STAT3 Interactors as Potential Therapeutic Targets for Cancer Treatment




Abstract
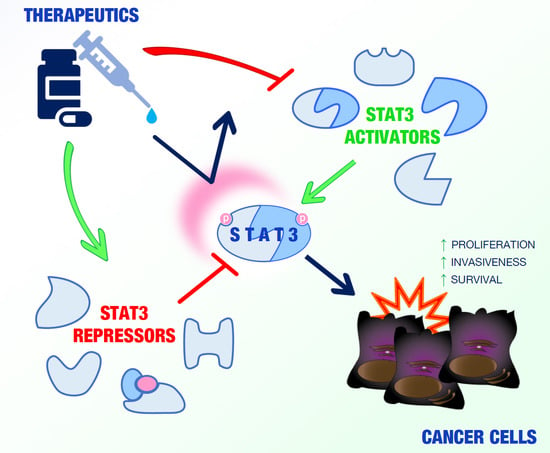
:
1. Introduction
2. STAT3-Interacting Proteins
2.1. STAT3 Activators
2.1.1. STAT3 Activators in the Cytosol
2.1.2. STAT3 Activators in the Nucleus
2.1.3. STAT3 Activators in the Mitochondrion
2.2. STAT3 Repressors
3. Conclusions
Author Contributions
Acknowledgments
Conflict of interest
References
- Abroun, S.; Saki, N.; Ahmadvand, M.; Asghari, F.; Salari, F.; Rahim, F. STATs: An Old Story, Yet Mesmerizing. Cell J. 2015, 17, 395–411. [Google Scholar] [PubMed]
- Miklossy, G.; Hilliard, T.S.; Turkson, J. Therapeutic modulators of STAT signalling for human diseases. Nat. Rev. Drug Discov. 2013, 12, 611–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demaria, M.; Camporeale, A.; Poli, V. STAT3 and metabolism: How many ways to use a single molecule? Int. J. Cancer 2014, 135, 1997–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.L.; Lo, H.W. STAT3 Target Genes Relevant to Human Cancers. Cancers 2014, 6, 897–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbers, C.; Aparicio-Siegmund, S.; Rose-John, S. The IL-6/gp130/STAT3 signaling axis: Recent advances towards specific inhibition. Curr. Opin. Immunol. 2015, 34, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117 Pt 8, 1281–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bromberg, J.; Darnell, J.E., Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000, 19, 2468–2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turkson, J.; Bowman, T.; Garcia, R.; Caldenhoven, E.; De Groot, R.P.; Jove, R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol. Cell. Biol. 1998, 18, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Coppo, P.; Dusanter-Fourt, I.; Millot, G.; Nogueira, M.M.; Dugray, A.; Bonnet, M.L.; Mitjavila-Garcia, M.T.; Le Pesteur, D.; Guilhot, F.; Vainchenker, W.; et al. Constitutive and specific activation of STAT3 by BCR-ABL in embryonic stem cells. Oncogene 2003, 22, 4102–4110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriuranpong, V.; Park, J.I.; Amornphimoltham, P.; Patel, V.; Nelkin, B.D.; Gutkind, J.S. Epidermal growth factor receptor-independent constitutive activation of STAT3 in head and neck squamous cell carcinoma is mediated by the autocrine/paracrine stimulation of the interleukin 6/gp130 cytokine system. Cancer Res. 2003, 63, 2948–2956. [Google Scholar] [PubMed]
- Coppo, P.; Flamant, S.; De Mas, V.; Jarrier, P.; Guillier, M.; Bonnet, M.L.; Lacout, C.; Guilhot, F.; Vainchenker, W.; Turhan, A.G. BCR-ABL activates STAT3 via JAK and MEK pathways in human cells. Br. J. Haematol. 2006, 134, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andres, R.M.; Hald, A.; Johansen, C.; Kragballe, K.; Iversen, L. Studies of Jak/STAT3 expression and signalling in psoriasis identifies STAT3-Ser727 phosphorylation as a modulator of transcriptional activity. Exp. Dermatol. 2013, 22, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhang, H.; Paddon, H.; Lee, G.; Cao, X.; Pelech, S. Phosphorylation of STAT3 serine-727 by cyclin-dependent kinase 1 is critical for nocodazole-induced mitotic arrest. Biochemistry 2006, 45, 5857–5867. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Rincon, M. Mitochondrial Stat3, the Need for Design Thinking. Int. J. Biol. Sci. 2016, 12, 532–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, J.A.; Larner, A.C. Toward a new STATe: The role of STATs in mitochondrial function. Semin. Immunol. 2014, 26, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, M.; Dermawan, J.K.; Willard, B.; Stark, G.R. STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc. Natl. Acad. Sci. USA 2015, 112, 3985–3990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, E.; Tsruya, R.; Levitsky, P.; Pomp, O.; Taller, M.; Weisberg, S.; Parris, W.; Kulkarni, S.; Malovani, H.; Pawson, T.; et al. TMF/ARA160 is a BC-box-containing protein that mediates the degradation of Stat3. Oncogene 2004, 23, 8908–8919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, M.; Li, J.; Xiao, M.; Chin, Y.E.; Cheng, J.; Yeh, E.T.; Yang, J.; Yi, J. SUMOylation and SENP3 regulate STAT3 activation in head and neck cancer. Oncogene 2016, 35, 5826–5838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butturini, E.; Darra, E.; Chiavegato, G.; Cellini, B.; Cozzolino, F.; Monti, M.; Pucci, P.; Dell’Orco, D.; Mariotto, S. S-Glutathionylation at Cys328 and Cys542 impairs STAT3 phosphorylation. ACS Chem. Biol. 2014, 9, 1885–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Won, J.S.; Singh, A.K.; Sharma, A.K.; Singh, I. STAT3 regulation by S-nitrosylation: Implication for inflammatory disease. Antioxid. Redox Signal. 2014, 20, 2514–2527. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Hanada, T.; Mitsuyama, K.; Yoshida, T.; Kamizono, S.; Hoshino, T.; Kubo, M.; Yamashita, A.; Okabe, M.; Takeda, K.; et al. CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J. Exp. Med. 2001, 193, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Liu, B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 2003, 3, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, H.; Sasaki, A.; Yoshimura, A. Negative regulation of cytokine signaling pathways. Annu. Rev. Immunol. 2000, 18, 143–164. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Lee, C.K. What does Stat3 do? J. Clin. Investig. 2002, 109, 1143–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Chatterjee-Kishore, M.; Staugaitis, S.M.; Nguyen, H.; Schlessinger, K.; Levy, D.E.; Stark, G.R. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005, 65, 939–947. [Google Scholar] [PubMed]
- Avalle, L.; Pensa, S.; Regis, G.; Novelli, F.; Poli, V. STAT1 and STAT3 in tumorigenesis: A matter of balance. Jak-Stat 2012, 1, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Nero, T.L.; Morton, C.J.; Holien, J.K.; Wielens, J.; Parker, M.W. Oncogenic protein interfaces: Small molecules, big challenges. Nat. Rev. Cancer 2014, 14, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Pott, S.; Lieb, J.D. What are super-enhancers? Nat. Genet. 2015, 47, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Schuijers, J.; Lin, C.Y.; Weintraub, A.S.; Abraham, B.J.; Lee, T.I.; Bradner, J.E.; Young, R.A. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell 2015, 58, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Mitra, S.; Spolski, R.; Oh, J.; Liao, W.; Tang, Z.; Mo, F.; Li, X.; West, E.E.; Gromer, D.; et al. STAT5-mediated chromatin interactions in superenhancers activate IL-2 highly inducible genes: Functional dissection of the Il2ra gene locus. Proc. Natl. Acad. Sci. USA 2017, 114, 12111–12119. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Yamamoto, T.; Sekine, Y.; Yumioka, T.; Junicho, A.; Fuse, H.; Matsuda, T. Involvement of heat-shock protein 90 in the interleukin-6-mediated signaling pathway through STAT3. Biochem. Biophys. Res. Commun. 2003, 300, 847–852. [Google Scholar] [CrossRef]
- Tsai, C.L.; Chao, A.; Jung, S.M.; Tsai, C.N.; Lin, C.Y.; Chen, S.H.; Sue, S.C.; Wang, T.H.; Wang, H.S.; Lai, C.H. Stress-induced phosphoprotein-1 maintains the stability of JAK2 in cancer cells. Oncotarget 2016, 7, 50548–50563. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, O.; Miyasaka, Y.; Sekine, Y.; Mizushima, A.; Muromoto, R.; Nanbo, A.; Yoshimura, A.; Matsuda, T. STAP-2 is phosphorylated at tyrosine-250 by Brk and modulates Brk-mediated STAT3 activation. Biochem. Biophys. Res. Commun. 2009, 384, 71–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudka, A.A.; Sweet, S.M.; Heath, J.K. Signal transducers and activators of transcription-3 binding to the fibroblast growth factor receptor is activated by receptor amplification. Cancer Res. 2010, 70, 3391–3401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, F.; Li, W.; Xiong, Q.; Yang, M.; Zheng, P.; Li, C.; Pei, J.; Ge, F. 14-3-3zeta interacts with stat3 and regulates its constitutive activation in multiple myeloma cells. PLoS ONE 2012, 7, e29554. [Google Scholar]
- Song, X.; Hao, J.; Wang, J.; Guo, C.; Wang, Y.; He, Q.; Tang, H.; Qin, X.; Li, Y.; Zhang, Y.; et al. The cancer/testis antigen MAGEC2 promotes amoeboid invasion of tumor cells by enhancing STAT3 signaling. Oncogene 2017, 36, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; McBride, K.M.; Reich, N.C. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc. Natl. Acad. Sci. USA 2005, 102, 8150–8155. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Cao, X. Regulation of Stat3 nuclear import by importin alpha5 and importin alpha7 via two different functional sequence elements. Cell. Signal. 2006, 18, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Muromoto, R.; Okabe, K.; Fujimuro, M.; Sugiyama, K.; Yokosawa, H.; Seya, T.; Matsuda, T. Physical and functional interactions between STAT3 and Kaposi’s sarcoma-associated herpesvirus-encoded LANA. FEBS Lett. 2006, 580, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Muromoto, R.; Ikeda, O.; Okabe, K.; Togi, S.; Kamitani, S.; Fujimuro, M.; Harada, S.; Oritani, K.; Matsuda, T. Epstein-Barr virus-derived EBNA2 regulates STAT3 activation. Biochem. Biophys. Res. Commun. 2009, 378, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Muromoto, R.; Sekine, Y.; Imoto, S.; Ikeda, O.; Okayama, T.; Sato, N.; Matsuda, T. BART is essential for nuclear retention of STAT3. Int. Immunol. 2008, 20, 395–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Togi, S.; Muromoto, R.; Hirashima, K.; Kitai, Y.; Okayama, T.; Ikeda, O.; Matsumoto, N.; Kon, S.; Sekine, Y.; Oritani, K.; et al. A New STAT3-binding Partner, ARL3, Enhances the Phosphorylation and Nuclear Accumulation of STAT3. J. Biol. Chem. 2016, 291, 11161–11171. [Google Scholar] [CrossRef] [PubMed]
- Ohbayashi, N.; Taira, N.; Kawakami, S.; Togi, S.; Sato, N.; Ikeda, O.; Kamitani, S.; Muromoto, R.; Sekine, Y.; Matsuda, T. An RNA biding protein, Y14 interacts with and modulates STAT3 activation. Biochem. Biophys. Res. Commun. 2008, 372, 475–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.S.; Zhang, H.X.; Zhang, Y.L.; Liu, T.T.; Ran, Y.; Chen, L.T.; Wang, Y.Y.; Shu, H.B. PASD1 promotes STAT3 activity and tumor growth by inhibiting TC45-mediated dephosphorylation of STAT3 in the nucleus. J. Mol. Cell Biol. 2016, 8, 221–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manavathi, B.; Nair, S.S.; Wang, R.A.; Kumar, R.; Vadlamudi, R.K. Proline-, glutamic acid-, and leucine-rich protein-1 is essential in growth factor regulation of signal transducers and activators of transcription 3 activation. Cancer Res. 2005, 65, 5571–5577. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, A.; Kugimiya, N.; Hosoyama, T.; Enoki, T.; Li, T.S.; Hamano, K. JAB1 regulates unphosphorylated STAT3 DNA-binding activity through protein-protein interaction in human colon cancer cells. Biochem. Biophys. Res. Commun. 2013, 438, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.E.; Kreimer, S.; Walker, S.R.; Emori, M.M.; Krystal, H.; Richardson, A.; Ivanov, A.R.; Frank, D.A. Granulin, a novel STAT3-interacting protein, enhances STAT3 transcriptional function and correlates with poorer prognosis in breast cancer. Genes Cancer 2015, 6, 153–168. [Google Scholar] [PubMed]
- Kwon, M.C.; Koo, B.K.; Moon, J.S.; Kim, Y.Y.; Park, K.C.; Kim, N.S.; Kwon, M.Y.; Kong, M.P.; Yoon, K.J.; Im, S.K.; et al. Crif1 is a novel transcriptional coactivator of STAT3. EMBO J. 2008, 27, 642–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.A.; Bai, S.; Grossman, G.; Titus, M.A.; Harris Ford, O.; Pop, E.A.; Smith, G.J.; Mohler, J.L.; Wilson, E.M.; French, F.S. Mechanism of androgen receptor corepression by CKbetaBP2/CRIF1, a multifunctional transcription factor coregulator expressed in prostate cancer. Mol. Cell. Endocrinol. 2014, 382, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, S.; Yue, P.; Paladino, D.C.; Bogdanovic, J.; Huo, Q.; Turkson, J. A functional nuclear epidermal growth factor receptor, SRC and Stat3 heteromeric complex in pancreatic cancer cells. PLoS ONE 2011, 6, e19605. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Cherukuri, P.; Luo, J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J. Biol. Chem. 2005, 280, 11528–11534. [Google Scholar] [CrossRef] [PubMed]
- Giraud, S.; Bienvenu, F.; Avril, S.; Gascan, H.; Heery, D.M.; Coqueret, O. Functional interaction of STAT3 transcription factor with the coactivator NcoA/SRC1a. J. Biol. Chem. 2002, 277, 8004–8011. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Han, J.J.; Stenson, M.; Wellik, L.; Witzig, T.E. Regulation of STAT3 by histone deacetylase-3 in diffuse large B-cell lymphoma: Implications for therapy. Leukemia 2012, 26, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, G.G.; Huang, L.; Alston, N.; Ouyang, N.; Vrankova, K.; Mattheolabakis, G.; Constantinides, P.P.; Rigas, B. Targeting mitochondrial STAT3 with the novel phospho-valproic acid (MDC-1112) inhibits pancreatic cancer growth in mice. PLoS ONE 2013, 8, e61532. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific inhibition of Stat3 signal transduction by PIAS3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Chung, J.; Kao, H.Y.; Yang, Y.C. Tip60 is a co-repressor for STAT3. J. Biol. Chem. 2003, 278, 11197–11204. [Google Scholar] [CrossRef] [PubMed]
- Muromoto, R.; Nakao, K.; Watanabe, T.; Sato, N.; Sekine, Y.; Sugiyama, K.; Oritani, K.; Shimoda, K.; Matsuda, T. Physical and functional interactions between Daxx and STAT3. Oncogene 2006, 25, 2131–2136. [Google Scholar] [CrossRef] [PubMed]
- Tsuruma, R.; Ohbayashi, N.; Kamitani, S.; Ikeda, O.; Sato, N.; Muromoto, R.; Sekine, Y.; Oritani, K.; Matsuda, T. Physical and functional interactions between STAT3 and KAP1. Oncogene 2008, 27, 3054–3059. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Yamamoto, Y.; Muromoto, R.; Ikeda, O.; Sekine, Y.; Grusby, M.J.; Kaisho, T.; Matsuda, T. PDLIM2 inhibits T helper 17 cell development and granulomatous inflammation through degradation of STAT3. Sci. Signal. 2011, 4, ra85. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhang, X.; Yang, J.; Wu, G.; Zhang, Y.; Yuan, Y.; Jin, C.; Chang, Z.; Wang, J.; Yang, X.; et al. Nuclear protein IkappaB-zeta inhibits the activity of STAT3. Biochem. Biophys. Res. Commun. 2009, 387, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zhang, K.; Li, S.; Li, H.; Yan, Z.; Huang, L.; Wu, J.; Han, X.; Jiang, W.; Mulatibieke, T.; et al. HIC1 attenuates invasion and metastasis by inhibiting the IL-6/STAT3 signalling pathway in human pancreatic cancer. Cancer Lett. 2016, 376, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.M.; Wang, C.M.; Jeng, J.C.; Leprince, D.; Shih, H.M. HIC1 interacts with and modulates the activity of STAT3. Cell Cycle 2013, 12, 2266–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, S.; Xu, F.; Chen, Y.; Ge, Y.; Zhang, F.; Huang, H.; Li, L.; Lin, D.; Luo, X.; Xu, J.; et al. Fbw7 regulates apoptosis in activated B-cell like diffuse large B-cell lymphoma by targeting Stat3 for ubiquitylation and degradation. J. Exp. Clin. Cancer Res. CR 2017, 36, 10. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Su, F.; Ning, H.; Wang, Y.; Geng, Y.; Feng, Y.; Wang, Y.; Zhang, Y.; Jin, Z.; Li, Y.; et al. SIPAR negatively regulates STAT3 signaling and inhibits progression of melanoma. Cell. Signal. 2013, 25, 2272–2280. [Google Scholar] [CrossRef] [PubMed]
- Icardi, L.; Mori, R.; Gesellchen, V.; Eyckerman, S.; De Cauwer, L.; Verhelst, J.; Vercauteren, K.; Saelens, X.; Meuleman, P.; Leroux-Roels, G.; et al. The Sin3a repressor complex is a master regulator of STAT transcriptional activity. Proc. Natl. Acad. Sci. USA 2012, 109, 12058–12063. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, J.; Roy, S.K.; Tininini, S.; Hu, J.; Bromberg, J.F.; Poli, V.; Stark, G.R.; Kalvakolanu, D.V. The cell death regulator GRIM-19 is an inhibitor of signal transducer and activator of transcription 3. Proc. Natl. Acad. Sci. USA 2003, 100, 9342–9347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Yang, M.; Hu, H.; Zhao, X.; Bao, L.; Huang, D.; Song, L.; Li, Y. Mitochondrial GRIM-19 as a potential therapeutic target for STAT3-dependent carcinogenesis of gastric cancer. Oncotarget 2016, 7, 41404–41420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavecchio, M.; Lisanti, S.; Lam, A.; Ghosh, J.C.; Martin, N.M.; O’Connell, M.; Weeraratna, A.T.; Kossenkov, A.V.; Showe, L.C.; Altieri, D.C. Cyclophilin D extramitochondrial signaling controls cell cycle progression and chemokine-directed cell motility. J. Biol. Chem. 2013, 288, 5553–5561. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Haslbeck, M.; Buchner, J. The heat shock response: Life on the verge of death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef] [PubMed]
- Odunuga, O.O.; Longshaw, V.M.; Blatch, G.L. Hop: More than an Hsp70/Hsp90 adaptor protein. BioEssays News Rev. Mol. Cell. Dev. Biol. 2004, 26, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Xing, B.; Sun, Y.; Du, X.; Lu, M.; Hao, C.; Lu, Z.; Mi, W.; Wu, S.; Wei, H.; et al. Proteome analysis of hepatocellular carcinoma by two-dimensional difference gel electrophoresis: Novel protein markers in hepatocellular carcinoma tissues. Mol. Cell. Proteom. MCP 2007, 6, 1798–1808. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.; O’Donovan, N.; Kennedy, S.; Henry, M.; Meleady, P.; Clynes, M.; Dowling, P. Identification of pancreatic cancer invasion-related proteins by proteomic analysis. Proteom. Sci. 2009, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, H.; Yamamoto, S.; Itoh, E.; Abe, Y.; Nakamura, A.; Izumi, Y.; Okada, H.; Iida, M.; Nanjo, H.; Itoh, H.; et al. Increased expression of co-chaperone HOP with HSP90 and HSC70 and complex formation in human colonic carcinoma. Cell Stress Chaperones 2010, 15, 1003–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, A.; Lai, C.H.; Tsai, C.L.; Hsueh, S.; Hsueh, C.; Lin, C.Y.; Chou, H.H.; Lin, Y.J.; Chen, H.W.; Chang, T.C.; et al. Tumor stress-induced phosphoprotein1 (STIP1) as a prognostic biomarker in ovarian cancer. PLoS ONE 2013, 8, e57084. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.; Larkin, A.; Swan, N.; Conlon, K.; Dowling, P.; McDermott, R.; Clynes, M. RNAi knockdown of Hop (Hsp70/Hsp90 organising protein) decreases invasion via MMP-2 down regulation. Cancer Lett. 2011, 306, 180–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minoguchi, M.; Minoguchi, S.; Aki, D.; Joo, A.; Yamamoto, T.; Yumioka, T.; Matsuda, T.; Yoshimura, A. STAP-2/BKS, an adaptor/docking protein, modulates STAT3 activation in acute-phase response through its YXXQ motif. J. Biol. Chem. 2003, 278, 11182–11189. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, O.; Sekine, Y.; Mizushima, A.; Nakasuji, M.; Miyasaka, Y.; Yamamoto, C.; Muromoto, R.; Nanbo, A.; Oritani, K.; Yoshimura, A.; et al. Interactions of STAP-2 with Brk and STAT3 participate in cell growth of human breast cancer cells. J. Biol. Chem. 2010, 285, 38093–38103. [Google Scholar] [CrossRef] [PubMed]
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araujo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. FGFR a promising druggable target in cancer: Molecular biology and new drugs. Crit. Rev. Oncol./Hematol. 2017, 113, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.; Subramanian, R.R.; Masters, S.C. 14-3-3 proteins: Structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 617–647. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K. The 14-3-3 proteins: Integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol. 2009, 19, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Simpson, A.J.; Caballero, O.L.; Jungbluth, A.; Chen, Y.T.; Old, L.J. Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer 2005, 5, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhou, X.; Miao, X.; Zhang, T.; Hang, X.; Tie, R.; Liu, N.; Tian, F.; Wang, F.; Yuan, J. MAGEC2, an epithelial-mesenchymal transition inducer, is associated with breast cancer metastasis. Breast Cancer Res. Treat. 2014, 145, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curioni-Fontecedro, A.; Nuber, N.; Mihic-Probst, D.; Seifert, B.; Soldini, D.; Dummer, R.; Knuth, A.; van den Broek, M.; Moch, H. Expression of MAGE-C1/CT7 and MAGE-C2/CT10 predicts lymph node metastasis in melanoma patients. PLoS ONE 2011, 6, e21418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koganti, S.; Hui-Yuen, J.; McAllister, S.; Gardner, B.; Grasser, F.; Palendira, U.; Tangye, S.G.; Freeman, A.F.; Bhaduri-McIntosh, S. STAT3 interrupts ATR-Chk1 signaling to allow oncovirus-mediated cell proliferation. Proc. Natl. Acad. Sci. USA 2014, 111, 4946–4951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, F.; Xiao, Y.; Qu, Z. Oncovirus Kaposi sarcoma herpesvirus (KSHV) represses tumor suppressor PDLIM2 to persistently activate nuclear factor kappaB (NF-kappaB) and STAT3 transcription factors for tumorigenesis and tumor maintenance. J. Biol. Chem. 2015, 290, 7362–7368. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Cunningham, L.; Marcus, A.I.; Li, Y.; Kahn, R.A. Arl2 and Arl3 regulate different microtubule-dependent processes. Mol. Biol. Cell 2006, 17, 2476–2487. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, D.; Tekmal, R.R.; Vadlamudi, R.K. PELP1: A novel therapeutic target for hormonal cancers. IUBMB Life 2010, 62, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Plowman, G.D.; Green, J.M.; Neubauer, M.G.; Buckley, S.D.; McDonald, V.L.; Todaro, G.J.; Shoyab, M. The epithelin precursor encodes two proteins with opposing activities on epithelial cell growth. J. Biol. Chem. 1992, 267, 13073–13078. [Google Scholar] [PubMed]
- Zhou, J.; Gao, G.; Crabb, J.W.; Serrero, G. Purification of an autocrine growth factor homologous with mouse epithelin precursor from a highly tumorigenic cell line. J. Biol. Chem. 1993, 268, 10863–10869. [Google Scholar] [PubMed]
- Lu, R.; Serrero, G. Inhibition of PC cell-derived growth factor (PCDGF, epithelin/granulin precursor) expression by antisense PCDGF cDNA transfection inhibits tumorigenicity of the human breast carcinoma cell line MDA-MB-468. Proc. Natl. Acad. Sci. USA 2000, 97, 3993–3998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.B.; Michener, C.M.; Blanchette, J.O.; Kuznetsov, V.A.; Raffeld, M.; Serrero, G.; Emmert-Buck, M.R.; Petricoin, E.F.; Krizman, D.B.; Liotta, L.A.; et al. The granulin-epithelin precursor/PC-cell-derived growth factor is a growth factor for epithelial ovarian cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 44–51. [Google Scholar]
- He, Z.; Ong, C.H.; Halper, J.; Bateman, A. Progranulin is a mediator of the wound response. Nat. Med. 2003, 9, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.K.; Yi, Y.W.; Jung, N.C.; Kim, D.; Suh, J.M.; Kim, H.; Park, K.C.; Song, J.H.; Kim, D.W.; Hwang, E.S.; et al. CR6-interacting factor 1 interacts with Gadd45 family proteins and modulates the cell cycle. J. Biol. Chem. 2003, 278, 28079–28088. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kwon, M.C.; Ryu, M.J.; Chung, H.K.; Tadi, S.; Kim, Y.K.; Kim, J.M.; Lee, S.H.; Park, J.H.; Kweon, G.R.; et al. CRIF1 is essential for the synthesis and insertion of oxidative phosphorylation polypeptides in the mammalian mitochondrial membrane. Cell Metab. 2012, 16, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Tomas, A.; Futter, C.E.; Eden, E.R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 2014, 24, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hsu, S.C.; Hung, M.C. EGFR signaling pathway in breast cancers: From traditional signal transduction to direct nuclear translocalization. Breast Cancer Res. Treat. 2006, 95, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hsu, S.C.; Ali-Seyed, M.; Gunduz, M.; Xia, W.; Wei, Y.; Bartholomeusz, G.; Shih, J.Y.; Hung, M.C. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005, 7, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Garama, D.J.; White, C.L.; Balic, J.J.; Gough, D.J. Mitochondrial STAT3: Powering up a potent factor. Cytokine 2016, 87, 20–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Raje, V.; Yakovlev, V.A.; Yacoub, A.; Szczepanek, K.; Meier, J.; Derecka, M.; Chen, Q.; Hu, Y.; Sisler, J.; et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J. Biol. Chem. 2013, 288, 31280–31288. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Liu, B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Ogata, Y.; Osaki, T.; Naka, T.; Iwahori, K.; Furukawa, M.; Nagatomo, I.; Kijima, T.; Kumagai, T.; Yoshida, M.; Tachibana, I.; et al. Overexpression of PIAS3 suppresses cell growth and restores the drug sensitivity of human lung cancer cells in association with PI3-K/Akt inactivation. Neoplasia 2006, 8, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Junicho, A.; Matsuda, T.; Yamamoto, T.; Kishi, H.; Korkmaz, K.; Saatcioglu, F.; Fuse, H.; Muraguchi, A. Protein inhibitor of activated STAT3 regulates androgen receptor signaling in prostate carcinoma cells. Biochem. Biophys. Res. Commun. 2000, 278, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Ikura, T.; Ogryzko, V.V.; Grigoriev, M.; Groisman, R.; Wang, J.; Horikoshi, M.; Scully, R.; Qin, J.; Nakatani, Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 2000, 102, 463–473. [Google Scholar] [CrossRef]
- Hlubek, F.; Lohberg, C.; Meiler, J.; Jung, A.; Kirchner, T.; Brabletz, T. Tip60 is a cell-type-specific transcriptional regulator. J. Biochem. 2001, 129, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Murr, R.; Loizou, J.I.; Yang, Y.G.; Cuenin, C.; Li, H.; Wang, Z.Q.; Herceg, Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Moschos, S.; Varanasi, S.; Kirkwood, J.M. Interferons in the treatment of solid tumors. Cancer Treat. Res. 2005, 126, 207–241. [Google Scholar] [PubMed]
- Stagg, J.; Loi, S.; Divisekera, U.; Ngiow, S.F.; Duret, H.; Yagita, H.; Teng, M.W.; Smyth, M.J. Anti-ErbB-2 mAb therapy requires type I and II interferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 7142–7147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisirak, V.; Faget, J.; Gobert, M.; Goutagny, N.; Vey, N.; Treilleux, I.; Renaudineau, S.; Poyet, G.; Labidi-Galy, S.I.; Goddard-Leon, S.; et al. Impaired IFN-alpha production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res. 2012, 72, 5188–5197. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Smyth, M.J.; Kroemer, G. Mechanism of action of conventional and targeted anticancer therapies: Reinstating immunosurveillance. Immunity 2013, 39, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, X.; Fu, M.L.; Weichselbaum, R.R.; Gajewski, T.F.; Guo, Y.; Fu, Y.X. Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell 2014, 25, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Pei, H.; Watson, D.K.; Papas, T.S. EAP1/Daxx interacts with ETS1 and represses transcriptional activation of ETS1 target genes. Oncogene 2000, 19, 745–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.C.; Lin, D.Y.; Fang, H.I.; Chen, R.H.; Shih, H.M. Daxx mediates the small ubiquitin-like modifier-dependent transcriptional repression of Smad4. J. Biol. Chem. 2005, 280, 10164–10173. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, J.H.; La, M.; Jang, M.J.; Chae, G.W.; Kim, S.B.; Tak, H.; Jung, Y.; Byun, B.; Ahn, J.K.; et al. Inhibition of NF-kappaB acetylation and its transcriptional activity by Daxx. J. Mol. Biol. 2007, 368, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Puto, L.A.; Reed, J.C. Daxx represses RelB target promoters via DNA methyltransferase recruitment and DNA hypermethylation. Genes Dev. 2008, 22, 998–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Z.; Zhong, Z.; Darnell, J.E., Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef]
- Wei, J.; Yuan, Y.; Jin, C.; Chen, H.; Leng, L.; He, F.; Wang, J. The ubiquitin ligase TRAF6 negatively regulates the JAK-STAT signaling pathway by binding to STAT3 and mediating its ubiquitination. PLoS ONE 2012, 7, e49567. [Google Scholar] [CrossRef] [PubMed]
- Romo-Tena, J.; Rajme-Lopez, S.; Aparicio-Vera, L.; Alcocer-Varela, J.; Gomez-Martin, D. Lys63-polyubiquitination by the E3 ligase casitas B-lineage lymphoma-b (Cbl-b) modulates peripheral regulatory T cell tolerance in patients with systemic lupus erythematosus. Clin. Exp. Immunol. 2018, 191, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Kanehira, K.; Okita, K.; Morimatsu, M.; Saito, M. MAIL, a novel nuclear I kappa B protein that potentiates LPS-induced IL-6 production. FEBS Lett. 2000, 485, 53–56. [Google Scholar] [CrossRef]
- Yamazaki, S.; Muta, T.; Takeshige, K. A novel IkappaB protein, IkappaB-zeta, induced by proinflammatory stimuli, negatively regulates nuclear factor-kappaB in the nuclei. J. Biol. Chem. 2001, 276, 27657–27662. [Google Scholar] [CrossRef] [PubMed]
- Rood, B.R.; Leprince, D. Deciphering HIC1 control pathways to reveal new avenues in cancer therapeutics. Expert Opin. Ther. Targets 2013, 17, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J.; Welcker, M.; Clurman, B.E. Tumor suppression by the Fbw7 ubiquitin ligase: Mechanisms and opportunities. Cancer Cell 2014, 26, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Geng, Y.; Minami, T.; Qiu, Y.; Feng, Y.; Liu, C.; Zhao, J.; Wang, Y.; Fan, X.; Wang, Y.; et al. Nuclear termination of STAT3 signaling through SIPAR (STAT3-Interacting Protein As a Repressor)-dependent recruitment of T cell tyrosine phosphatase TC-PTP. FEBS Lett. 2015, 589, 1890–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, M.J.; Liu, J.; Libby, E.F.; Lee, M.; Crawford, N.P.; Hurst, D.R. SIN3A and SIN3B differentially regulate breast cancer metastasis. Oncotarget 2016, 7, 78713–78725. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; David, G.; Farias, E.; Waxman, S. Emerging Roles of Epigenetic Regulator Sin3 in Cancer. Adv. Cancer Res. 2016, 130, 113–135. [Google Scholar] [PubMed]
- Tammineni, P.; Anugula, C.; Mohammed, F.; Anjaneyulu, M.; Larner, A.C.; Sepuri, N.B. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J. Biol. Chem. 2013, 288, 4723–4732. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Hilfiker-Kleiner, D.; Heusch, G.; Schulz, R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res. Cardiol. 2010, 105, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avalle, L.; Camporeale, A.; Camperi, A.; Poli, V. STAT3 in cancer: A double edged sword. Cytokine 2017, 98, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-kappaB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Kesanakurti, D.; Chetty, C.; Rajasekhar Maddirela, D.; Gujrati, M.; Rao, J.S. Essential role of cooperative NF-kappaB and Stat3 recruitment to ICAM-1 intronic consensus elements in the regulation of radiation-induced invasion and migration in glioma. Oncogene 2013, 32, 5144–5155. [Google Scholar] [CrossRef] [PubMed]
- Kamitani, S.; Togi, S.; Ikeda, O.; Nakasuji, M.; Sakauchi, A.; Sekine, Y.; Muromoto, R.; Oritani, K.; Matsuda, T. Kruppel-associated box-associated protein 1 negatively regulates TNF-alpha-induced NF-kappaB transcriptional activity by influencing the interactions among STAT3, p300, and NF-kappaB/p65. J. Immunol. 2011, 187, 2476–2483. [Google Scholar] [CrossRef] [PubMed]
- Togi, S.; Shiga, K.; Muromoto, R.; Kato, M.; Souma, Y.; Sekine, Y.; Kon, S.; Oritani, K.; Matsuda, T. Y14 positively regulates TNF-alpha-induced NF-kappaB transcriptional activity via interacting RIP1 and TRADD beyond an exon junction complex protein. J. Immunol. 2013, 191, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Sekine, Y.; Yumioka, T.; Yamamoto, T.; Muromoto, R.; Imoto, S.; Sugiyma, K.; Oritani, K.; Shimoda, K.; Minoguchi, M.; Akira, S.; et al. Modulation of TLR4 signaling by a novel adaptor protein signal-transducing adaptor protein-2 in macrophages. J. Immunol. 2006, 176, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Grusby, M.J.; Kaisho, T. PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat. Immunol. 2007, 8, 584–591. [Google Scholar] [CrossRef] [PubMed]


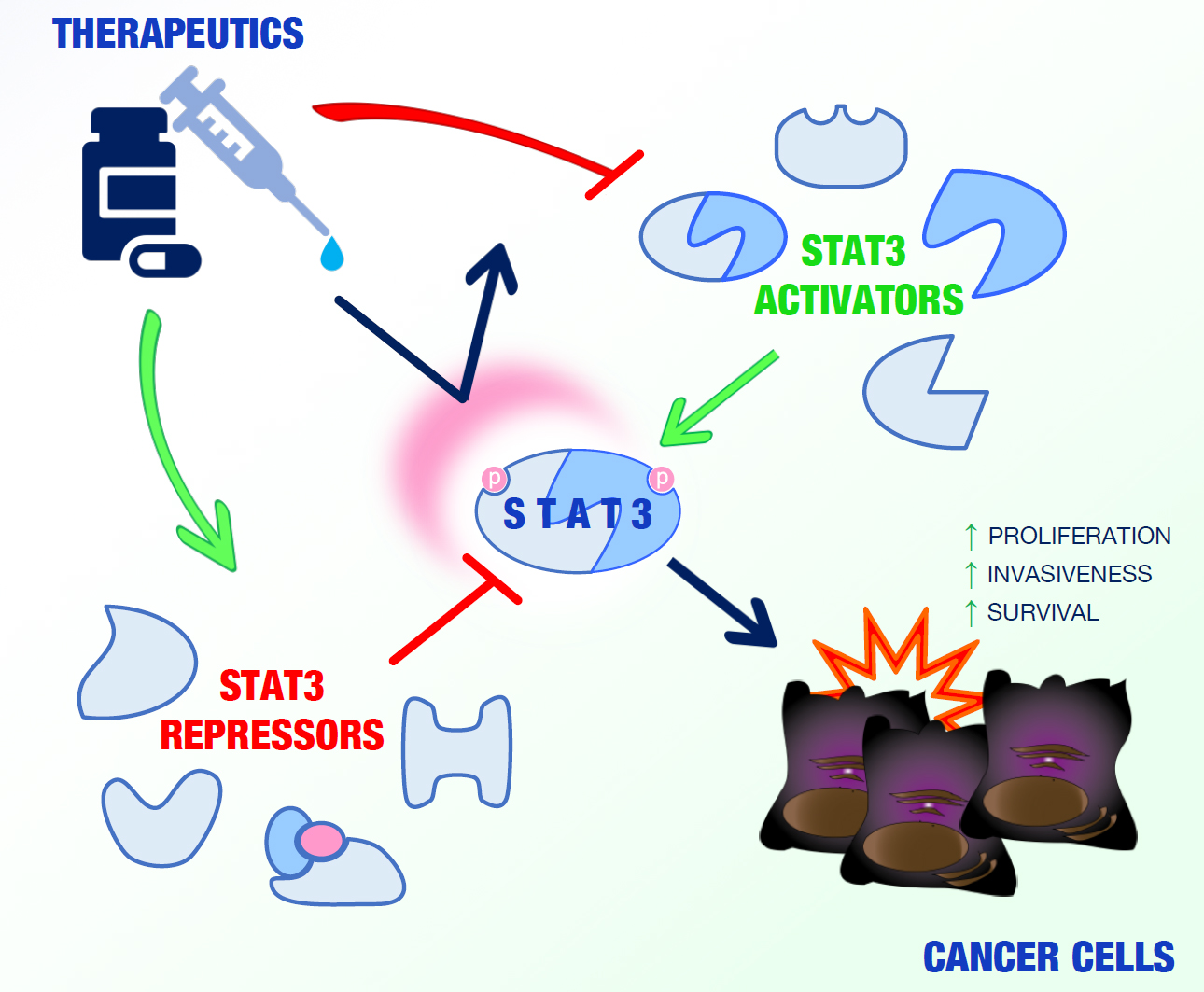{kind=link}
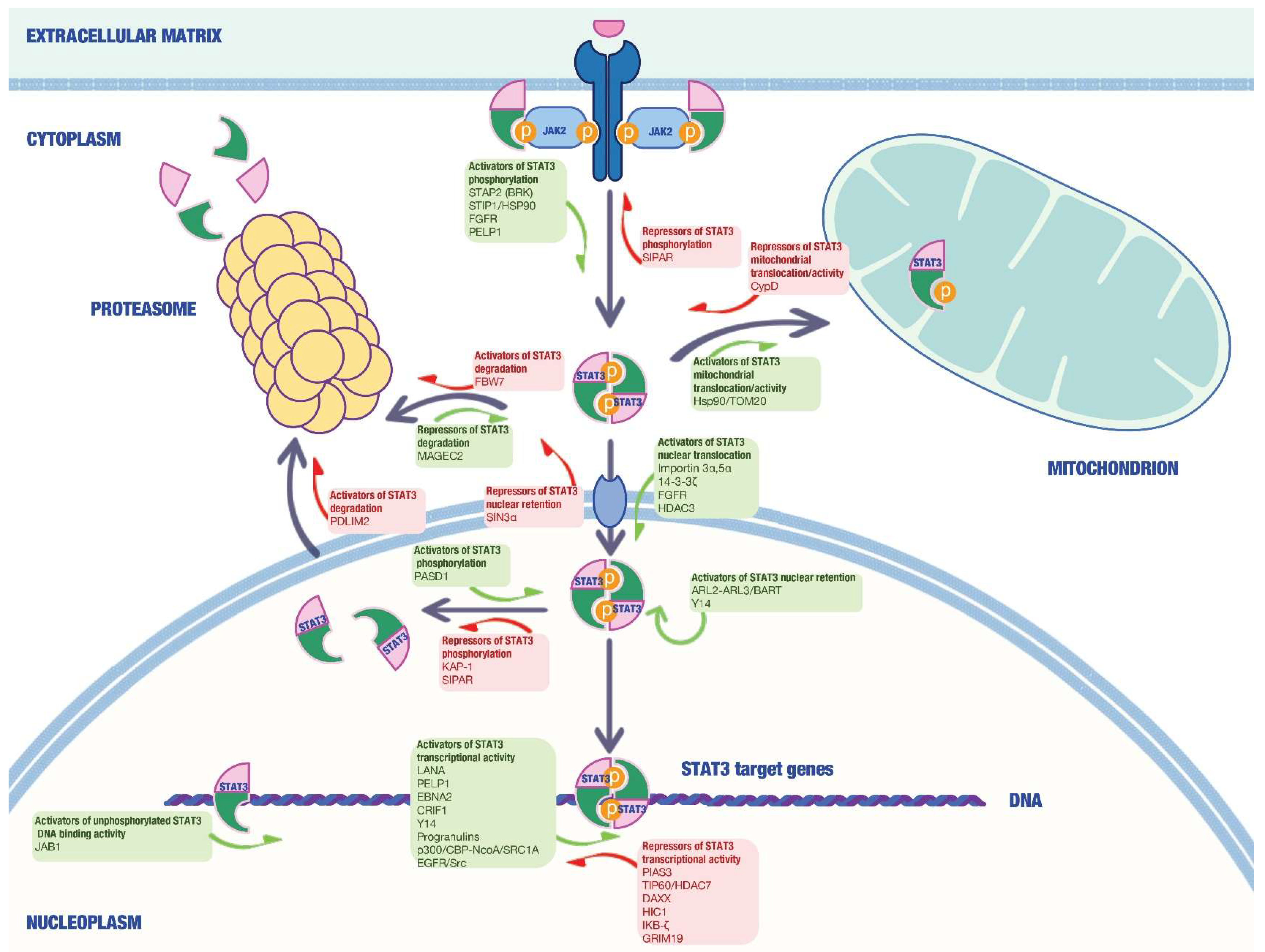{kind=link}
| Name | Mechanism | Cancer Type | Ref. |
|---|---|---|---|
| Hsp90 | Chaperone activity via interaction with STAT3 N-terminal region | Hep3B cells | [37] |
| STIP1 | Stabilization of JAK2 through its N-terminal domain, thereby promoting STAT3 phosphorylation, signal transduction and JAK2-HSP90-STAT3 complex assembly | MDAH2774, SKOV3 and ARK2 cells, primary ovarian tissues | [38] |
| STAP-2 | Modulation of STAT3 activity upon phosphorylation by BRK | HeLa, MCF-7 and T47D cells | [39] |
| FGFR | Induction of STAT3 phosphorylation on tyrosine residues, nuclear translocation and activation of STAT3 target genes | HeLa and SUM-52PE cells | [40] |
| 14-3-3ζ | Interacts with and prevents pSTAT3 Ser727 dephosphorylation by PP2A phosphatase | Multiple myeloma cells | [41] |
| MAGEC2 | Interacts with and inhibits STAT3 polyubiquitination and proteasomal degradation | Human (A375) and mouse (B16) melanoma cell lines | [42] |
| Importin 3α | Binding to the nuclear localization sequence in the coiled-coil domain of unphosphorylated STAT3 | HeLa and Hep3B cells | [43] |
| Importin 5α | Binding to tyrosine phosphorylated STAT3 at the N-terminal domain and maintenance of appropriate conformation | HepG2, MCF7 and HeLa cells | [44] |
| LANA | Interaction with STAT3 C-terminal domain and increased STAT3 transcriptional activity | KSHV-negative B lymphoma, DG75 and KSHV-positive BC3 cells | [45] |
| EBNA2 | Increased STAT3 DNA-binding activity | HeLa cells | [46] |
| ARL2/BART | ARL2-GTP-BART complex is critical for STAT3 activation and nuclear translocation | Hep3B and HeLa cells | [47] |
| ARL3 | Interaction with STAT3 DNA-binding domain and C-terminal domain, with consequent STAT3 phosphorylation | HeLa cells | [48] |
| Y14 | Association with STAT3 C-terminal domain and positive regulation of STAT3 function | Hep3B cells | [49] |
| PASD1 | Impairment of nuclear STAT3 dephosphorylation by TC45 | HeLa cells, HeLa-derived xenografts in nude mice | [50] |
| PELP1 | Increases STAT3 phosphorylation on Ser727 residue, facilitates STAT3 recruitment/retention in the target gene promoters | HeLa and MCF-7 cells | [51] |
| JAB1 | Increases unphosphorylated STAT3 DNA-binding activity | Colo205 colon cancer cells | [52] |
| Progranulin | Regulation of STAT3 phosphorylation, nuclear translocation and transcriptional activity | TNBC cell lines. Primary breast cancer samples | [53] |
| CRIF1 | Association with STAT3 C-terminal coiled-coil domain and positive regulation of STAT3 transcription activity | HeLa, HCT-116, SNU387 and MDA-MB 468 cells | [54] |
| Binding and suppression of the androgen receptor transcription activity and coactivation of STAT3 | CWR-R1 cells and surgical specimens of prostate | [55] | |
| EGFR | Acts as a transcriptional co-activator of STAT3 | Panc-1 and Colo-357 pancreatic cancer cell lines | [56] |
| p300/CREB | Regulation of Lys 685 acetylation, critical for STAT3 to form stable dimers and required for DNA binding | HeLa, MCF-7 and HepG2 cells | [21,57] |
| NcoA/SRC1a | Associates with p300/CBP and acts as a cofactor to potentiate STAT3 transcriptional activity | HepG2 cells | [58] |
| HDAC3 | Modulation of STAT3 Lys685 acetylation and Tyr705 phosphorylation | Ly3 and DHL2 DLBCL cell lines | [59] |
| TOM20 | Regulates STAT3 mitochondrial import and oncogenic functions | Human pancreatic cell lines. BxPC-3-and MIA PaCa-2-derived xenografts in nude mice | [60] |
| Name | Mechanism | Cancer Type | Ref. |
|---|---|---|---|
| PIAS3 | Inhibition of STAT3-mediated gene activation | HepG2 and MCF-7 cells | [61] |
| TIP60 | Repression of STAT3 activity upon HDAC7 recruitment with its central domain | HepG2 and TS1 cells | [62] |
| DAXX | Binding and down-regulation of nuclear STAT3 in response to type I IFN signaling by impairing STAT3-binding to the consensus DNA sequence | HeLa and Hep3B cells | [63] |
| KAP-1 | Impairment of STAT3 phosphorylation status on Ser727 residue by competing with p300 | Hep3B cells | [64] |
| PDLIM2 | Degradation of STAT3 in a proteasome-dependent manner | Hep3B cells | [65] |
| IKB-ζ | Binding to STAT3 coil-coiled domain | HeLa cells | [66] |
| HIC1 | Interacts with the DNA binding domain of STAT3 via its C-terminal domain thus suppressing the binding of STAT3 to its target gene promoters | MDA-MB-231 breast cancer cells, pancreatic cancer cell lines | [67,68] |
| Fbw7 | STAT3 and pSTAT3 Tyr705 stability reduction | ABC-DLBCL cell lines SU-DHL-2 and OCI-LY-3 | [69] |
| SIPAR | Accelerates STAT3 dephosphorylation by enhancing the interaction of STAT3 with the tyrosine phosphatase TC45 | B16 mouse melanoma cells | [70] |
| Sin3A | Modulates STAT3 acetylation pattern and nucleocytoplasmic distribution | HepG2 and MCF-7 cells | [71] |
| GRIM-19 | Binding to Ser727 residue on STAT3 and inhibition of STAT3-mediated gene expression | MCF-7, T47D and BT-20 cells | [72] |
| Attenuation of STAT3 nuclear translocation | SGC-7901 and BGC-823 gastric cancer cells | [73] | |
| CypD | Interacts with mitochondrial STAT3 to regulate the MPTP | Glioblastoma and breast cancer cell lines | [74] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laudisi, F.; Cherubini, F.; Monteleone, G.; Stolfi, C. STAT3 Interactors as Potential Therapeutic Targets for Cancer Treatment. Int. J. Mol. Sci. 2018, 19, 1787. https://doi.org/10.3390/ijms19061787
Laudisi F, Cherubini F, Monteleone G, Stolfi C. STAT3 Interactors as Potential Therapeutic Targets for Cancer Treatment. International Journal of Molecular Sciences. 2018; 19(6):1787. https://doi.org/10.3390/ijms19061787
Chicago/Turabian StyleLaudisi, Federica, Fabio Cherubini, Giovanni Monteleone, and Carmine Stolfi. 2018. "STAT3 Interactors as Potential Therapeutic Targets for Cancer Treatment" International Journal of Molecular Sciences 19, no. 6: 1787. https://doi.org/10.3390/ijms19061787
APA StyleLaudisi, F., Cherubini, F., Monteleone, G., & Stolfi, C. (2018). STAT3 Interactors as Potential Therapeutic Targets for Cancer Treatment. International Journal of Molecular Sciences, 19(6), 1787. https://doi.org/10.3390/ijms19061787