SeqSVM: A Sequence-Based Support Vector Machine Method for Identifying Antioxidant Proteins


Abstract
:1. Introduction
- (1)
- A computational method called (SeqSVM) is proposed to predict antioxidant proteins, which is based on the primary sequence features proposed in [17]. The features are described by the physicochemical properties and sequence information of the protein, the dimensionality of the extracted features is 188, so the feature used here is called 188D.
- (2)
- There is redundancy in the 188D feature. In the manuscript, the features are selected by maximum relevance maximum distance method [28]. The features will be kept which can maximize the Pearson’s correlation coefficient and the distance between attributes. The experimental results show that the performance of the method using selected features is competitive, or even better than that of the method using 188D.
- (3)
- The proposed method uses support vector machine for antioxidant protein prediction. The experiments demonstrated that our proposed method performs better than existing methods with the accuracy of 89.46%. The best result of existing work is 74.79% proposed by Lin et al. [9].
2. Results and Discussion
2.1. Comparison with Existing Methods
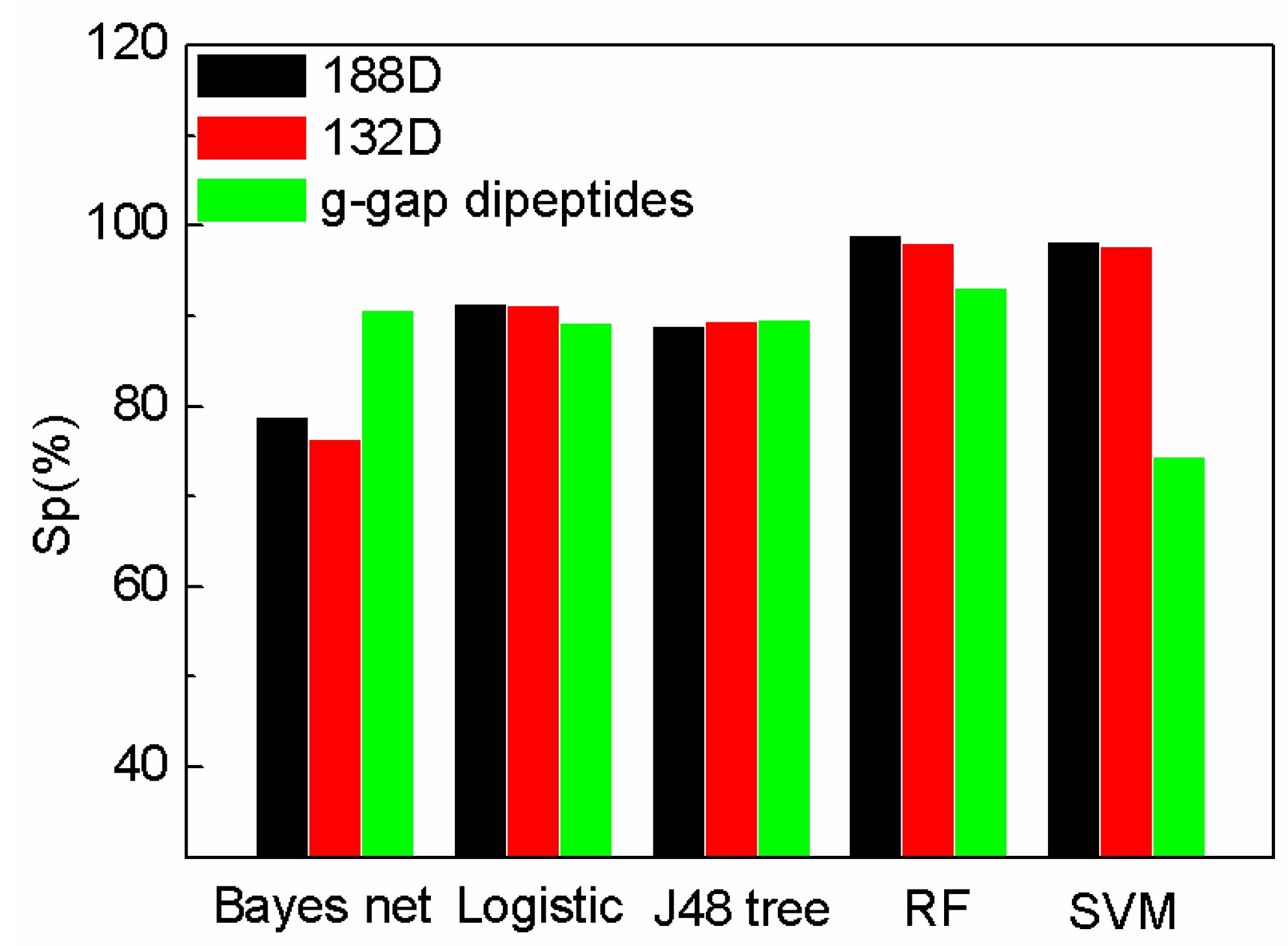
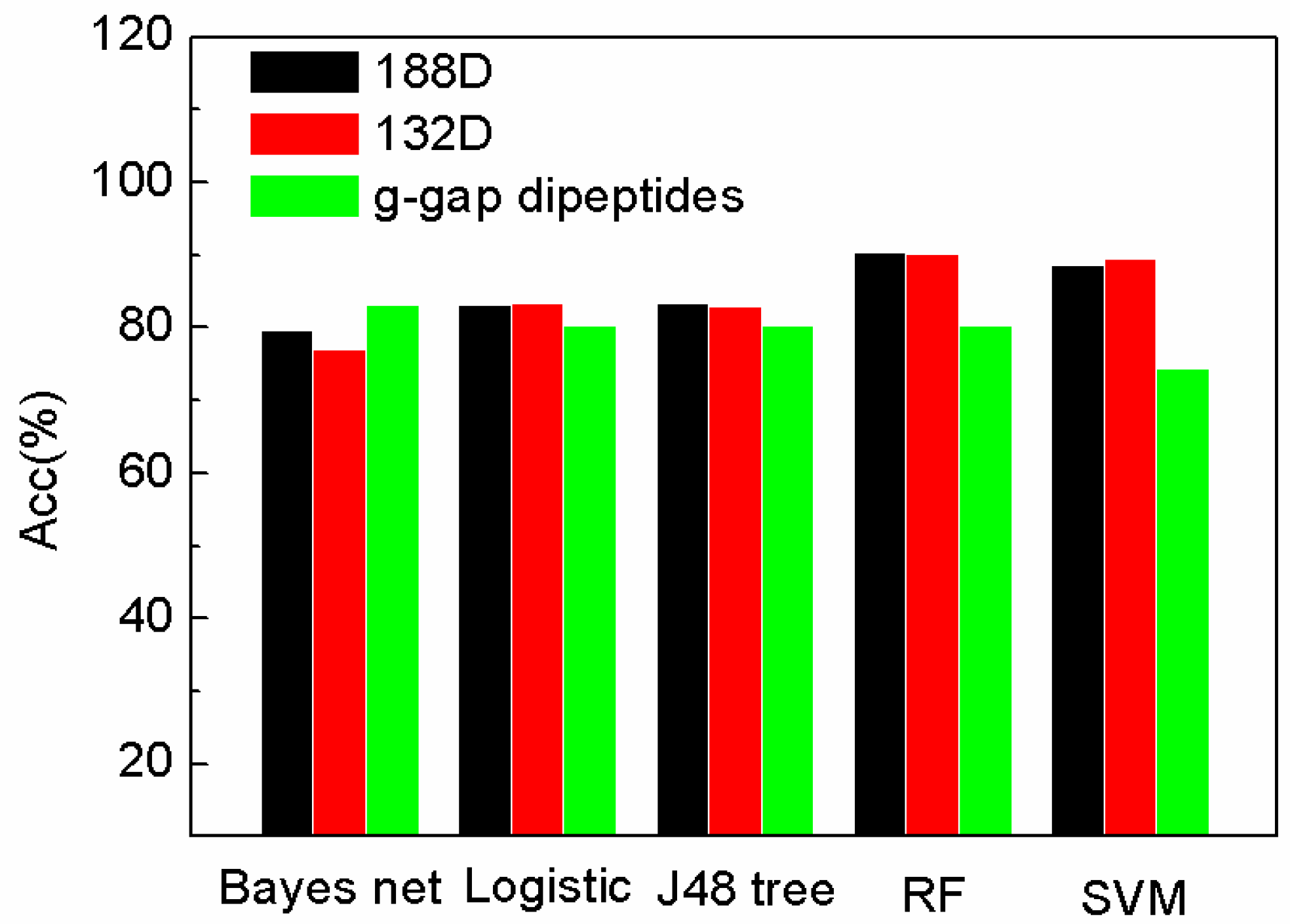
2.2. The Comparison of Performance Evaluation on Feature Selection Methods
2.3. The Comparison of SeqSVM
3. Materials and Methods
3.1. Benchmark Dataset
3.2. Support Vector Machine
3.3. SMOTE Processing
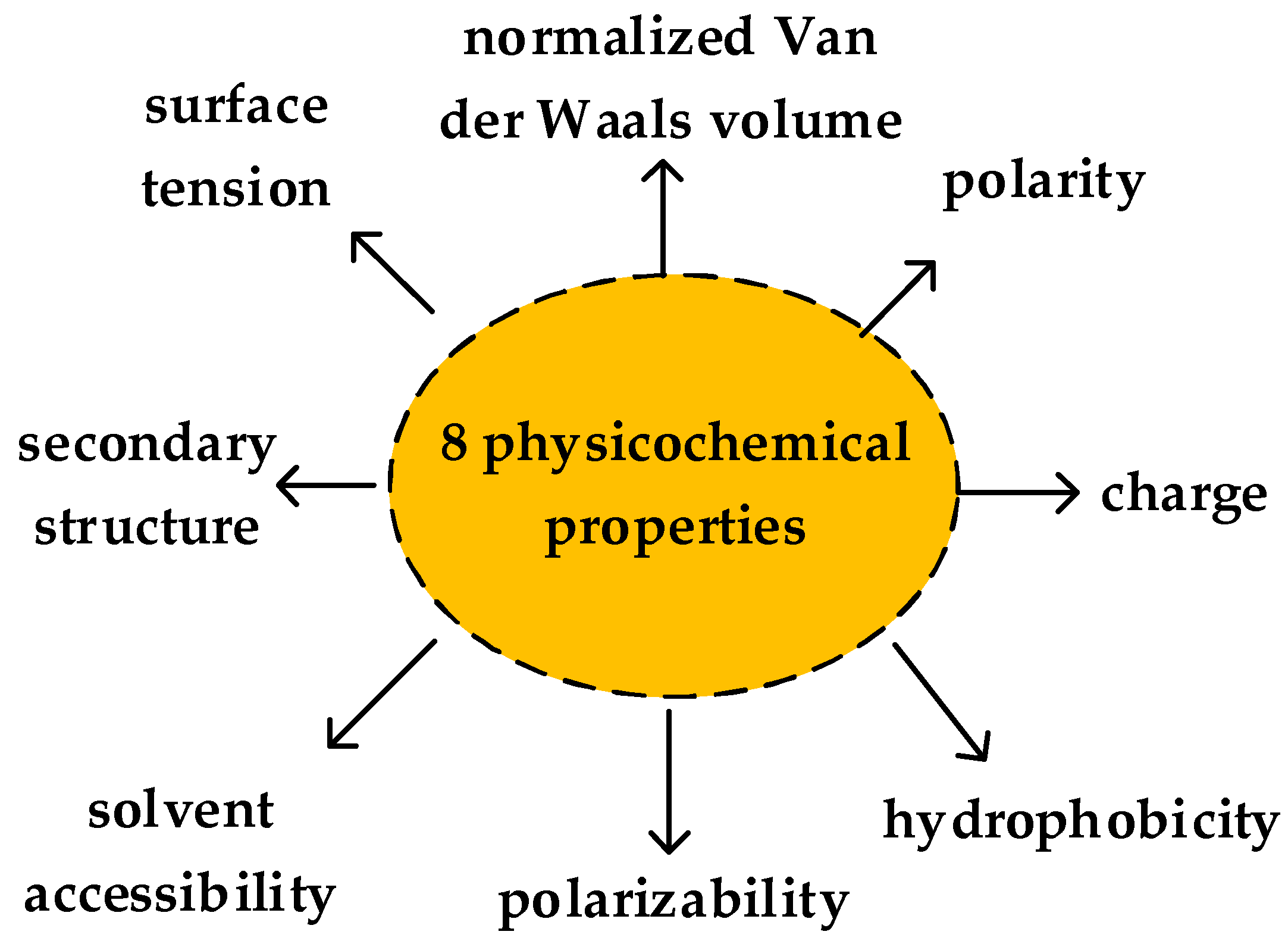
3.4. Sequence Representation
3.5. Performance Evaluation
3.6. Feature Selection
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Barbusinki, K. Fenton reaction-controversy concerning the chemistry. Ecol. Chem. Eng. 2009, 16, 347–358. [Google Scholar]
- Phamhuy, L.A.; He, H.; Phamhuy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. IJBS 2008, 4, 89–96. [Google Scholar]
- Fernández-Blanco, E.; Aguiar-Pulido, V.; Munteanu, C.R.; Dorado, J. Random forest classification based on star graph topological indices for antioxidant proteins. J. Theor. Biol. 2013, 317, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.M.; Channon, K.M. Free radicals and redox signalling in cardiovascular disease. Heart 2004, 90, 486–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.-C.; Zhang, Z. SNPdryad: Predicting deleterious non-synonymous human SNPs using only orthologous protein sequences. Bioinformatics 2014, 30, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.M.; Lin, H.; Chen, W. Identification of antioxidants from sequence information using naïve bayes. Comput. Math. Methods Med. 2013, 2013, 567529. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Chen, W.; Lin, H. Identifying antioxidant proteins by using optimal dipeptide compositions. Interdiscip. Sci. Comput. Life Sci. 2016, 8, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Liu, B. BioSeq-Analysis: A platform for DNA, RNA, and protein sequence analysis based on machine learning approaches. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, F.; Wang, X.; Chen, J.; Fang, L.; Chou, K.-C. Pse-in-One: A web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015, 43, W65–W71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.T.; Chou, K.C. An optimization approach to predicting protein structural class from amino acid composition. Protein Sci. 1992, 1, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, H.; Nishikawa, K.; Ooi, T. The folding type of a protein is relevant to the amino acid composition. J. Biochem. 1986, 99, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.P. An intriguing controversy over protein structural class prediction. J. Protein Chem. 1998, 17, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.M.; Lin, H.; Chou, K.C. iRSpot-PseDNC: Identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res. 2013, 41, e68. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Liang, G.; Wang, L.; Liao, C. A novel hybrid sequence-based model for identifying anticancer peptides. Genes 2018, 9, 158. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.Z.; Han, L.Y.; Ji, Z.L.; Chen, X.; Chen, Y.Z. SVM-Prot: Web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res. 2003, 31, 3692–3697. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins Struct. Funct. Bioinform. 2001, 44, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ding, H.; Feng, P.; Lin, H.; Kuo-Chen, C. iACP: A sequence-based tool for identifying anticancer peptides. Oncotarget 2016, 7, 16895–16909. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L.; Lumini, A.; Gupta, D.; Garg, A. Identifying bacterial virulent proteins by fusing a set of classifiers based on variants of Chou’s pseudo amino acid composition and on evolutionary information. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, W.; Zhang, Q.; Li, G.Z. MultiP-SChlo: Multi-label protein subchloroplast localization prediction with Chou’s pseudo amino acid composition and a novel multi-label classifier. Bioinformatics 2015, 31, 2639–2645. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.B.; Chen, C.; Li, Z.C.; Zou, X.Y. Using Chou’s amphiphilic pseudo-amino acid composition and support vector machine for prediction of enzyme subfamily classes. J. Theor. Biol. 2007, 248, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Mukhopadhyay, A.; Maulik, U. Prediction of protein subcellular localization by incorporating multiobjective PSO-based feature subset selection into the general form of Chou’s PseAAC. Med. Biol. Eng. Comput. 2015, 53, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, M.; Mohabatkar, H.; Mohsenzadeh, S. Using the concept of Chou’s pseudo amino acid composition for risk type prediction of human papillomaviruses. J. Theor. Biol. 2010, 263, 203–209. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Jia, C.; Duan, Y.; Zou, Q. 70ProPred: A predictor for discovering sigma70 promoters based on combining multiple features. BMC Syst. Biol. 2018, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, D.; Xu, R.; Xu, J.; Wang, X.; Chen, Q.; Dong, Q.; Chou, K.C. Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics 2014, 30, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Wan, S.; Ju, Y.; Tang, J.; Zeng, X. Pretata: Predicting TATA binding proteins with novel features and dimensionality reduction strategy. BMC Syst. Biol. 2016, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Zeng, J.; Cao, L.; Ji, R. A novel features ranking metric with application to scalable visual and bioinformatics data classification. Neurocomputing 2016, 173, 346–354. [Google Scholar] [CrossRef]
- Frank, E.; Hall, M.; Trigg, L.; Holmes, G.; Witten, I.H. Data mining in bioinformatics using WEKA. Bioinformatics 2004, 20, 2479–2481. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Hui, D.; Hao, L.; Wei, C. AOD: The antioxidant protein database. Sci. Rep. 2017, 7, 7449. [Google Scholar] [CrossRef] [PubMed]
- Dreher, D.; Junod, A.F. Role of oxygen free radicals in cancer development. Eur. J. Cancer 1996, 32A, 30–38. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.H.; Deng, E.Z.; Xu, L.Q.; Ding, H.; Lin, H.; Chen, W.; Chou, K.C. iNuc-PseKNC: A sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition. Bioinformatics 2014, 30, 1522–1529. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.R.; Xiao, X.; Lin, W.Z.; Chou, K.C. iUbiq-Lys: Prediction of lysine ubiquitination sites in proteins by extracting sequence evolution information via a gray system model. J. Biomol. Struct. Dyn. 2015, 33, 1731–1742. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, L.; Liu, F.; Wang, X.; Chen, J.; Chou, K.C. Identification of real microRNA precursors with a pseudo structure status composition approach. PLoS ONE 2015, 10, e0121501. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Deng, E.Z.; Ding, H.; Chen, W.; Chou, K.C. iPro54-PseKNC: A sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo K-tuple nucleotide composition. Nucleic Acids Res. 2014, 42, 12961–12972. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.M.; Chen, W.; Lin, H.; Chou, K.C. iHSP-PseRAAAC: Identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem. 2013, 442, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.-M.; Lin, H.; Chou, K.-C. iSS-PseDNC: Identifying Splicing Sites Using Pseudo Dinucleotide Composition. BioMed Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tang, H.; Chen, X.X.; Zhang, C.J.; Zhu, P.P.; Ding, H.; Chen, W.; Lin, H. Identification of secretory proteins in mycobacterium tuberculosis using pseudo amino acid composition. BioMed Res. Int. 2016, 2016, 5413903. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.X.; Tang, H.; Li, W.C.; Wu, H.; Chen, W.; Ding, H.; Lin, H. Identification of bacterial cell wall lyases via pseudo amino acid composition. BioMed Res. Int. 2016, 2016, 1654623. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Ding, C.; Song, Q.; Yang, P.; Ding, H.; Deng, K.J.; Chen, W. The prediction of protein structural class using averaged chemical shifts. J. Biomol. Struct. Dyn. 2012, 29, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xu, J.; Lan, X.; Xu, R.; Zhou, J.; Wang, X.; Chou, K.-C. iDNA-Prot|dis: Identifying DNA-binding proteins by incorporating amino acid distance-pairs and reduced alphabet profile into the general pseudo amino acid composition. PLoS ONE 2014, 9, e106691. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhang, J.; Deng, L. Prediction of lncRNA-protein interactions using HeteSim scores based on heterogeneous networks. Sci. Rep. 2017, 7, 3664. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Tao, X.W.; Zhao, J.; Feng, Y.M.; Cai, Y.D.; Zhang, N. Computational prediction of protein epsilon lysine acetylation sites based on a feature selection method. Comb. Chem. High Throughput Screen. 2017, 20, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yu, S.; Guo, Y.; Wang, L.; Wang, P.; Feng, Y. Discriminating ramos and jurkat cells with image textures from diffraction imaging flow cytometry based on a support vector machine. Curr. Bioinform. 2017, 11, 1. [Google Scholar] [CrossRef]
- Zhang, N.; Duan, G.; Gao, S.; Ruan, J.; Zhang, T. Prediction of the parallel/antiparallel orientation of beta-strands using amino acid pairing preferences and support vector machines. J. Theor. Biol. 2010, 263, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Cai, Y.D. Using functional domain composition and support vector machines for prediction of protein subcellular location. J. Biol. Chem. 2002, 277, 45765–45769. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.D.; Zhou, G.P.; Chou, K.C. Support vector machines for predicting membrane protein types by using functional domain composition. Biophys. J. 2003, 84, 3257–3263. [Google Scholar] [CrossRef]
- Cristianini, N.; Shawe-Taylor, J. An Introduction to Support Vector Machines: And Other Kernel-Based Learning Methods; Cambridge University Press: Cambridge, UK, 2000; pp. 1–28. [Google Scholar]
- Chang, C.C.; Lin, C.J. LIBSVM: A Library for Support Vector Machines; ACM: New York, NY, USA, 2011; pp. 1–27. [Google Scholar]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar]
- Chou, K.C. Using subsite coupling to predict signal peptides. Protein Eng. 2001, 14, 75–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, K.C. Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol. 2011, 273, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.Y.; Chen, X.X.; Chen, W.; Tang, H.; Lin, H. Sequence-based predictive modeling to identify cancerlectins. Oncotarget 2017, 8, 28169–28175. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yang, F.; Chou, K.-C. 2L-piRNA: A two-layer ensemble classifier for identifying PIWI-interacting RNAs and their function. Mol. Ther. Nucleic Acids 2017, 7, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Zhang, C.T. Review: Prediction of protein structral classes. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 275–349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Li, B.Q.; Gao, S.; Ruan, J.S.; Cai, Y.D. Computational prediction and analysis of protein γ-carboxylation sites based on a random forest method. Mol. Biosyst. 2012, 8, 2946–2955. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Zhang, Q.C.; Chen, Z.; Meng, Y.; Guan, J.; Zhou, S. PredHS: A web server for predicting protein–protein interaction hot spots by using structural neighborhood properties. Nucleic Acids Res. 2014, 42, W290–W295. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, Z.; Zhan, W.; Deng, L. Computational identification of binding energy hot spots in protein-RNA complexes using an ensemble approach. Bioinformatics 2017, 34, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liu, D.; Wang, Z.; Wen, T.; Deng, L. A boosting approach for prediction of protein-RNA binding residues. BMC Bioinform. 2017, 18, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, Z.; Meng, W.; Zhang, P.; Tao, H. Classification of cancers based on copy number variation landscapes. Biochim. Biophys. Acta 2016, 1860, 2750–2755. [Google Scholar]
- Tang, W.; Wan, S.; Yang, Z.; Teschendorff, A.E.; Zou, Q. Tumor Origin Detection with Tissue-Specific miRNA and DNA methylation Markers. Bioinformatics 2017, 34, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Tang, H.; Lin, H. MethyRNA: A web-server for identification of N6-methyladenosine sites. J. Biomol. Struct. Dyn. 2017, 35, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Tang, H.; Ye, J.; Lin, H.; Chou, K.C. iRNA-PseU: Identifying RNA pseudouridine sites. Mol. Ther. Nucleic Acids 2016, 5, e332. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yang, H.; Feng, P.; Ding, H.; Lin, H. iDNA4mC: Identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 2017, 33, 3518–3523. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Ding, H.; Yang, H.; Chen, W.; Lin, H.; Chou, K.C. iRNA-PseColl: Identifying the occurrence sites of different RNA modifications by incorporating collective effects of nucleotides into PseKNC. Mol. Ther. Nucleic Acids 2017, 7, 155–163. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Performance Evaluation | SeqSVM (132D) | AodPred | Nave Bayes |
|---|---|---|---|
| Accuracy | 89.46% | 74.49% | 66.88% |
| Performance Evaluation | SeqSVM (Non-SMOTE) | SeqSVM (SMOTE) | SeqSVM (SMOTE + MRMD) |
|---|---|---|---|
| Accuracy | 85.98% | 88.68% | 89.46% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, L.; Liang, G.; Shi, S.; Liao, C. SeqSVM: A Sequence-Based Support Vector Machine Method for Identifying Antioxidant Proteins. Int. J. Mol. Sci. 2018, 19, 1773. https://doi.org/10.3390/ijms19061773
Xu L, Liang G, Shi S, Liao C. SeqSVM: A Sequence-Based Support Vector Machine Method for Identifying Antioxidant Proteins. International Journal of Molecular Sciences. 2018; 19(6):1773. https://doi.org/10.3390/ijms19061773
Chicago/Turabian StyleXu, Lei, Guangmin Liang, Shuhua Shi, and Changrui Liao. 2018. "SeqSVM: A Sequence-Based Support Vector Machine Method for Identifying Antioxidant Proteins" International Journal of Molecular Sciences 19, no. 6: 1773. https://doi.org/10.3390/ijms19061773
APA StyleXu, L., Liang, G., Shi, S., & Liao, C. (2018). SeqSVM: A Sequence-Based Support Vector Machine Method for Identifying Antioxidant Proteins. International Journal of Molecular Sciences, 19(6), 1773. https://doi.org/10.3390/ijms19061773