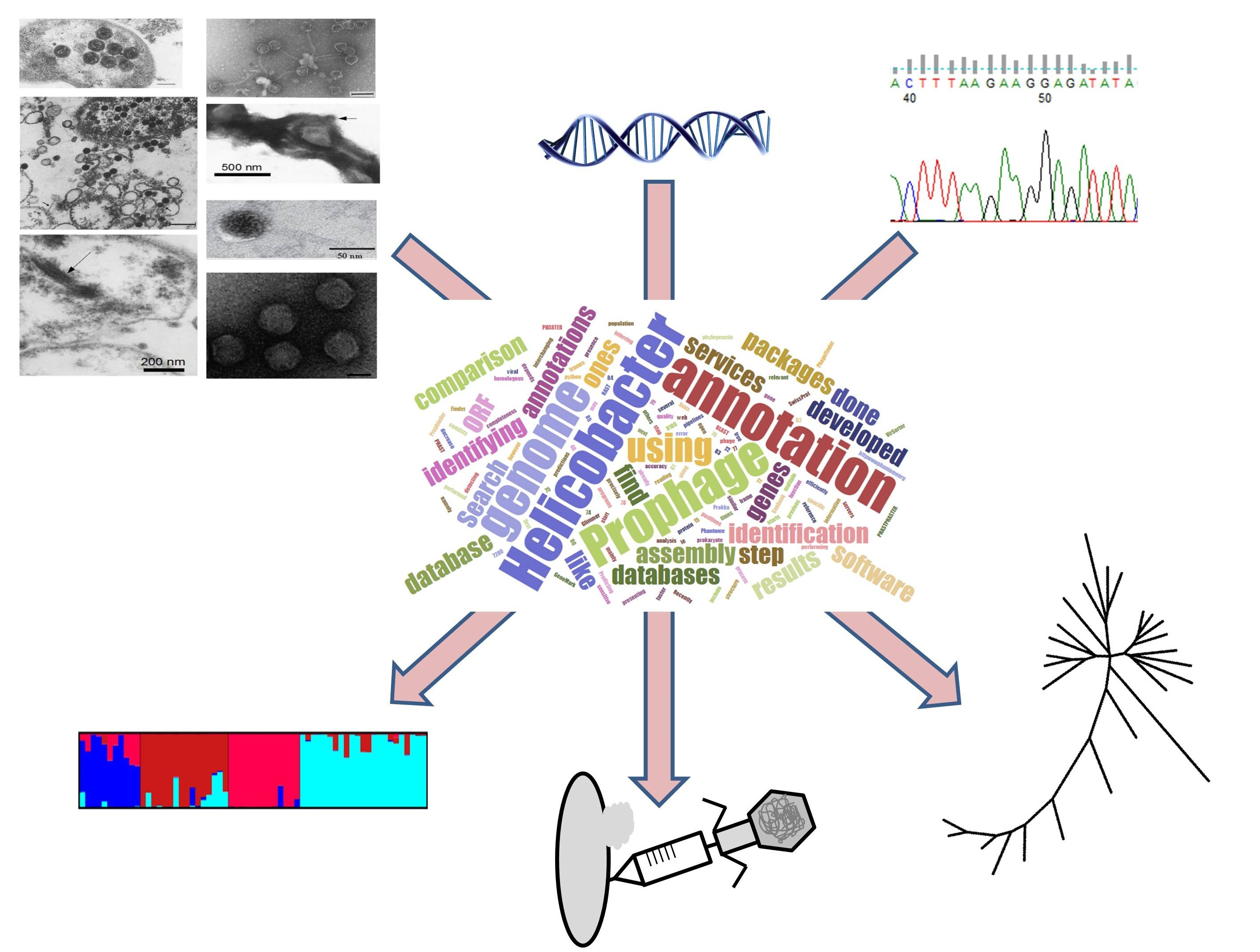
Relating Phage Genomes to Helicobacter pylori Population Structure: General Steps Using Whole-Genome Sequencing Data



Abstract
:
1. Introduction
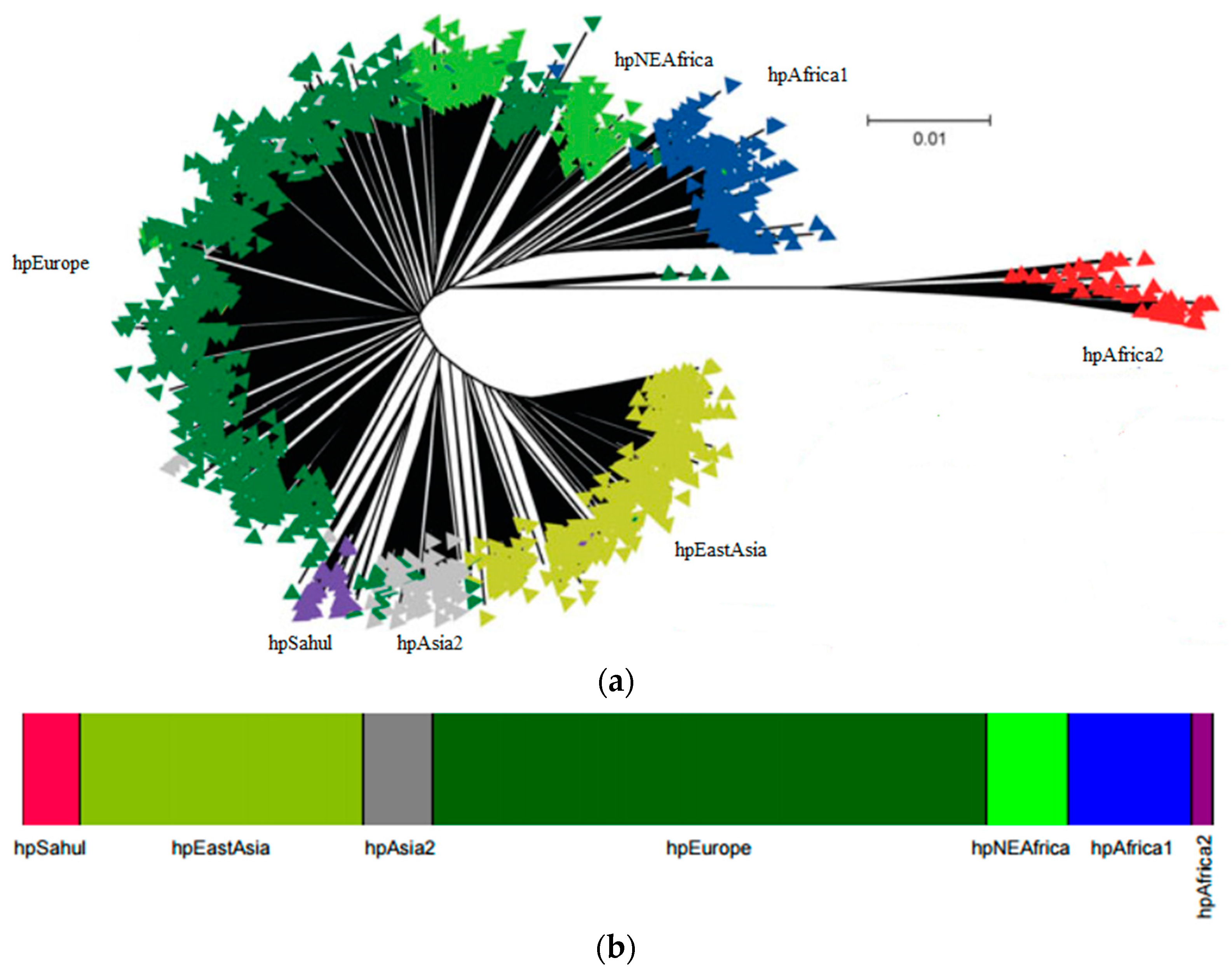
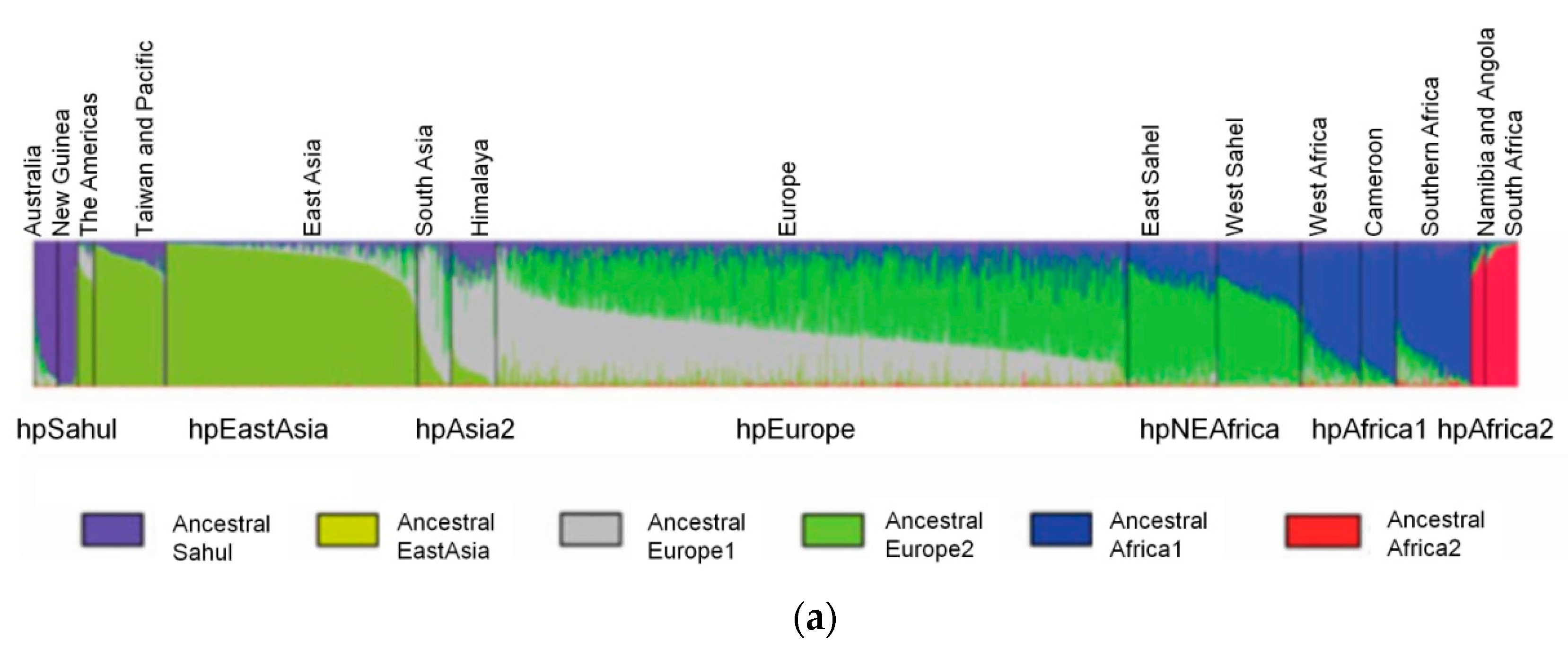
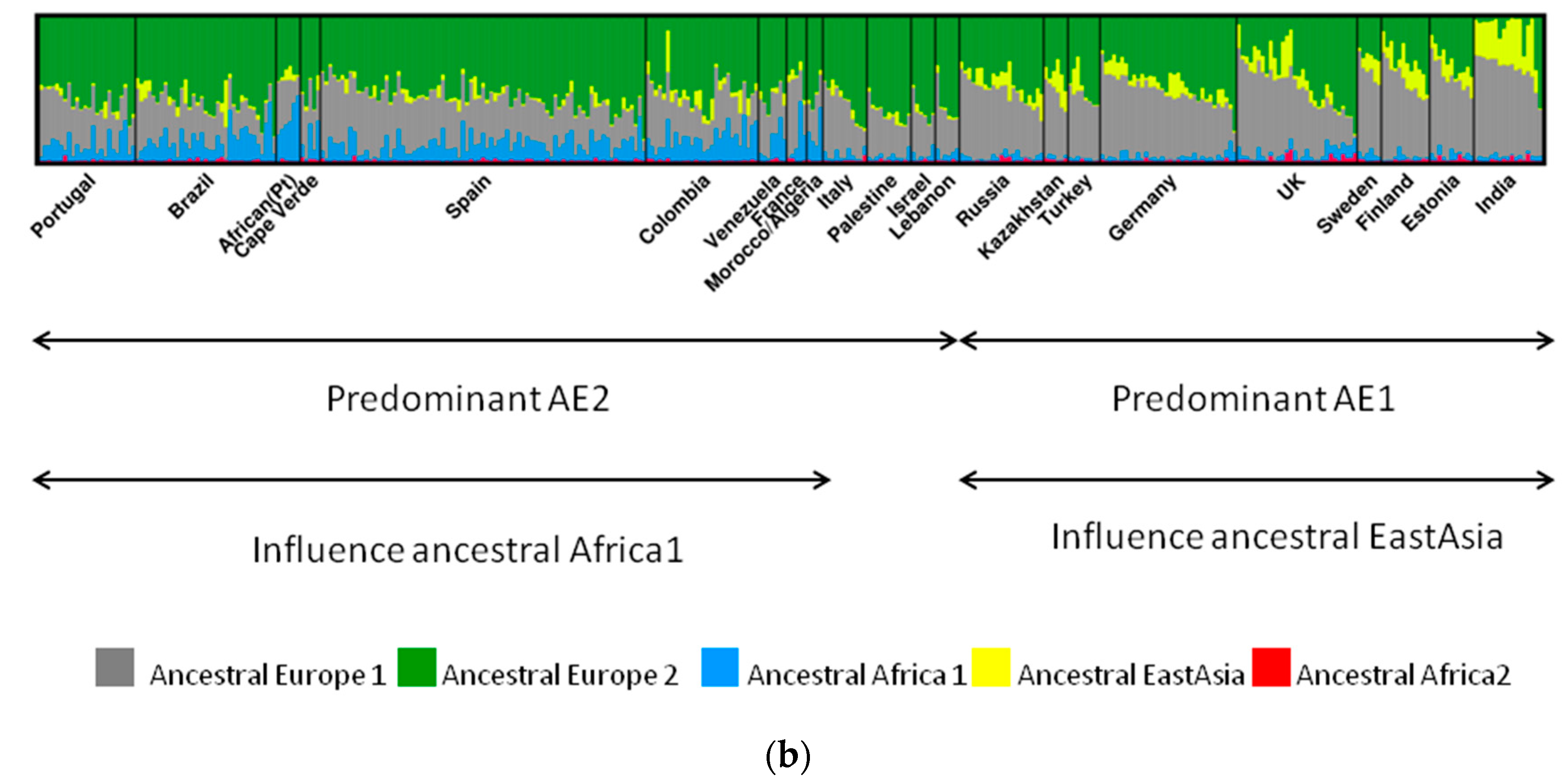
2. H. pylori Population Structure and Human Migrations
Ancestral H. pylori Populations
3. Bacteriophages
3.1. H. pylori Phages and Prophages
3.2. Contribution of Prophages Genomes to H. pylori Population Structure
3.3. Phage Detection and Annotation
3.4. Potential Use of Phages to Eradicate H. pylori
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MALT | Mucosa Associated Lymphoid Tissue |
| MLST | Multilocus Sequence Typing |
| NGS | Next Generation Sequencing |
| ORF | Open Reading Frame |
| PST | Prophage Sequence Typing |
| SNP | Single Nucleotide Polymorphism |
References
- Kusters, J.G.; van Vliet, A.H.; Kuipers, E.J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [PubMed]
- Falush, D.; Wirth, T.; Linz, B.; Pritchard, J.K.; Stephens, M.; Kidd, M.; Blaser, M.J.; Graham, D.Y.; Vacher, S.; Perez-Perez, G.I.; et al. Traces of human migrations in Helicobacter pylori populations. Science 2003, 299, 1582–1585. [Google Scholar] [CrossRef] [PubMed]
- Moodley, Y.; Linz, B.; Yamaoka, Y.; Windsor, H.M.; Breurec, S.; Wu, J.; Maady, A.; Bernhöft, S.; Thiberge, J.; Phuanukoonnon, S.; et al. The peopling of the Pacific from a bacterial perspective. Science 2009, 323, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Suerbaum, S.; Josenhans, C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat. Rev. Microbiol. 2007, 5, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Yahara, K.; Furuta, Y.; Oshima, K.; Yoshida, M.; Azuma, T.; Hattori, M.; Uchiyama, I.; Kobayashi, I. Chromosome painting in silico in a bacterial species reveals fine population structure. Mol. Biol. Evol. 2013, 30, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Vale, F.F.; Vitor, J.M. Genomic Methylation: A Tool for Typing Helicobacter pylori Isolates. Appl. Environ. Microbiol. 2007, 73, 4243–4249. [Google Scholar] [CrossRef] [PubMed]
- Vale, F.F.; Encarnacao, P.; Vitor, J.M. A new algorithm for cluster analysis of genomic methylation: The Helicobacter pylori case. Bioinformatics 2008, 24, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Bubendorfer, S.; Krebes, J.; Yang, I.; Hage, E.; Schulz, T.F.; Bahlawane, C.; Didelot, X.; Suerbaum, S. Genome-wide analysis of chromosomal import patterns after natural transformation of Helicobacter pylori. Nat. Commun. 2016, 7, 11995. [Google Scholar] [CrossRef] [PubMed]
- Berthenet, E.; Sheppard, S.; Vale, F.F. Recent “omics” advances in Helicobacter pylori. Helicobacter 2016, 21 (Suppl. 1), 14–18. [Google Scholar] [CrossRef] [PubMed]
- Vale, F.F.; Vadivelu, J.; Oleastro, M.; Breurec, S.; Engstrand, L.; Perets, T.T.; Megraud, F.; Lehours, P. Dormant phages of Helicobacter pylori reveal distinct populations in Europe. Sci. Rep. 2015, 5, 14333. [Google Scholar] [CrossRef] [PubMed]
- Olbermann, P.; Josenhans, C.; Moodley, Y.; Uhr, M.; Stamer, C.; Vauterin, M.; Suerbaum, S.; Achtman, M.; Linz, B. A global overview of the genetic and functional diversity in the Helicobacter pylori cag pathogenicity island. PLoS Genet. 2010, 6, e1001069. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, A.; Rice, P.A. Transposable phages, DNA reorganization and transfer. Curr. Opin. Microbiol. 2017, 38, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Achtman, M.; Azuma, T.; Berg, D.E.; Ito, Y.; Morelli, G.; Pan, Z.J.; Suebaum, S.; Thompson, S.A.; van der Ende, A.; van Doom, L.J. Recombination and clonal groupings within Helicobacter pylori from different geographical regions. Mol. Microbiol. 1999, 32, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Linz, B.; Balloux, F.; Moodley, Y.; Manica, A.; Liu, H.; Roumagnac, P.; Falush, D.; Stamer, C.; Prugnolle, F.; van der Merwe, S.W.; et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature 2007, 445, 915–918. [Google Scholar] [CrossRef] [PubMed]
- Suerbaum, S.; Achtman, M. Helicobacter pylori: Recombination, population structure and human migrations. Int. J. Med. Microbiol. 2004, 294, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Shiota, S.; Yamaoka, Y. Molecular epidemiology, population genetics, and pathogenic role of Helicobacter pylori. Infect. Genet. Evol. 2012, 12, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Megraud, F.; Lehours, P.; Vale, F.F. The history of Helicobacter pylori: From phylogeography to paleomicrobiology. Clin. Microbiol. Infect. 2016, 22, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Cavalli-Sforza, L.L.; Menozzi, P.; Piazza, A. The History and Geography of Human Genes; Abridged Paperback Edition; Princeton University Press: Princeton, NJ, USA, 1996. [Google Scholar]
- DeMenocal, P.B.; Stringer, C. Human migration: Climate and the peopling of the world. Nature 2016, 538, 49–50. [Google Scholar] [CrossRef] [PubMed]
- Moodley, Y.; Linz, B.; Bond, R.P.; Nieuwoudt, M.; Soodyall, H.; Schlebusch, C.M.; Bernhöft, S.; Hale, J.; Suerbaum, S.; Mugisha, L.; et al. Age of the association between Helicobacter pylori and man. PLoS Pathog. 2012, 8, e1002693. [Google Scholar] [CrossRef] [PubMed]
- Suerbaum, S.; Smith, J.M.; Bapumia, K.; Morelli, G.; Smith, N.H.; Kunstmann, E.; Dyrek, I.; Achtman, M. Free recombination within Helicobacter pylori. Proc. Natl. Acad. Sci. USA 1998, 95, 12619–12624. [Google Scholar] [CrossRef] [PubMed]
- Moodley, Y. Helicobacter pylori: Genetics, recombination, population structure, and human migrations. In Helicobacter pylori Research from Bench to Bedside; Backert, S., Yamaoka, Y., Eds.; Springer: Berlin, Germany, 2016; pp. 3–27. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- El-Metwally, S.; Hamza, T.; Zakaria, M.; Helmy, M. Next-generation sequence assembly: Four stages of data processing and computational challenges. PLoS Comput. Biol. 2013, 9, e1003345. [Google Scholar] [CrossRef] [PubMed]
- Deurenberg, R.H.; Bathoorn, E.; Chlebowicz, M.A.; Couto, N.; Ferdous, M.; García-Cobos, S.; Kooistra-Smid, A.M.; Raangs, E.C.; Rosema, S.; Veloo, A.C.; et al. Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 2017, 243, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.J.; Hellenthal, G.; Myers, S.; Falush, D. Inference of population structure using dense haplotype data. PLoS Genet. 2012, 8, e1002453. [Google Scholar] [CrossRef] [PubMed]
- Thorell, K.; Yahara, K.; Berthenet, E.; Lawson, D.J.; Mikhail, J.; Kato, I.; Mendez, A.; Rizzato, C.; Bravo, M.M.; Suzuki, R.; et al. Rapid evolution of distinct Helicobacter pylori subpopulations in the Americas. PLoS Genet. 2017, 13, e1006546. [Google Scholar] [CrossRef] [PubMed]
- Breurec, S.; Guillard, B.; Hem, S.; Brisse, S.; Dieye, F.B.; Huerre, M.; Oung, C.; Raymond, J.; Tan, T.S.; Thiberge, J.; et al. Evolutionary history of Helicobacter pylori sequences reflect past human migrations in Southeast Asia. PLoS ONE 2011, 6, e22058. [Google Scholar] [CrossRef] [PubMed]
- Oleastro, M.; Rocha, R.; Vale, F.F. Population genetic structure of Helicobacter pylori strains from Portuguese-speaking countries. Helicobacter 2017, 22, 12382. [Google Scholar] [CrossRef] [PubMed]
- Torroni, A.; Bandelt, H.J.; D’Urbano, L.; Lahermo, P.; Moral, P.; Sellitto, D.; Rengo, C.; Forster, P.; Savontaus, M.L.; Bonné-Tamir, B.; et al. mtDNA analysis reveals a major late Paleolithic population expansion from southwestern to northeastern Europe. Am. J. Hum. Genet. 1998, 62, 1137–1152. [Google Scholar] [CrossRef] [PubMed]
- Maixner, F.; Krause-Kyora, B.; Turaev, D.; Herbig, A.; Hoopmann, M.R.; Hallows, J.L.; Kusebauch, U.; Vigl, E.E.; Malfertheiner, P.; Megraud, F.; et al. The 5300-year-old Helicobacter pylori genome of the Iceman. Science 2016, 351, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Brussow, H.; Canchaya, C.; Hardt, W.D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed]
- Feiner, R.; Argov, T.; Rabinovich, L.; Sigal, N.; Borovok, I.; Herskovits, A.A. A new perspective on lysogeny: Prophages as active regulatory switches of bacteria. Nat. Rev. Microbiol. 2015, 13, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Los, M.; Wegrzyn, G. Pseudolysogeny. Adv. Virus Res. 2012, 82, 339–349. [Google Scholar] [PubMed]
- Golais, F.; Holly, J.; Vitkovska, J. Coevolution of bacteria and their viruses. Folia Microbiol. (Praha) 2013, 58, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kim, Y.; Ma, Q.; Hong, S.H.; Pokusaeva, K.; Sturino, J.M.; Wood, T.K. Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. 2010, 1, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wood, T.K. Cryptic prophages as targets for drug development. Drug Resist. Updat. 2016, 27, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Gama, J.A.; Reis, A.M.; Domingues, I.; Mendes-Soares, H.; Matos, A.M.; Dionisio, F. Temperate bacterial viruses as double-edged swords in bacterial warfare. PLoS ONE 2013, 8, e59043. [Google Scholar] [CrossRef] [PubMed]
- Bobay, L.M.; Touchon, M.; Rocha, E.P. Pervasive domestication of defective prophages by bacteria. Proc. Natl. Acad. Sci. USA 2014, 111, 12127–12132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touchon, M.; Bernheim, A.; Rocha, E.P. Genetic and life-history traits associated with the distribution of prophages in bacteria. ISME J. 2016, 10, 2744–2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitoriano, I.; Vitor, J.M.; Oleastro, M.; Roxo-Rosa, M.; Vale, F.F. Proteome variability among Helicobacter pylori isolates clustered according to genomic methylation. J. Appl. Microbiol. 2013, 114, 1817–1832. [Google Scholar] [CrossRef] [PubMed]
- Vale, F.F.; Megraud, F.; Vitor, J.M. Geographic distribution of methyltransferases of Helicobacter pylori: evidence of human host population isolation and migration. BMC Microbiol. 2009, 9, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backert, S.; Kwok, T.; Konig, W. Conjugative plasmid DNA transfer in Helicobacter pylori mediated by chromosomally encoded relaxase and TraG-like proteins. Microbiology 2005, 151, 3493–3503. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Aljaro, C.; Balleste, E.; Muniesa, M. Beyond the canonical strategies of horizontal gene transfer in prokaryotes. Curr. Opin. Microbiol. 2017, 38, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Gonzalez, E.; Backert, S. DNA transfer in the gastric pathogen Helicobacter pylori. J. Gastroenterol. 2014, 49, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Lood, R.; Erturk, G.; Mattiasson, B. Revisiting Antibiotic Resistance Spreading in Wastewater Treatment Plants—Bacteriophages as a Much Neglected Potential Transmission Vehicle. Front. Microbiol. 2017, 8, 2298. [Google Scholar] [CrossRef] [PubMed]
- Von Wintersdorff, C.J.; Penders, J.; van Niekerk, J.M.; Mills, N.D.; Majumder, S.; van Alphen, L.B.; Savelkoul, P.H.M.; Wolffs, P.F.G. Dissemination of Antimicrobial Resistance in Microbial Ecosystems through Horizontal Gene Transfer. Front. Microbiol. 2016, 7, 173. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, E.J.; Israel, D.A.; Kusters, J.G.; Blaser, M.J. Evidence for a conjugation-like mechanism of DNA transfer in Helicobacter pylori. J. Bacteriol. 1998, 180, 2901–2905. [Google Scholar] [PubMed]
- Haas, R.; Meyer, T.F.; van Putten, J.P. Aflagellated mutants of Helicobacter pylori generated by genetic transformation of naturally competent strains using transposon shuttle mutagenesis. Mol. Microbiol. 1993, 8, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Stingl, K.; Muller, S.; Scheidgen-Kleyboldt, G.; Clausen, M.; Maier, B. Composite system mediates two-step DNA uptake into Helicobacter pylori. Proc. Natl. Acad. Sci. USA 2010, 107, 1184–1189. [Google Scholar] [CrossRef] [PubMed]
- Brussow, H.; Kutter, E. Phage ecology. In Bacteriophages Biology and Applications; Kutter, E., Sulakvelidze, A., Eds.; CRC Press: London, UK, 2005; pp. 129–163. [Google Scholar]
- Marshall, B.J.; Armstrong, J.A.; Francis, G.J.; Nokes, N.T.; Wee, S.H. Antibacterial action of bismuth in relation to Campylobacter pyloridis colonization and gastritis. Digestion 1987, 37 (Suppl. 2), 16–30. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.S.; Armstrong, J.A.; Peters, M. Microbiology of C. pylori. In Campylobacter Pylori in Gastritis and Peptic Ulcer Disease; Blaser, M.J., Ed.; MD.IGAKU-SHOIN: New York, NY, USA, 1989; pp. 25–49. [Google Scholar]
- Vale, F.F.; Alves Matos, A.P.; Carvalho, P.; Vitor, J.M. Helicobacter pylori phage screening. Microsc. Microanal. 2008, 14 (Suppl. 3), 150–151. [Google Scholar] [CrossRef]
- Schmid, E.N.; von, R.G.; Ansorg, R. Bacteriophages in Helicobacter (Campylobacter) pylori. J. Med. Microbiol. 1990, 32, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Heintschel von, H.E.; Nalik, H.P.; Schmid, E.N. Characterisation of a Helicobacter pylori phage (HP1). J. Med. Microbiol. 1993, 38, 245–249. [Google Scholar]
- Thiberge, J.M.; Lehours, P.; Bouchier, C.; Ma, L.; Creno, S.; Medigue, C.; Vallene, D.; Ecobichon, C.; Boursaux-Eude, C.; Labigne, A. Sequence of the first Helicobacter pylori strains involved in low-grade Mucosa-Associated Lymphoid Tissue (MALT) Lymphoma. Helicobacter 2006, 11, 02.01. [Google Scholar]
- Lehours, P.; Vale, F.F.; Bjursell, M.K.; Melefors, O.; Advani, R.; Glavas, S.; Guegueniat, J.; Gontier, E.; Lacomme, S.; Alves Matos, A.; et al. Genome sequencing reveals a phage in Helicobacter pylori. MBio 2011, 2, e00239-11. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.H.; Chiou, P.Y.; Yang, C.Y.; Lin, N.T. Genome, integration and transduction of a novel temperate phage of Helicobacter pylori. J. Virol. 2012, 86, 8781–8792. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, J.; Takeuchi, H.; Kato, S.; Takemura-Uchiyamaa, I.; Ujiharae, T.; Daibataa, M.; Matsuzakia, S. Complete Genome Sequences of Two Helicobacter pylori Bacteriophages Isolated from Japanese Patients. J. Virol. 2012, 86, 11400–11401. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, J.; Takeuchi, H.; Kato, S.; Gamoh, K.; Takemura-Uchiyama, I.; Ujihara, T.; Daibata, M.; Matsuzaki, S. Characterization of Helicobacter pylori bacteriophage KHP30. Appl. Environ. Microbiol. 2013, 79, 3176–3184. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; He, L.; Zhang, M.; Zhang, J. Comparative genomics of a Helicobacter pylori isolate from a Chinese Yunnan Naxi ethnic aborigine suggests high genetic divergence and phage insertion. PLoS ONE 2015, 10, e0120659. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Li, Y.; He, R.; Li, Q.; He, W. Comparative analysis of prophage-like elements in Helicobacter sp. genomes. PeerJ 2016, 4, e2012. [Google Scholar] [CrossRef] [PubMed]
- Kyrillos, A.; Arora, G.; Murray, B.; Rosenwald, A.G. The Presence of Phage Orthologous Genes in Helicobacter pylori Correlates with the Presence of the Virulence Factors CagA and VacA. Helicobacter 2016, 21, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Eppinger, M.; Baar, C.; Linz, B.; Raddatz, G.; Lanz, C.; Keller, H.; Morelli, G.; Gressmann, H.; Achtman, M.; Schuster, S.C. Who ate whom? Adaptive Helicobacter genomic changes that accompanied a host jump from early humans to large felines. PLoS Genet. 2006, 2, e120. [Google Scholar] [CrossRef]
- Arnold, I.C.; Zigova, Z.; Holden, M.; Lawley, T.D.; Rad, R.; Dougan, G.; Falkow, S.; Bentley, S.D.; Müller, A. Comparative whole genome sequence analysis of the carcinogenic bacterial model pathogen Helicobacter felis. Genome Biol. Evol. 2011, 3, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Schott, T.; Kondadi, P.K.; Hanninen, M.L.; Rossi, M. Comparative genomics of Helicobacter pylori and the human-derived Helicobacter bizzozeronii CIII-1 strain reveal the molecular basis of the zoonotic nature of non-pylori gastric Helicobacter infections in humans. BMC Genom. 2011, 12, 534. [Google Scholar] [CrossRef] [PubMed]
- Thiberge, J.M.; Boursaux-Eude, C.; Lehours, P.; Dillies, M.A.; Creno, S.; Coppée, J.Y.; Rouy, Z.; Lajus, A.; Ma, L.; Burucoa, C.; et al. From array-based hybridization of Helicobacter pylori isolates to the complete genome sequence of an isolate associated with MALT lymphoma. BMC Genom. 2010, 11, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchiyama, J.; Takemura-Uchiyama, I.; Kato, S.; Takeuchi, H.; Sakaguchi, Y.; Ujihara, T.; Daibata, M.; Shimakura, H.; Okamoto, N.; Sakaguchi, M.; et al. Screening of KHP30-like prophages among Japanese Helicobacter pylori strains, and genetic analysis of a defective KHP30-like prophage sequence integrated in the genome of the H. pylori strain NY40. FEMS Microbiol. Lett. 2016, 363. [Google Scholar] [CrossRef] [PubMed]
- Kersulyte, D.; Rossi, M.; Berg, D.E. Sequence divergence and conservation in genomes of Helicobacter cetorum strains from a dolphin and a whale. PLoS ONE 2013, 8, e83177. [Google Scholar] [CrossRef] [PubMed]
- Secka, O.; Vale, F.F.; Buissonniere, A.; Thomas, J.E.; Megraud, F.; Lehours, P. Phylogeographic agreement between prophage and bacterial housekeeping genes in Helicobacter pylori strains from The Gambia. Helicobacter 2017, 22, 12394. [Google Scholar] [CrossRef] [PubMed]
- Siezen, R.J.; van Hijum, S.A. Genome (re-)annotation and open-source annotation pipelines. Microb. Biotechnol. 2010, 3, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Harmon, D.; Kasif, S.; White, O.; Salzberg, S.L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukashin, A.V.; Borodovsky, M. GeneMark.hmm: New solutions for gene finding. Nucleic Acids Res. 1998, 26, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2017, 45, D37–D42. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 2011, 39, D214–D219. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Dutilh, B.E.; Backus, L.; Edwards, R.A.; Wels, M.; Bayjanov, J.R.; van Hijum, S.A. Explaining microbial phenotypes on a genomic scale: GWAS for microbes. Brief. Funct. Genom. 2013, 12, 366–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef] [PubMed]
- Bose, M.; Barber, R.D. Prophage Finder: A prophage loci prediction tool for prokaryotic genome sequences. In Silico Biol. 2006, 6, 223–227. [Google Scholar] [PubMed]
- Fouts, D.E. Phage_Finder: Automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006, 34, 5839–5851. [Google Scholar] [CrossRef] [PubMed]
- Lima-Mendez, G.; Van, H.J.; Toussaint, A.; Leplae, R. Prophinder: A computational tool for prophage prediction in prokaryotic genomes. Bioinformatics 2008, 24, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Akhter, S.; Aziz, R.K.; Edwards, R.A. PhiSpy: A novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucleic Acids Res. 2012, 40, e126. [Google Scholar] [CrossRef] [PubMed]
- Oliver, S.L.; Lenards, A.J.; Barthelson, R.A.; Merchant, N.; McKay, S.J. Using the iPlant collaborative discovery environment. Curr. Protoc. Bioinform. 2013. [Google Scholar] [CrossRef] [PubMed]
- Vale, F.F.; Nunes, A.; Oleastro, M.; Gomes, J.P.; Sampaio, D.A.; Rocha, R.; Vítor, J.M.; Engstrand, L.; Pascoe, B.; Berthenet, E.; et al. Genomic structure and insertion sites of Helicobacter pylori prophages from various geographic origins. Sci. Rep. 2017, 7, 42471. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F. Bacteriophage genomics. Curr. Opin. Microbiol. 2008, 11, 447–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [PubMed]
- Cresawn, S.G.; Bogel, M.; Day, N.; Jacobs-Sera, D.; Hendrix, R.W.; Hatfull, G.F. Phamerator: A bioinformatic tool for comparative bacteriophage genomics. BMC Bioinform. 2011, 12, 395. [Google Scholar] [CrossRef] [PubMed]
- McNair, K.; Bailey, B.A.; Edwards, R.A. PHACTS, a computational approach to classifying the lifestyle of phages. Bioinformatics 2012, 28, 614–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Roxo-Rosa, M.; Oleastro, M.; Vale, F.F. Helicobacter pylori eradication—The alternatives beyond antibiotics. In Microbial Pathogens and Strategies for Combating Them: Science, Technology and Education; Méndez-Vilas, A., Ed.; Formatex: Badajoz, Spain, 2013; pp. 1656–1667. [Google Scholar]
- Cisek, A.A.; Dabrowska, I.; Gregorczyk, K.P.; Wyzewski, Z. Phage Therapy in Bacterial Infections Treatment: One Hundred Years after the Discovery of Bacteriophages. Curr. Microbiol. 2017, 74, 277–283. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
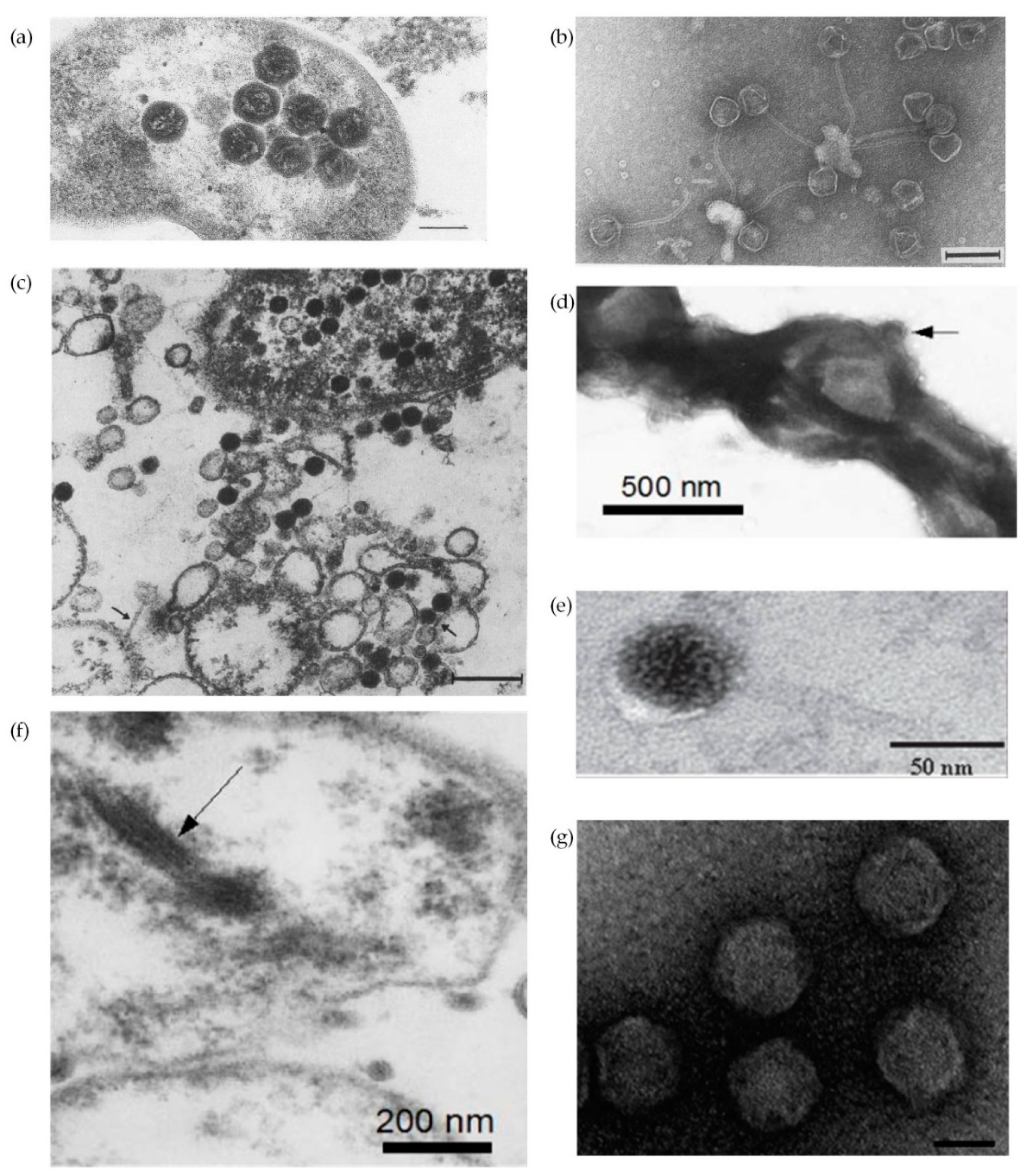{kind=link}
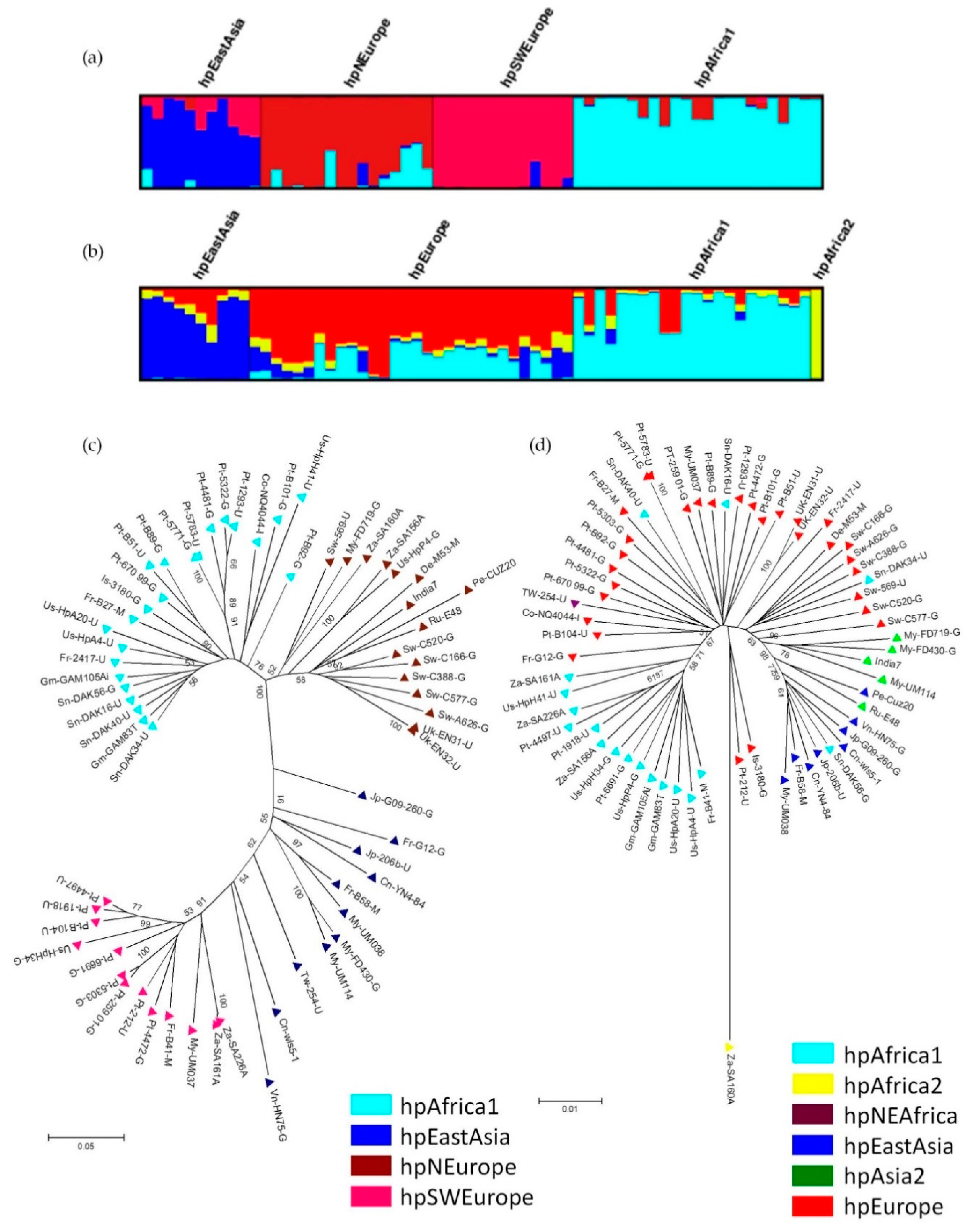{kind=link}
| Name | Family | Gene Number | Genome Size (kb) | Gene Functions | Head/Tail Size (nm) | Reference |
|---|---|---|---|---|---|---|
| HP1 | Siphoviridae | nd | 22 | nd | 50–60/170 × 9.5 | [56] |
| phiHP33 | Siphoviridae | 27 | 24.6 | Integration | 55–70/92 × 6 | [58] |
| regulation, replication, structural, lysis | ||||||
| KHP30 | Corticoviridae/Tectiviridae * | 30 | 26.2 | Integration | 67–71/absent | [61] |
| replication, structural, lysis | ||||||
| 1961P | Podoviridae | 33 | 26.8 | Integration | 68–74/23 × 13.3 | [59] |
| replication, structural, lysis |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vale, F.F.; Lehours, P. Relating Phage Genomes to Helicobacter pylori Population Structure: General Steps Using Whole-Genome Sequencing Data. Int. J. Mol. Sci. 2018, 19, 1831. https://doi.org/10.3390/ijms19071831
Vale FF, Lehours P. Relating Phage Genomes to Helicobacter pylori Population Structure: General Steps Using Whole-Genome Sequencing Data. International Journal of Molecular Sciences. 2018; 19(7):1831. https://doi.org/10.3390/ijms19071831
Chicago/Turabian StyleVale, Filipa F., and Philippe Lehours. 2018. "Relating Phage Genomes to Helicobacter pylori Population Structure: General Steps Using Whole-Genome Sequencing Data" International Journal of Molecular Sciences 19, no. 7: 1831. https://doi.org/10.3390/ijms19071831
APA StyleVale, F. F., & Lehours, P. (2018). Relating Phage Genomes to Helicobacter pylori Population Structure: General Steps Using Whole-Genome Sequencing Data. International Journal of Molecular Sciences, 19(7), 1831. https://doi.org/10.3390/ijms19071831