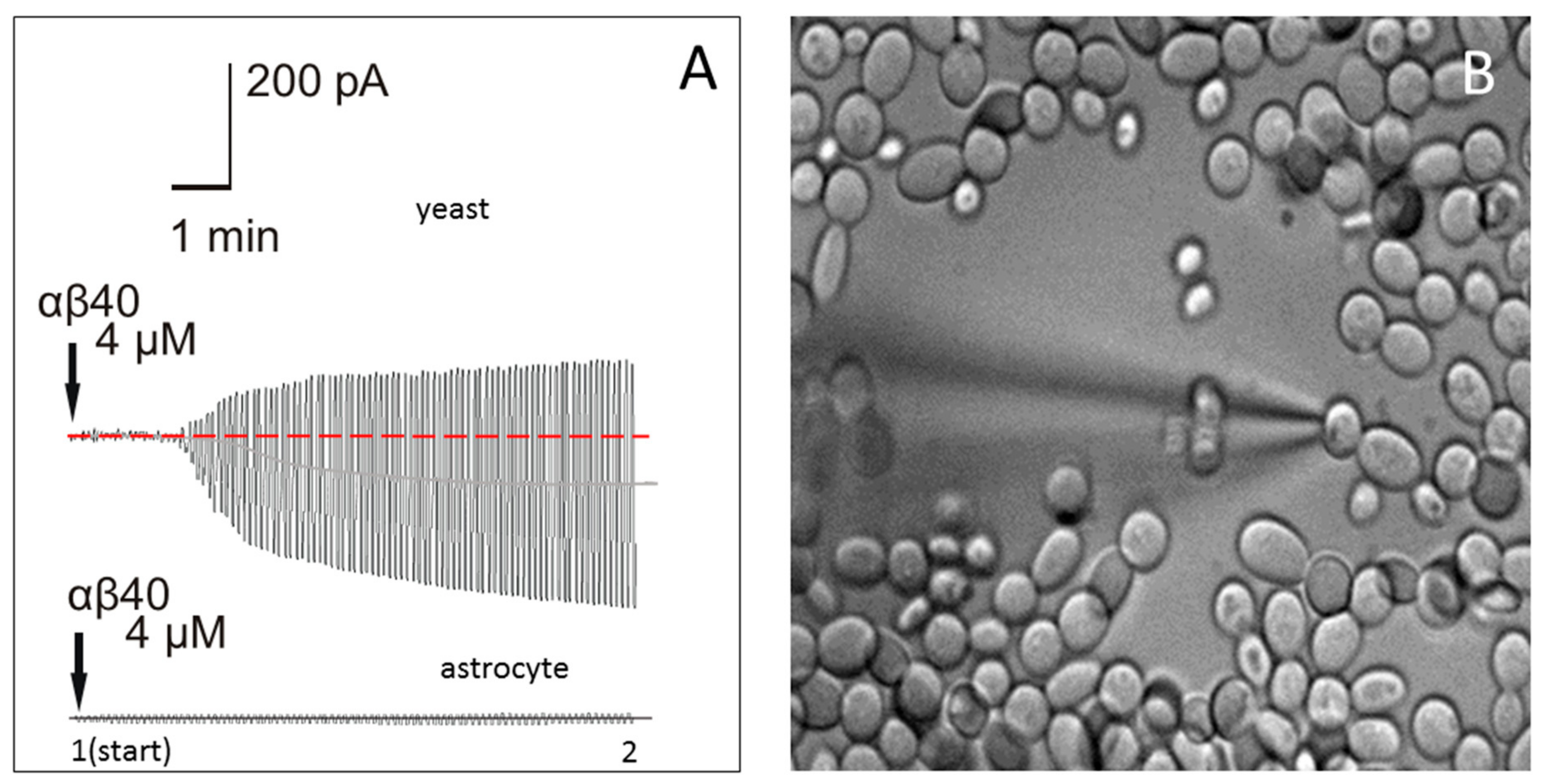
2.3. Application of Aβ40 to the External Membrane of Yeast Cells Visibly Augmented Membrane Conductance
2.4. Application of Aβ40 to the External Membrane of Mouse Astrocytes Did Not Significantly Affect Membrane Conductance
3. Discussion
4. Materials and Methods
4.1. Photothrombosis Model
4.2. Mouse Brain Slice Preparation
4.3. Preparation of Fungi (Yeast) for Patch-Clamping of Plasma Membrane
4.4. Whole-Cell Recordings in Astrocytes
4.5. Preparation of Aβ Peptide Solution
4.6. Immunohistochemistry and Confocal Microscopy
4.7. Enzyme-Linked Immunosorbent Assay (ELISA) Measurements
4.8. Statistics and Measurements
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | amyloid beta peptide |
| AD | Alzheimer’s disease |
References
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Askanas, V.; Engel, W.K.; Alvarez, R.B. Light and electron microscopic localization of β-amyloid protein in muscle biopsies of patients with inclusion-body myositis. Am. J. Pathol. 1992, 141, 31–36. [Google Scholar] [PubMed]
- Askanas, V.; Alvarez, R.B.; Engel, W.K. β-Amyloid precursor epitopes in muscle fibers of inclusion body myositis. Ann. Neurol. 1993, 34, 551–560. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, S.J.; Lehman, D.M.; Kerrigan-Baumrind, L.A.; Merges, C.A.; Pease, M.E.; Kerrigan, D.F.; Ransom, N.L.; Tahzib, N.G.; Reitsamer, H.A.; Levkovitch-Verbin, H.; et al. Caspase activation and amyloid precursor protein cleavage in rat ocular hypertension. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1077–1087. [Google Scholar]
- Ito, Y.; Shimazawa, M.; Tsuruma, K.; Mayama, C.; Ishii, K.; Onoe, H.; Aihara, M.; Araie, M.; Hara, H. Induction of amyloid-β(1-42) in the retina and optic nerve head of chronic ocular hypertensive monkeys. Mol. Vis. 2012, 18, 2647–2657. [Google Scholar] [PubMed]
- Van Wijngaarden, P.; Hadoux, X.; Alwan, M.; Keel, S.; Dirani, M. Emerging ocular biomarkers of Alzheimer disease. Clin. Exp. Ophthalmol. 2017, 45, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Troncone, L.; Luciani, M.; Coggins, M.; Wilker, E.H.; Ho, C.Y.; Codispoti, K.E.; Frosch, M.P.; Kayed, R.; del Monte, F. Aβ amyloid pathology affects the hearts of patients with Alzheimer’s disease: Mind the heart. J. Am. Coll. Cardiol. 2016, 68, 2395–2407. [Google Scholar] [CrossRef] [PubMed]
- Joachim, C.L.; Mori, H.; Selkoe, D.J. Amyloid β-protein deposition in tissues other than brain in Alzheimer’s disease. Nature 1989, 341, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, O.; Soininen, H.; Syrjänen, S.; Neittaanmäki, H.; Paljärvi, L.; Kosunen, O.; Syrjänen, K.; Riekkinen, P., Sr. β-Amyloid protein immunoreactivity in skin is not a reliable marker of Alzheimer’s disease. An autopsy-controlled study. Arch. Neurol. 1994, 51, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Ono, S.; Matsuno, S.; Shimizu, N.; Shoji, S.; Tamaoka, A. Amyloid β protein in skin of patients with amyotrophic lateral sclerosis. Lancet 1998, 351, 956–957. [Google Scholar] [CrossRef]
- Kucheryavykh, L.Y.; Dávila-Rodríguez, J.; Rivera-Aponte, D.E.; Zueva, L.V.; Washington, A.V.; Sanabria, P.; Inyushin, M.Y. Platelets are responsible for the accumulation of β-amyloid in blood clots inside and around blood vessels in mouse brain after thrombosis. Brain Res. Bull. 2017, 128, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Martins, R.N.; Rumble, B.; Moir, R.; Fuller, S.; Milward, E.; Currie, J.; David, A.; Andreas, W.; Fischer, R.; et al. The amyloid precursor protein of Alzheimer’s disease is released by human platelets. J. Biol. Chem. 1990, 265, 15977–15983. [Google Scholar] [PubMed]
- Van Nostrand, W.E.; Schmaier, A.H.; Farrow, J.S.; Cunningham, D.D. Protease nexin-II (amyloid β-protein precursor): A platelet α-granule protein. Science 1990, 248, 745–748. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.N.; Baskin, F.; Fosmire, J.A.; Risser, R.; Adams, P.; Svetlik, D.; Honig, L.S.; Cullum, C.M.; Weiner, M.F. Altered amyloid protein processing in platelets of patients with Alzheimer disease. Arch. Neurol. 1997, 54, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Baskin, F.; Rosenberg, R.N.; Iyer, L.; Hynan, L.; Cullum, C.M. Platelet APP isoform ratios correlate with declining cognition in AD. Neurology 2000, 54, 1907–1909. [Google Scholar] [CrossRef] [PubMed]
- Padovani, A.; Pastorino, L.; Borroni, B.; Colciaghi, F.; Rozzini, L.; Monastero, R.; Perez, J.; Pettenati, C.; Mussi, M.; Parrinello, G.; et al. Amyloid precursor protein in platelets: A peripheral marker for the diagnosis of sporadic AD. Neurology 2001, 57, 2243–2248. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, W.E.; Schmaier, A.H.; Farrow, J.S.; Cines, D.B.; Cunningham, D.D. Protease nexin-2/amyloid β-protein precursor in blood is a platelet-specific protein. Biochem. Biophys. Res. Commun. 1991, 175, 15–21. [Google Scholar] [CrossRef]
- Ledesma, M.D.; da Silva, J.S.; Crassaerts, K.; Delacourte, A.; de Strooper, B.; Dotti, C.G. Brain plasmin enhances APP α-cleavage and Aβ degradation and is reduced in Alzheimer’s disease brains. EMBO Rep. 2000, 1, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Schmaier, A.H. Alzheimer disease is in part a thrombohemorrhagic disorder. J. Thromb. Haemost. 2016, 14, 991–994. [Google Scholar] [CrossRef] [PubMed]
- Zamolodchikov, D.; Renné, T.; Strickland, S. The Alzheimer’s disease peptide β-amyloid promotes thrombin generation through activation of coagulation factor XII. J. Thromb. Haemost. 2016, 14, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Inestrosa, C.C.; Ross, G.S.; Fernandez, H.L. Platelets are the primary source of amyloid P-peptide in human blood. Biochem. Biophys. Res. Comrnun. 1995, 213, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid P-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Casoli, T.; di Stefano, G.; Giorgetti, B.; Grossi, Y.; Balietti, M.; Fattoretti, P.; Bertoni-Freddari, C. Release of β-amyloid from high-density platelets: Implications for Alzheimer’s disease pathology. Ann. N. Y. Acad. Sci. 2007, 1096, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s disease-associated amyloid β-protein is an antimicrobial peptide. PLoS ONE 2010, 5, e9505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukiw, W.J.; Cui, J.G.; Yuan, L.Y.; Bhattacharjee, P.S.; Corkern, M.; Clement, C.; Kammerman, E.M.; Ball, M.J.; Zhao, Y.; Sullivan, P.M.; et al. Acyclovir or Aβ42 peptides attenuate HSV-1-induced miRNA-146a levels in human primary brain cells. Neuroreport 2010, 21, 922–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, M.R.; Kandel, R.; Tripathi, S.; Condon, D.; Qi, L.; Taubenberger, J.; Hartshorn, K.L. Alzheimer’s associated β-Amyloid protein inhibits influenza a virus and modulates viral interactions with phagocytes. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgade, K.; Garneau, H.; Giroux, G.; Le Page, A.Y.; Bocti, C.; Dupuis, G.; Frost, E.H.; Fülöp, T., Jr. β-Amyloid peptides display protective activity against the human Alzheimer’s disease-associated herpes simplex virus-1. Biogerontology 2014, 16, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.K.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Inyushin, M.Y.; Sanabria, P.; Rojas, L.; Kucheryavykh, Y.; Kucheryavykh, L. Aβ peptide originated from platelets promises new strategy in anti-Alzheimer’s drug development. BioMed Res. Int. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Tang, Y.-Q.; Shen, A.J.; Bayer, A.S.; Selsted, M.E. Purification and in vitro activities of rabbit platelet microbicidal proteins. Infect. Immun. 1997, 65, 1023–1031. [Google Scholar] [PubMed]
- Yeaman, M.R.; Ibrahim, A.S.; Edwards, J.E., Jr.; Bayer, A.S.; Ghannoum, M.A. Thrombin-induced rabbit platelet microbicidal protein is fungicidal in vitro. Antimicrob. Agents Chemother. 1993, 37, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Krijgsveld, J.; Zaat, S.A.; Meeldijk, J.; van Veelen, P.A.; Fang, G.; Poolman, B.; Brandt, E.; Ehlert, J.E.; Kuijpers, A.J.; Engbers, G.H.; et al. Thrombocidins, microbicidal proteins from human blood platelets, are C-terminal deletion products of CXC chemokines. J. Biol. Chem. 2000, 275, 20374–20381. [Google Scholar] [CrossRef] [PubMed]
- Kupferwasser, L.I.; Yeaman, M.R.; Shapiro, S.M.; Nast, C.C.; Bayer, A.S. In vitro susceptibility to thrombin-induced platelet microbicidal protein is associated with reduced disease progression and complication rates in experimental Staphylococcus aureus endocarditis: Microbiological, histopathologic, and echocardiographic analyses. Circulation 2002, 105, 746–752. [Google Scholar] [PubMed]
- Tang, Y.Q.; Yeaman, M.R.; Selsted, M.E. Antimicrobial peptides from human platelets. Infect. Immun. 2002, 70, 6524–6533. [Google Scholar] [CrossRef] [PubMed]
- Yount, N.Y.; Gank, K.D.; Xiong, Y.Q.; Bayer, A.S.; Pender, T.; Welch, W.H.; Yeaman, M.R. Platelet microbicidal protein 1: Structural themes of a multifunctional antimicrobial peptide. Antimicrob. Agents Chemother. 2004, 48, 4395–4404. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Arispe, N.; Kuroda, Y.; Rojas, E. Alzheimer’s disease amyloid β-protein forms Zn2+-sensitive, cation-selective channels across excised membrane patches from hypothalamic neurons. Biophys. J. 1997, 73, 67–75. [Google Scholar] [CrossRef]
- Lin, H.; Bhatia, R.; Lal, R. Amyloid β protein forms ion channels: Implications for Alzheimer’s disease pathophysiology. FASEB J. 2001, 15, 2433–2444. [Google Scholar] [CrossRef] [PubMed]
- Lal, R.; Lin, H.; Quist, A.P. Amyloid β ion channel: 3D structure and relevance to amyloid channel paradigm. Biochim. Biophys. Acta 2007, 1768, 1966–1975. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M. Neurotoxicity of β-amyloid protein: Oligomerization, channel formation, and calcium dyshomeostasis. Curr. Pharm. Des. 2010, 16, 2779–2789. [Google Scholar] [CrossRef] [PubMed]
- Harder, J.; Bartels, J.; Christophers, E.; Schröder, J.-M. A peptide antibiotic from human skin. Nature 1997, 387, 861. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Chapple, D.S. Peptide antibiotics. Antimicrob. Agents Chemother. 1999, 43, 1317–1323. [Google Scholar] [CrossRef]
- Lan, J.; Liu, J.; Zhao, Z.; Xue, R.; Zhang, N.; Zhang, P.; Zhao, P.; Zheng, F.; Sun, X. The peripheral blood of Aβ binding RBC as a biomarker for diagnosis of Alzheimer’s disease. Age Ageing 2015, 44, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, J.G.; Eckley, D.M.; Williamson, J.D.; Launer, L.J.; Rifkind, J.M. Do red blood cell-β-amyloid interactions alter oxygen delivery in Alzheimer’s disease? Adv. Exp. Med. Biol. 2008, 614, 29–35. [Google Scholar] [PubMed]
- Inyushin, M.Y.; Tsytsarev, V.Y.; Ignashchenkova, A.Y.; Lenkov, D.N. Use of artificial ion channels for quasi-intracellular recording of cerebral cortex neuron activity. Neurosci. Behav. Physiol. 1997, 27, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Miloshevsky, G.V.; Jordan, P.C. The open state gating mechanism of gramicidin a requires relative opposed monomer rotation and simultaneous lateral displacement. Structure 2006, 14, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Watson, B.D.; Dietrich, W.D.; Busto, R.; Wachtel, M.S.; Ginsberg, M.D. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann. Neurol. 1985, 17, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Youmans, K.L.; Tai, L.M.; Kanekiyo, T.; Stine, W.B., Jr.; Michon, S.C.; Nwabuisi-Heath, E.; Manelli, A.M.; Fu, Y.; Riordan, S.; Eimer, W.A.; et al. Intraneuronal Aβ detection in 5xFAD mice by a new Aβ-specific antibody. Mol. Neurodegener. 2012, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, J.M.; King, A.E.; Woodhouse, A.; Kirkcaldie, M.T.; Vickers, J.C. The effect of focal brain injury on β-amyloid plaque deposition, inflammation and synapses in the APP/PS1 mouse model of Alzheimer’s disease. Exp. Neurol. 2015, 267, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Ismail, J.A.; Poppa, V.; Kemper, L.E.; Scatena, M.; Giachelli, C.M.; Coffin, J.D.; Murry, C.E. Immunohistologic labeling of murine endothelium. Cardiovasc. Pathol. 2003, 12, 82–90. [Google Scholar] [CrossRef]
- DeLisser, H.M.; Newman, P.J.; Albelda, S.M. Platelet endothelial cell adhesion molecule (CD31). Curr. Top. Microbiol. Immunol. 1993, 184, 37–45. [Google Scholar] [PubMed]
- Bryckaert, M.; Rosa, J.P.; Denis, C.V.; Lenting, P.J. Of von Willebrand factor and platelets. Cell. Mol. Life Sci. 2015, 72, 307–326. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, L. Griffonia simplicifolia lectins bind specifically to endothelial cells and some epithelial cells in mouse tissues. Histochem. J. 1987, 19, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Hansen-Smith, F.M.; Watson, L.; Lu, D.Y.; Goldstein, I. Griffonia simplicifolia I: Fluorescent tracer for microcirculatory vessels in nonperfused thin muscles and sectioned muscle. Microvasc. Res. 1988, 36, 199–215. [Google Scholar] [CrossRef]
- Hirabayashi, J. Lectins: Methods and Protocols, Methods in Molecular Biology; Springer: New York, NY, USA, 2014; Volume 1200, pp. 1–667. [Google Scholar]
- Cho, S.M.; Lee, S.; Yang, S.H.; Kim, H.Y.; Lee, M.J.; Kim, H.V.; Kim, J.; Baek, S.; Yun, J.; Kim, D.; et al. Age-dependent inverse correlations in CSF and plasma amyloid-β(1-42) concentrations prior to amyloid plaque deposition in the brain of 3xTg-AD mice. Sci. Rep. 2016, 6, 20185. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Pesini, P.; Espinosa, A.; Pérez-Grijalba, V.; Valero, S.; Sotolongo-Grau, O.; Alegret, M.; Monleón, I.; Lafuente, A.; Buendía, M.; et al. Blood amyloid β levels in healthy, mild cognitive impairment and Alzheimer’s disease individuals: Replication of diastolic blood pressure correlations and analysis of critical covariates. PLoS ONE 2013, 8, e81334. [Google Scholar] [CrossRef] [PubMed]
- Emadi, S.; Aghababaee, L.; Maghbooli, M. Serum concentration of β amyloid peptide and the activity of angiotensin converting enzyme in Alzheimer’s disease patients: Search for a potential biomarker. Int. J. Res. Med. Sci. 2016, 4, 421–427. [Google Scholar] [CrossRef]
- Fukumoto, H.; Tennis, M.; Locascio, J.J.; Hyman, B.T.; Growdon, J.H.; Irizarry, M.C. Age but not diagnosis is the main predictor of plasma amyloid β-protein levels. Arch. Neurol. 2003, 60, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Aluise, C.D.; Sowell, R.A.; Butterfield, D.A. Peptides and proteins in plasma and cerebrospinal fluid as biomarkers for the prediction, diagnosis, and monitoring of therapeutic efficacy of Alzheimer’s disease. Biochim. Biophys. Acta 2008, 1782, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Bertl, A.; Bihler, H.; Kettner, C.; Slayman, C.L. Electrophysiology in the eukaryotic model cell Saccharomyces cerevisiae. Pflugers Arch. 1998, 436, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Farkas, V. Structure and biosynthesis of fungal cell walls: Methodological approaches. Folia Microbiol. 2003, 48, 469–478. [Google Scholar] [CrossRef]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer disease amyloid β protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, F.J.; Parodi, J.; Peoples, R.W.; Opazo, C.; Aguayo, L.G. Synaptotoxicity of Alzheimer β amyloid can be explained by its membrane perforating property. PLoS ONE 2010, 5, e11820. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Hernandez, A.; Urbanke, H.; Gillman, A.L.; Lee, J.; Ryazanov, S.; Agbemenyah, H.Y.; Benito, E.; Jain, G.; Kaurani, L.; et al. The diphenylpyrazole compound anle138b blocks Aβ channels and rescues disease phenotypes in a mouse model for amyloid pathology. EMBO Mol. Med. 2018, 10, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Morioka, S.; Shigemori, T.; Hara, K.; Morisaka, H.; Kuroda, K.; Ueda, M. Effect of sterol composition on the activity of the yeast G-protein-coupled receptor Ste2. Appl. Microbiol. Biotechnol. 2013, 97, 4013–4020. [Google Scholar] [CrossRef] [PubMed]
- Arispe, N.; Doh, M. Plasma membrane cholesterol controls the cytotoxicity of Alzheimer’s disease AβP (1-40) and (1-42) peptides. FASEB J. 2002, 16, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- de Planque, M.R.; Raussens, V.; Contera, S.A.; Rijkers, D.T.; Liskamp, R.M.; Ruysschaert, J.M.; Ryan, J.F.; Separovic, F.; Watts, A. β-Sheet structured β-amyloid(1-40) perturbs phosphatidylcholine model membranes. J. Mol. Biol. 2007, 368, 982–997. [Google Scholar] [CrossRef] [PubMed]
- Kagan, B.L.; Thundimadathil, J. Amyloid peptide pores and the β sheet conformation. Adv. Exp. Med. Biol. 2010, 677, 150–167. [Google Scholar] [PubMed]
- Ryan, T.M.; Caine, J.; Mertens, H.D.; Kirby, N.; Nigro, J.; Breheney, K.; Waddington, L.J.; Streltsov, V.A.; Curtain, C.; Masters, C.L.; et al. Ammonium hydroxide treatment of Aβ produces an aggregate free solution suitable for biophysical and cell culture characterization. PeerJ 2013, 1, e73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porzoor, A.; Caine, J.M.; Macreadie, I.G. Pretreatment of chemically-synthesized Aβ42 affects its biological activity in yeast. Prion 2014, 8, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.M.; Kokjohn, T.A.; Kalback, W.; Luehrs, D.; Galasko, D.R.; Chevallier, N.; Koo, E.H.; Emmerling, M.R.; Roher, A.E. Amyloid-β peptides interact with plasma proteins and erythrocytes: Implications for their quantitation in plasma. Biochem. Biophys. Res. Commun. 2000, 268, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Esh, C.L.; Kokjohn, T.A.; Castaño, E.M.; van Vickle, G.D.; Kalback, W.M.; Patton, R.L.; Luehrs, D.C.; Daugs, I.D.; Kuo, Y.M.; et al. Amyloid β peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimers Dement. 2009, 5, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Toledo, J.B.; Shaw, L.M.; Trojanowski, J.Q. Plasma amyloid β measurements—A desired but elusive Alzheimer’s disease biomarker. Alzheimers Res. Ther. 2013, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Sanan, D.A.; Weisgraber, K.H.; Russell, S.J.; Mahley, R.W.; Huang, D.; Saunders, A.; Schmechel, D.; Wisniewski, T.; Frangione, B.; Roses, A.D. Apolipoprotein E associates with β amyloid peptide of Alzheimer’s disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J. Clin. Investig. 1994, 94, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Arispe, N.; Pollard, H.B.; Rojas, E. β-Amyloid Ca2+-channel hypothesis for neuronal death in Alzheimer disease. Mol. Cell. Biochem. 1994, 140, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Fenjves, E.S.; Gordon, D.A.; Pershing, L.K.; Williams, D.L.; Taichman, L.B. Systemic distribution of apolipoprotein E secreted by grafts of epidermal keratinocytes: Implications for epidermal function and gene therapy. Proc. Natl. Acad. Sci. USA 1989, 86, 8803–8807. [Google Scholar] [CrossRef] [PubMed]
- Barra, R.M.; Fenjves, E.S.; Taichman, L.B. Secretion of apolipoprotein E by basal cells in cultures of epidermal keratinocytes. J. Investig. Dermatol. 1994, 102, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Vigo-Pelfrey, C.; Teplow, D.B.; Miller, C.; Schenk, D.; Johnston, J.; Winblad, B.; Venizelos, N.; Lannfelt, L.; Selkoe, D.J. Excessive production of amyloid β-protein by peripheral cells of symptomatic and presymptomatic patients carrying the Swedish familial Alzheimer disease mutation. Proc. Natl. Acad. Sci. USA 1994, 91, 11993–11997. [Google Scholar] [CrossRef] [PubMed]
- Furumoto, H.; Shimizu, T.; Asagami, C.; Muto, M.; Takahashi, M.; Hoshii, Y.; Ishihara, T.; Nakamura, K. Apolipoprotein E is present in primary localized cutaneous amyloidosis. J. Investig. Dermatol. 1998, 111, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Ito, K.; Taniguchi, Y.; Yang, F.; Youngberg, G. Keratin in cutaneous amyloidoses. Clin. Dermatol. 1990, 8, 55–65. [Google Scholar] [CrossRef]
- Furumoto, H.; Hashimoto, Y.; Muto, M.; Shimizu, T.; Nakamura, K. Apolipoprotein E4 is associated with primary localized cutaneous amyloidosis. J. Investig. Dermatol. 2002, 119, 532–533. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Lee, B.I.; Park, C.B. Photo-induced inhibition of Alzheimer’s β-amyloid aggregation in vitro by rose Bengal. Biomaterials 2015, 38, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, C. Elan-Wyeth Alzheimer’s Trial Suspended Due to Skin Lesion. BioWorld Today. 21 April 2008. Available online: www.bioworld.com (accessed on 23 April 2015).
- Bu, X.L.; Xiang, Y.; Jin, W.S.; Wang, J.; Shen, L.L.; Huang, Z.L.; Zhang, K.; Liu, Y.H.; Zeng, F.; Liu, J.H.; et al. Blood-derived amyloid-β protein induces Alzheimer’s disease pathologies. Mol. Psychiatry 2017. [Google Scholar] [CrossRef] [PubMed]
- Tincu, J.A.; Taylor, S.W. Antimicrobial peptides from marine invertebrates. Antimicrob. Agents Chemother. 2004, 48, 3645–3654. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kucheryavykh, L.Y.; Kucheryavykh, Y.V.; Washington, A.V.; Inyushin, M.Y. Amyloid Beta Peptide Is Released during Thrombosis in the Skin. Int. J. Mol. Sci. 2018, 19, 1705. https://doi.org/10.3390/ijms19061705
Kucheryavykh LY, Kucheryavykh YV, Washington AV, Inyushin MY. Amyloid Beta Peptide Is Released during Thrombosis in the Skin. International Journal of Molecular Sciences. 2018; 19(6):1705. https://doi.org/10.3390/ijms19061705
Chicago/Turabian StyleKucheryavykh, Lilia Y., Yuriy V. Kucheryavykh, A. Valance Washington, and Mikhail Y. Inyushin. 2018. "Amyloid Beta Peptide Is Released during Thrombosis in the Skin" International Journal of Molecular Sciences 19, no. 6: 1705. https://doi.org/10.3390/ijms19061705
APA StyleKucheryavykh, L. Y., Kucheryavykh, Y. V., Washington, A. V., & Inyushin, M. Y. (2018). Amyloid Beta Peptide Is Released during Thrombosis in the Skin. International Journal of Molecular Sciences, 19(6), 1705. https://doi.org/10.3390/ijms19061705