Human Papilloma Virus and Autophagy




Abstract
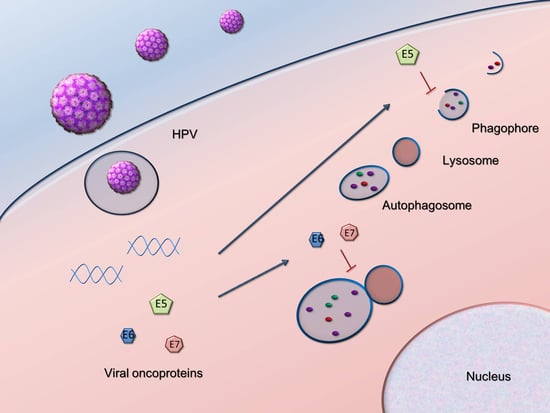
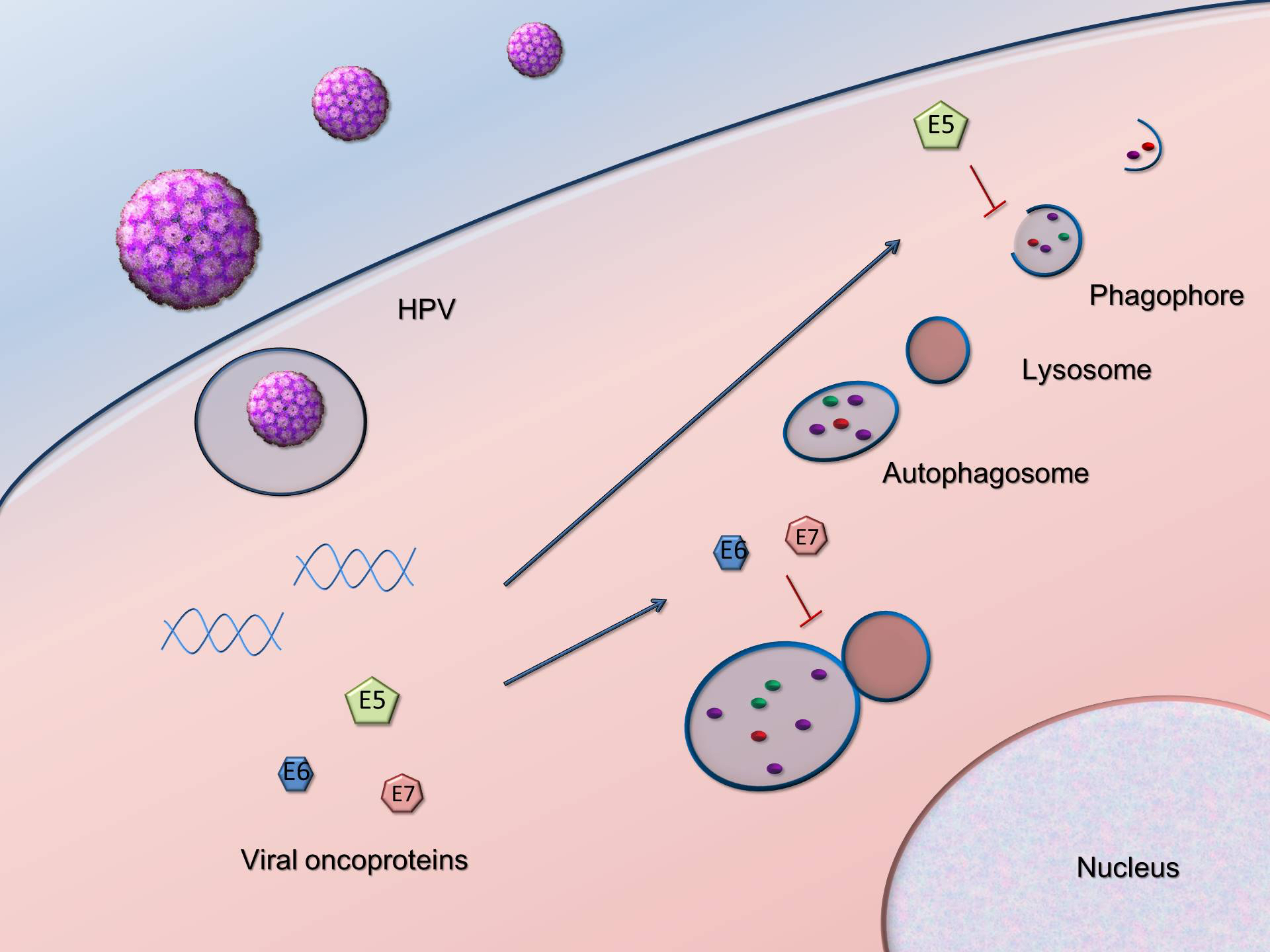
:
1. Introduction
2. HPV Manipulation of Host Autophagy
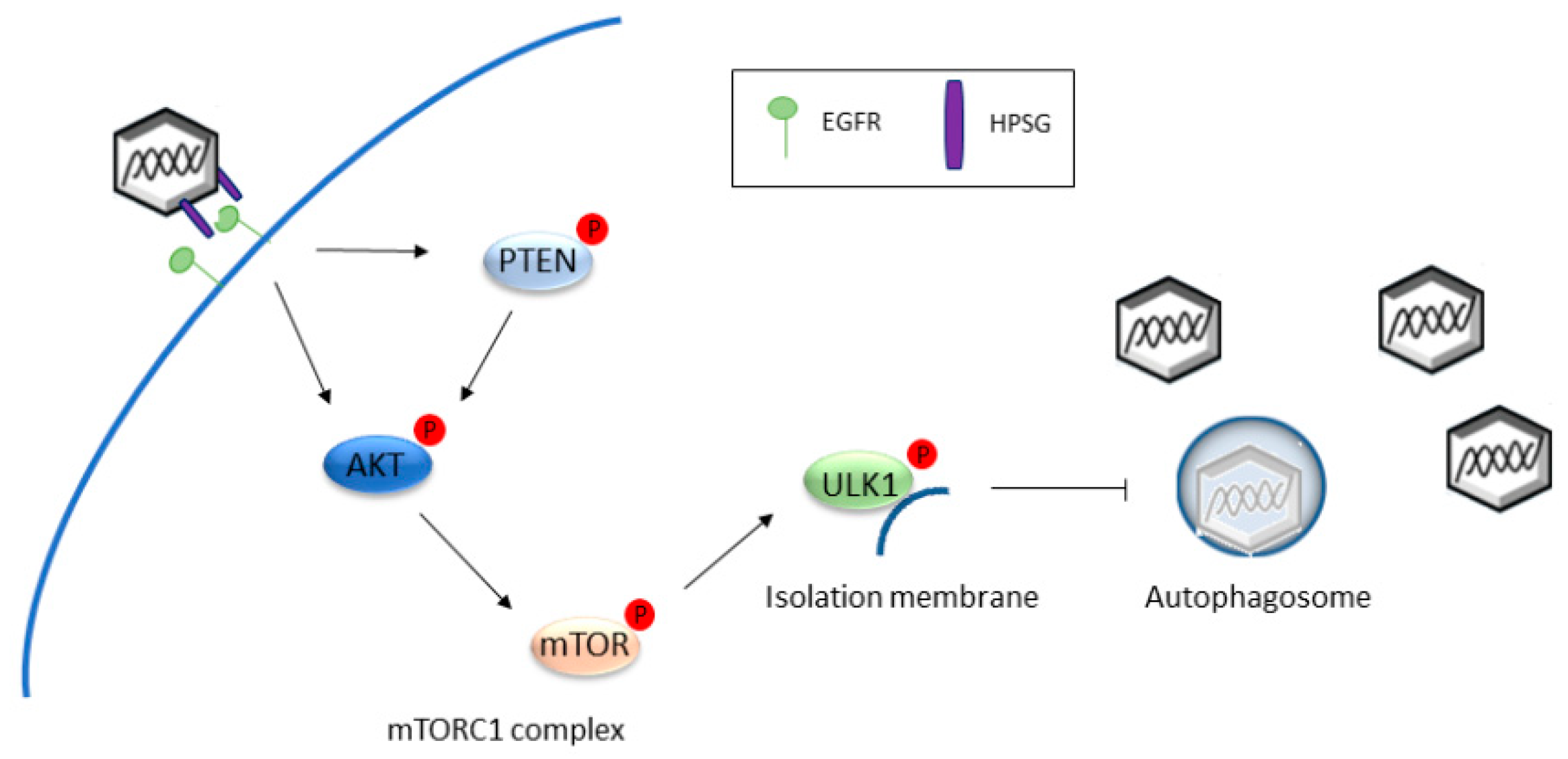
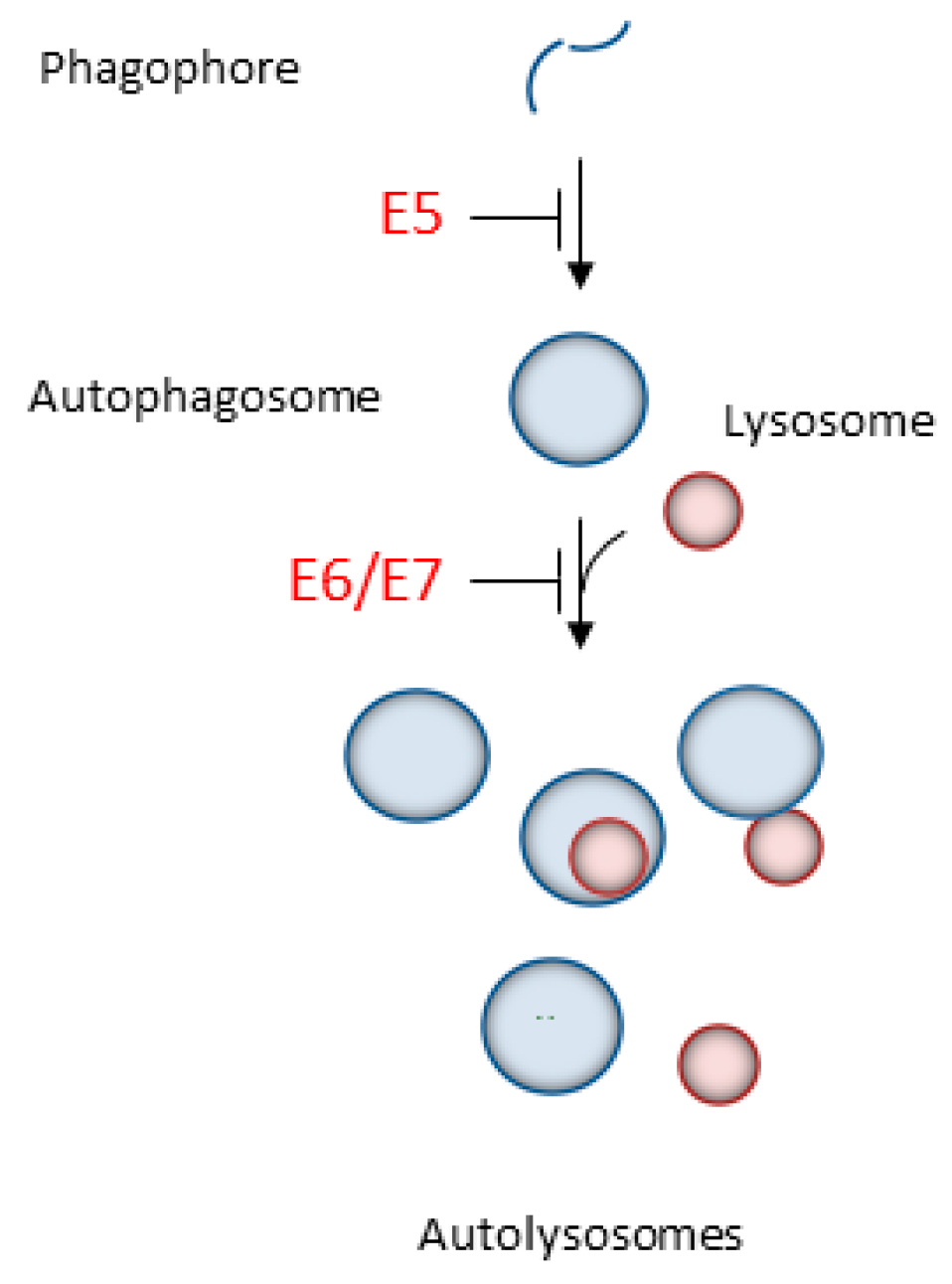
2.1. Autophagy Inhibition Promotes HPV Infectivity
2.2. HPV16 Dampens Autophagy to Promote Cancer Progression
2.3. Tumor Viruses Usurp Host Autophagy during Their Infection Cycle
3. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Forman, D.; de Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M.; et al. Global burden of human papillomavirus and related diseases. Vaccine 2012, 30, F12–F23. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, A.R.; Nyitray, A.G.; Kreimer, A.R.; Pierce Campbell, C.M.; Goodman, M.T.; Sudenga, S.L.; Monsonego, J.; Franceschi, S. EUROGIN 2014 roadmap: Differences in human papillomavirus infection natural history, transmission and human papillomavirus-related cancer incidence by gender and anatomic site of infection. Int. J. Cancer 2015, 136, 2752–2760. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Tan, J.; Miao, Y.; Zhang, Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell. Physiol. 2018, 233, 3867–3874. [Google Scholar] [CrossRef] [PubMed]
- Hamacher-Brady, A.; Brady, N.R. Mitophagy programs: Mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell. Mol. Life Sci. 2016, 73, 775–795. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.; Amadio, M.; Viiri, J.; Pascale, A.; Salminen, A.; Kaarniranta, K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res. Rev. 2014, 18, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Kimmey, J.M.; Stallings, C.L. Bacterial Pathogens versus Autophagy: Implications for Therapeutic Interventions. Trends Mol. Med. 2016, 22, 1060–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.A.; Salmani, J.M.M.; Jiang, Z.; Feng, L.; Song, J.; Jia, X.; Chen, B. Autophagy: An overview and its roles in cancer and obesity. Clin. Chim. Acta 2017, 468, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Melville, D.; Zhang, M.; Schekman, R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. eLife 2013, 2, e00947. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Stanley, R.E.; Ragusa, M.J.; Hurley, J.H. The beginning of the end: How scaffolds nucleate autophagosome biogenesis. Trends Cell Biol. 2014, 24, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Lee, J.S.; Inn, K.S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, W.T. Viruses and the autophagy pathway. Virology 2015, 479–480, 450–456. [Google Scholar] [CrossRef] [PubMed]
- DiGiuseppe, S.; Bienkowska-Haba, M.; Sapp, M. Human Papillomavirus Entry: Hiding in a Bubble. J. Virol. 2016, 90, 8032–8035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surviladze, Z.; Dziduszko, A.; Ozbun, M.A. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog. 2012, 8, e1002519. [Google Scholar] [CrossRef] [PubMed]
- Surviladze, Z.; Sterk, R.T.; DeHaro, S.A.; Ozbun, M.A. Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy. J. Virol. 2013, 87, 2508–2517. [Google Scholar] [CrossRef] [PubMed]
- Papinski, D.; Kraft, C. Regulation of Autophagy By Signaling Through the Atg1/ULK1 Complex. J. Mol. Biol. 2016, 428 Pt A, 1725–1741. [Google Scholar] [CrossRef] [PubMed]
- Seglen, P.O.; Gordon, P.B. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, L.M.; Cicchini, L.; Pyeon, D. Human papillomavirus infection is inhibited by host autophagy in primary human keratinocytes. Virology 2013, 437, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y. Electron microscopic visualization of autophagosomes induced by infection of human papillomavirus pseudovirions. Biochem. Biophys. Res. Commun. 2013, 433, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Sapp, M.; Bienkowska-Haba, M. Viral entry mechanisms: Human papillomavirus and a long journey from extracellular matrix to the nucleus. FEBS J. 2009, 276, 7206–7216. [Google Scholar] [CrossRef] [PubMed]
- Pinidis, P.; Tsikouras, P.; Iatrakis, G.; Zervoudis, S.; Koukouli, Z.; Bothou, A.; Galazios, G.; Vladareanu, S. Human Papilloma Virus’ Life Cycle and Carcinogenesis. Maedica 2016, 11, 48–54. [Google Scholar] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Meisels, A.; Fortin, R. Condylomatous lesions of the cervix and vagina. I. Cytologic patterns. Acta Cytol. 1976, 20, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Venuti, A.; Paolini, F.; Nasir, L.; Corteggio, A.; Roperto, S.; Campo, M.S.; Borzacchiello, G. Papillomavirus E5: The smallest oncoprotein with many functions. Mol. Cancer 2011, 10, 140–158. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar] [PubMed]
- Darragh, T.M.; Colgan, T.J.; Thomas Cox, J.; Heller, D.S.; Henry, M.R.; Luff, R.D.; McCalmont, T.; Nayar, R.; Palefsky, J.M.; Stoler, M.H.; et al. The Lower Anogenital Squamous Terminology Standardization project for HPV-associated lesions: Background and consensus recommendations from the College of American Pathologists and the American Society for Colposcopy and Cervical Pathology. Int. J. Gynecol. Pathol. 2013, 32, 76–115. [Google Scholar] [CrossRef] [PubMed]
- Belleudi, F.; Nanni, M.; Raffa, S.; Torrisi, M.R. HPV16 E5 deregulates the autophagic process in human keratinocytes. Oncotarget 2015, 6, 9370–9386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattoscio, D.; Casadio, C.; Miccolo, C.; Maffini, F.; Raimondi, A.; Tacchetti, C.; Gheit, T.; Tagliabue, M.; Galimberti, V.E.; De Lorenzi, F.; et al. Autophagy regulates UBC9 levels during viral-mediated tumorigenesis. PLoS Pathog. 2017, 13, e1006262. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Munger, K. Expression of the human papillomavirus type 16 E7 oncoprotein induces an autophagy-related process and sensitizes normal human keratinocytes to cell death in response to growth factor deprivation. Virology 2009, 385, 192–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanning, J.E.; Saini, H.K.; Murray, M.J.; Caffarel, M.M.; van Dongen, S.; Ward, D.; Barker, E.M.; Scarpini, C.G.; Groves, I.J.; Stanley, M.A.; et al. Depletion of HPV16 early genes induces autophagy and senescence in a cervical carcinogenesis model, regardless of viral physical state. J. Pathol. 2013, 231, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Carchman, E.H.; Matkowskyj, K.A.; Meske, L.; Lambert, P.F. Dysregulation of Autophagy Contributes to Anal Carcinogenesis. PLoS ONE 2016, 11, e0164273. [Google Scholar] [CrossRef] [PubMed]
- Rademacher, B.L.; Matkowskyj, K.A.; Meske, L.M.; Romero, A.; Sleiman, H.; Carchman, E.H. The role of pharmacologic modulation of autophagy on anal cancer development in an HPV mouse model of carcinogenesis. Virology 2017, 507, 82–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.Y.; Zhang, Y.N.; Wu, Q.L.; Sun, X.M.; Sun, J.R.; Huang, X. Expression of beclin 1, an autophagy-related protein, in human cervical carcinoma and its clinical significance. Eur. J. Gynaecol. Oncol. 2012, 33, 15–20. [Google Scholar] [PubMed]
- Wang, Z.H.; Xu, L.; Wang, Y.; Cao, M.Q.; Li, L.; Bai, T. Clinicopathologic correlations between human papillomavirus 16 infection and Beclin 1 expression in human cervical cancer. Int. J. Gynecol. Pathol. 2011, 30, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Shu, S.; Yongmei, L.; Endong, Z.; Lirong, Y.; Bei, S. miR-224-3p inhibits autophagy in cervical cancer cells by targeting FIP200. Sci. Rep. 2016, 6, 33229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.Y.; Yang, G.F.; Huang, Y.H.; Huang, Q.W.; Gao, J.; Zhao, X.D.; Huang, L.M.; Chen, H.L. Reduced expression of autophagy markers correlates with high-risk human papillomavirus infection in human cervical squamous cell carcinoma. Oncol. Lett. 2014, 8, 1492–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.C.; Hung, Y.C.; Lin, T.Y.; Chang, H.W.; Chiang, I.P.; Chen, Y.Y.; Chow, K.C. Human papillomavirus infection and expression of ATPase family AAA domain containing 3A, a novel anti-autophagy factor, in uterine cervical cancer. Int. J. Mol. Med. 2011, 28, 689–696. [Google Scholar] [PubMed]
- Wang, F.; Shan, S.; Huo, Y.; Xie, Z.; Fang, Y.; Qi, Z.; Chen, F.; Li, Y.; Sun, B. MiR-155-5p inhibits PDK1 and promotes autophagy via the mTOR pathway in cervical cancer. Int. J. Biochem. Cell Biol. 2018, 99, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.; Natsume, T.; Guan, J.L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Leo, A.; Colavita, F.; Ciccosanti, F.; Fimia, G.M.; Lieberman, P.M.; Mattia, E. Inhibition of autophagy in EBV-positive Burkitt’s lymphoma cells enhances EBV lytic genes expression and replication. Cell Death Dis. 2015, 6, e1876. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Santarelli, R.; Farina, A.; Gonnella, R.; Lotti, L.V.; Faggioni, A.; Cirone, M. Epstein-barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J. Virol. 2014, 88, 12715–12726. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.P.; Kok, K.H.; Jin, D.Y. Human T-Cell Leukemia Virus Type 1 Infection and Adult T-Cell Leukemia. Adv. Exp. Med. Biol. 2017, 1018, 147–166. [Google Scholar] [PubMed]
- Tang, S.W.; Chen, C.Y.; Klase, Z.; Zane, L.; Jeang, K.T. The cellular autophagy pathway modulates human T-cell leukemia virus type 1 replication. J. Virol. 2013, 87, 1699–1707. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Takahashi, Y.; Liu, X.; Loughran, T.P.; Sun, S.C.; Wang, H.G.; Cheng, H. HTLV-1 Tax deregulates autophagy by recruiting autophagic molecules into lipid raft microdomains. Oncogene 2015, 34, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, J.; Shi, J.; Zhang, Y.; Liu, S.; Liu, Y.; Zheng, D. Human T-cell leukemia virus type 1 Tax-deregulated autophagy pathway and c-FLIP expression contribute to resistance against death receptor-mediated apoptosis. J. Virol. 2014, 88, 2786–2798. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, P.H.; Ziegelbauer, J.; Uldrick, T.S.; Yarchoan, R. Kaposi sarcoma herpesvirus-associated cancers and related diseases. Curr. Opin. HIV AIDS 2017, 12, 47–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leidal, A.M.; Lee, P.W.; McCormick, C. Viral subversion of autophagy impairs oncogene-induced senescence. Autophagy 2012, 8, 1138–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leidal, A.M.; Cyr, D.P.; Hill, R.J.; Lee, P.W.; McCormick, C. Subversion of autophagy by Kaposi’s sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell Host Microbe 2012, 11, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.J.; Yang, Z.; Zhou, Y.; Wood, C. Enhancement of autophagy during lytic replication by the Kaposi’s sarcoma-associated herpesvirus replication and transcription activator. J. Virol. 2010, 84, 7448–7458. [Google Scholar] [CrossRef] [PubMed]
- Michielsen, P.; Ho, E. Viral hepatitis B and hepatocellular carcinoma. Acta Gastro-Enterol. Belg. 2011, 74, 4–8. [Google Scholar]
- Sir, D.; Tian, Y.; Chen, W.L.; Ann, D.K.; Yen, T.S.; Ou, J.H. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl. Acad. Sci. USA 2010, 107, 4383–4388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Sir, D.; Kuo, C.F.; Ann, D.K.; Ou, J.H. Autophagy required for hepatitis B virus replication in transgenic mice. J. Virol. 2011, 85, 13453–13456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.T.; Chen, G.G.; Hu, B.G.; Zhang, Z.Y.; Yun, J.P.; He, M.L.; Lai, P.B. Hepatitis B virus x protein induces autophagy via activating death-associated protein kinase. J. Viral Hepat. 2014, 21, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Jin, M.; Ding, Y.; Zhang, Y.; Sun, Y.; Huang, S.; Xie, Q.; Xu, C.; Cai, W. Hepatitis B virus dampens autophagy maturation via negative regulation of Rab7 expression. Biosci. Trends 2016, 10, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, M.; Hu, Y.; Huang, B.; Li, N.; Chang, C.; Huang, R.; Xu, X.; Yang, Z.; Chen, Z.; et al. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy 2014, 10, 416–430. [Google Scholar] [CrossRef] [PubMed]
- Doring, T.; Zeyen, L.; Bartusch, C.; Prange, R. Hepatitis B Virus Subverts the Autophagy Elongation Complex Atg5-12/16L1 and Does Not Require Atg8/LC3 Lipidation for Viral Maturation. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.D.; Johnsen, I.B.; Stiberg, K.A.; Sherstova, T.; Wakita, T.; Richard, G.M.; Kandasamy, R.K.; Meurs, E.F.; Anthonsen, M.W. Hepatitis C virus triggers Golgi fragmentation and autophagy through the immunity-related GTPase M. Proc. Natl. Acad. Sci. USA 2017, 114, E3462–E3471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava, S.; Bhanja Chowdhury, J.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J. Virol. 2012, 86, 8705–8712. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tian, Y.; Ou, J.H. HCV induces the expression of Rubicon and UVRAG to temporally regulate the maturation of autophagosomes and viral replication. PLoS Pathog. 2015, 11, e1004764. [Google Scholar] [CrossRef] [PubMed]
- Taguwa, S.; Kambara, H.; Fujita, N.; Noda, T.; Yoshimori, T.; Koike, K.; Moriishi, K.; Matsuura, Y. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J. Virol. 2011, 85, 13185–13194. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Wang, L.; Lee, J.; Ou, J.J. Hepatitis C Virus Induces the Localization of Lipid Rafts to Autophagosomes for Its RNA Replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Devhare, P.; Sujijantarat, N.; Steele, R.; Kwon, Y.C.; Ray, R.; Ray, R.B. Knockdown of Autophagy Inhibits Infectious Hepatitis C Virus Release by the Exosomal Pathway. J. Virol. 2016, 90, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Kudchodkar, S.B.; Levine, B. Viruses and autophagy. Rev. Med. Virol. 2009, 19, 359–378. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.M.; Jung, J.U. Modulation of the autophagy pathway by human tumor viruses. Semin. Cancer Biol. 2013, 23, 323–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hundeshagen, P.; Hamacher-Brady, A.; Eils, R.; Brady, N.R. Concurrent detection of autolysosome formation and lysosomal degradation by flow cytometry in a high-content screen for inducers of autophagy. BMC Biol. 2011, 9, 38. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Tumor | Autophagic Protein Altered | Putative Use as Biomarker | Refs. |
|---|---|---|---|
| Anal dysplasia | LC3↑, p62↑ | Diagnosis, progression | [39] |
| Cervical dysplasia | p62↑ | Diagnosis, progression | [36] |
| Beclin 1↓ | Diagnosis, progression, prognosis | [41,42] | |
| miR-224-3p↑, FIP200↓ | Diagnosis, progression | [43] | |
| Cervical cancer | LC3↓ | HPV infection, diagnosis, prognosis | [44] |
| ATAD3A↑ | HPV infection, diagnosis, prognosis, therapeutic predictivity | [45] | |
| miR-224-3p↑, FIP200↓ | Diagnosis | [43] | |
| miR-155-5p↓ | Diagnosis | [46] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mattoscio, D.; Medda, A.; Chiocca, S. Human Papilloma Virus and Autophagy. Int. J. Mol. Sci. 2018, 19, 1775. https://doi.org/10.3390/ijms19061775
Mattoscio D, Medda A, Chiocca S. Human Papilloma Virus and Autophagy. International Journal of Molecular Sciences. 2018; 19(6):1775. https://doi.org/10.3390/ijms19061775
Chicago/Turabian StyleMattoscio, Domenico, Alessandro Medda, and Susanna Chiocca. 2018. "Human Papilloma Virus and Autophagy" International Journal of Molecular Sciences 19, no. 6: 1775. https://doi.org/10.3390/ijms19061775
APA StyleMattoscio, D., Medda, A., & Chiocca, S. (2018). Human Papilloma Virus and Autophagy. International Journal of Molecular Sciences, 19(6), 1775. https://doi.org/10.3390/ijms19061775