Targeting mTOR as a Therapeutic Approach in Medulloblastoma


Abstract
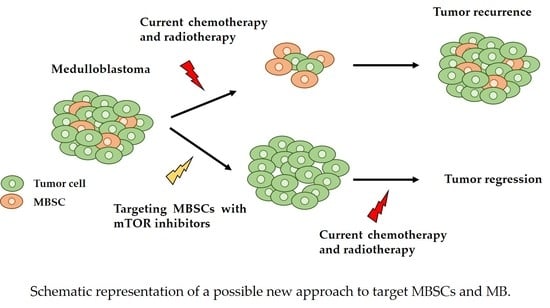
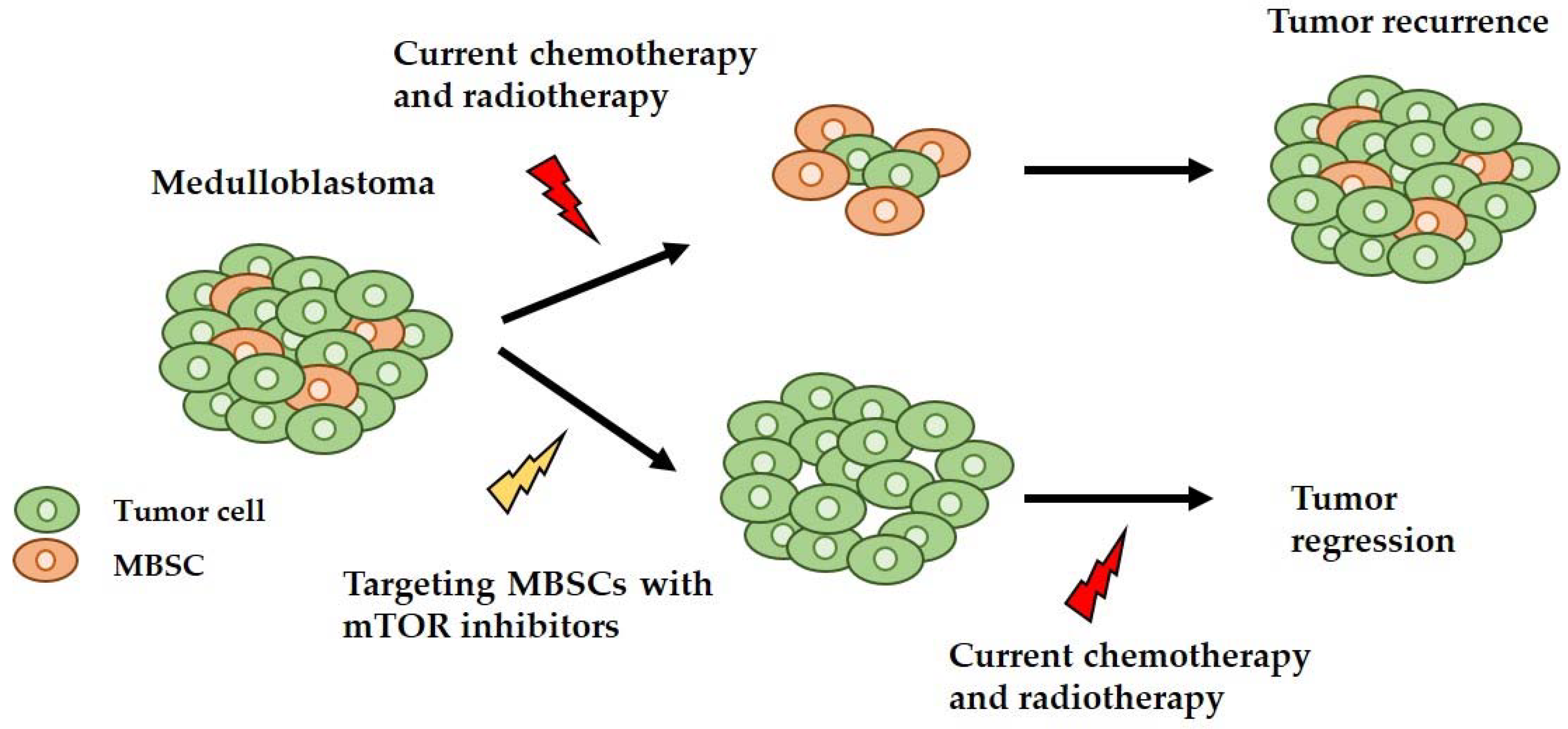
:
1. Mechanistic Target of Rapamycin (mTOR)
2. mTOR in Central Nervous System (CNS) Development
3. Medulloblastoma
4. Medulloblastoma Subgroups
4.1. WNT subgroup
4.2. SHH Subgroup
4.3. Group 3
4.4. Group 4
5. Medulloblastoma Stem Cells (MBSCs)
6. mTOR in Cancer
7. mTOR in Medulloblastoma
8. mTOR Signaling Pathway in MBSCs
9. Targeted Therapy
9.1. Targeting WNT Medulloblastomas
9.2. Targeting SHH Medulloblastomas
9.3. Targeting Group 3 and 4 Medulloblastomas
9.4. Targeting the mTOR Pathway
10. Concluding Remarks
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Laplante, M.; Sabatini, D.M. Mtor signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Grammer, T.C.; Lemon, K.P.; Kazlauskas, A.; Blenis, J. Pdgf- and insulin-dependent pp70s6k activation mediated by phosphatidylinositol-3-OH kinase. Nature 1994, 370, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Long, F. Mtor signaling in skeletal development and disease. Bone Res. 2018, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mtor in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Oh, W.J.; Jacinto, E. Mtor complex 2 signaling and functions. Cell Cycle 2011, 10, 2305–2316. [Google Scholar] [CrossRef] [PubMed]
- Pocza, T.; Sebestyen, A.; Turanyi, E.; Krenacs, T.; Mark, A.; Sticz, T.B.; Jakab, Z.; Hauser, P. Mtor pathway as a potential target in a subset of human medulloblastoma. Pathol. Oncol. Res. 2014, 20, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mtor-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Julien, L.A.; Carriere, A.; Moreau, J.; Roux, P.P. Mtorc1-activated s6k1 phosphorylates rictor on threonine 1135 and regulates mtorc2 signaling. Mol. Cell. Biol. 2010, 30, 908–921. [Google Scholar] [CrossRef] [PubMed]
- Bockaert, J.; Marin, P. Mtor in brain physiology and pathologies. Physiol. Rev. 2015, 95, 1157–1187. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mtorc2 assembly and akt/pkb. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Takei, N.; Nawa, H. Mtor signaling and its roles in normal and abnormal brain development. Front. Mol. Neurosci. 2014, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Ichisaka, T.; Maeda, M.; Oshiro, N.; Hara, K.; Edenhofer, F.; Kiyama, H.; Yonezawa, K.; Yamanaka, S. Mtor is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol. Cell. Biol. 2004, 24, 6710–6718. [Google Scholar] [CrossRef] [PubMed]
- Hentges, K.E.; Sirry, B.; Gingeras, A.C.; Sarbassov, D.; Sonenberg, N.; Sabatini, D.; Peterson, A.S. Frap/mtor is required for proliferation and patterning during embryonic development in the mouse. Proc. Natl. Acad. Sci. USA 2001, 98, 13796–13801. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y. Roles of mtor signaling in brain development. Exp. Neurobiol. 2015, 24, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, K.; Fu, Z.; Yu, D.; Huang, H.; Zang, X.; Mo, X. Brain development and akt signaling: The crossroads of signaling pathway and neurodevelopmental diseases. J. Mol. Neurosci. 2017, 61, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Mainwaring, L.A.; Kenney, A.M. Divergent functions for eif4e and s6 kinase by sonic hedgehog mitogenic signaling in the developing cerebellum. Oncogene 2011, 30, 1784–1797. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, F.; Kortmann, R.; Saran, F. Medulloblastoma. Clin. Oncol. 2013, 25, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Butts, T.; Green, M.J.; Wingate, R.J. Development of the cerebellum: Simple steps to make a ‘little brain’. Development 2014, 141, 4031–4041. [Google Scholar] [CrossRef] [PubMed]
- Wechsler-Reya, R.; Scott, M.P. The developmental biology of brain tumors. Annu. Rev. Neurosci. 2001, 24, 385–428. [Google Scholar] [CrossRef] [PubMed]
- Haegele, L.; Ingold, B.; Naumann, H.; Tabatabai, G.; Ledermann, B.; Brandner, S. Wnt signalling inhibits neural differentiation of embryonic stem cells by controlling bone morphogenetic protein expression. Mol. Cell. Neurosci. 2003, 24, 696–708. [Google Scholar] [CrossRef]
- Grimmer, M.R.; Weiss, W.A. Childhood tumors of the nervous system as disorders of normal development. Curr. Opin. Pediatr. 2006, 18, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Gibson, P.; Tong, Y.; Robinson, G.; Thompson, M.C.; Currle, D.S.; Eden, C.; Kranenburg, T.A.; Hogg, T.; Poppleton, H.; Martin, J.; et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 2010, 468, 1095–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodrich, L.V.; Milenkovic, L.; Higgins, K.M.; Scott, M.P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997, 277, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Dey, J.; Ditzler, S.; Knoblaugh, S.E.; Hatton, B.A.; Schelter, J.M.; Cleary, M.A.; Mecham, B.; Rorke-Adams, L.B.; Olson, J.M. A distinct smoothened mutation causes severe cerebellar developmental defects and medulloblastoma in a novel transgenic mouse model. Mol. Cell. Biol. 2012, 32, 4104–4115. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kumar, V.; McGuire, T.; Coulter, D.W.; Sharp, J.G.; Mahato, R.I. Challenges and recent advances in medulloblastoma therapy. Trends Pharmacol. Sci. 2017, 38, 1061–1084. [Google Scholar] [CrossRef] [PubMed]
- Coluccia, D.; Figuereido, C.; Isik, S.; Smith, C.; Rutka, J.T. Medulloblastoma: Tumor biology and relevance to treatment and prognosis paradigm. Curr. Neurol. Neurosci. Rep. 2016, 16, 43. [Google Scholar] [CrossRef] [PubMed]
- Saury, J.M.; Emanuelson, I. Cognitive consequences of the treatment of medulloblastoma among children. Pediatr. Neurol. 2011, 44, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Musial-Bright, L.; Fengler, R.; Henze, G.; Hernaiz Driever, P. Carboplatin and ototoxicity: Hearing loss rates among survivors of childhood medulloblastoma. Childs Nerv. Syst. 2011, 27, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Kadota, R.P.; Mahoney, D.H.; Doyle, J.; Duerst, R.; Friedman, H.; Holmes, E.; Kun, L.; Zhou, T.; Pollack, I.F. Dose intensive melphalan and cyclophosphamide with autologous hematopoietic stem cells for recurrent medulloblastoma or germinoma. Pediatr. Blood Cancer 2008, 51, 675–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, T.S.; Hoffman, H.J.; Hendrick, E.B.; Humphreys, R.P.; Becker, L.E. Medulloblastoma: Clinical presentation and management. Experience at the hospital for sick children, Toronto, 1950–1980. J. Neurosurg. 1983, 58, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Rochkind, S.; Blatt, I.; Sadeh, M.; Goldhammer, Y. Extracranial metastases of medulloblastoma in adults: Literature review. J. Neurol. Neurosurg. Psychiatry 1991, 54, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 who classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma comprises four distinct molecular variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalli, F.M.G.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell 2017, 31, 737–754.e6. [Google Scholar] [CrossRef] [PubMed]
- Clifford, S.C.; Lusher, M.E.; Lindsey, J.C.; Langdon, J.A.; Gilbertson, R.J.; Straughton, D.; Ellison, D.W. Wnt/wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle 2006, 5, 2666–2670. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Zhang, Y.; Wang, J.; Du, J.; Raynald; Qiu, X.; Wang, Y.; Li, C. A retrospective study of progression-free and overall survival in pediatric medulloblastoma based on molecular subgroup classification: A single-institution experience. Front. Neurol. 2017, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Grammel, D.; Warmuth-Metz, M.; von Bueren, A.O.; Kool, M.; Pietsch, T.; Kretzschmar, H.A.; Rowitch, D.H.; Rutkowski, S.; Pfister, S.M.; Schuller, U. Sonic hedgehog-associated medulloblastoma arising from the cochlear nuclei of the brainstem. Acta Neuropathol. 2012, 123, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Moore, C.E.; Wang, J.; Tewari, A.K.; Eroshkin, A.; Cho, Y.J.; Witt, H.; Korshunov, A.; Read, T.A.; Sun, J.L.; et al. An animal model of myc-driven medulloblastoma. Cancer Cell 2012, 21, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, D.; Robinson, G.; Uziel, T.; Gibson, P.; Rehg, J.; Gao, C.; Finkelstein, D.; Qu, C.; Pounds, S.; Ellison, D.W.; et al. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell 2012, 21, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: Aacr workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed]
- Manoranjan, B.; Venugopal, C.; McFarlane, N.; Doble, B.W.; Dunn, S.E.; Scheinemann, K.; Singh, S.K. Medulloblastoma stem cells: Where development and cancer cross pathways. Pediatr. Res. 2012, 71, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Populo, H.; Lopes, J.M.; Soares, P. The mtor signalling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed]
- Osaki, M.; Oshimura, M.; Ito, H. Pi3k-akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An integrative genomic and proteomic analysis of pik3ca, pten, and akt mutations in breast cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [PubMed]
- Danielsen, S.A.; Eide, P.W.; Nesbakken, A.; Guren, T.; Leithe, E.; Lothe, R.A. Portrait of the pi3k/akt pathway in colorectal cancer. Biochim. Biophys. Acta 2015, 1855, 104–121. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, A.; Graff, J.R. Eif-4e expression and its role in malignancies and metastases. Oncogene 2004, 23, 3189–3199. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mtor pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed]
- Parathath, S.R.; Mainwaring, L.A.; Fernandez, L.A.; Campbell, D.O.; Kenney, A.M. Insulin receptor substrate 1 is an effector of sonic hedgehog mitogenic signaling in cerebellar neural precursors. Development 2008, 135, 3291–3300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, G.; Pedone, C.A.; Del Valle, L.; Reiss, K.; Holland, E.C.; Fults, D.W. Sonic hedgehog and insulin-like growth factor signaling synergize to induce medulloblastoma formation from nestin-expressing neural progenitors in mice. Oncogene 2004, 23, 6156–6162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malaguarnera, R.; Belfiore, A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front. Endocrinol. 2014, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, V.; Arcaro, A. Targeting the pi3k/akt/mtor signaling pathway in medulloblastoma. Curr. Mol. Med. 2015, 15, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Truitt, M.L.; Ruggero, D. New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 2016, 16, 288–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.C.; Hou, S.; Orr, B.A.; Kuo, B.R.; Youn, Y.H.; Ong, T.; Roth, F.; Eberhart, C.G.; Robinson, G.W.; Solecki, D.J.; et al. Mtorc1-mediated inhibition of 4ebp1 is essential for hedgehog signaling-driven translation and medulloblastoma. Dev. Cell 2017, 43, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; Wojnowski, L.; Specht, K.; Kappler, R.; Calzada-Wack, J.; Potter, D.; Zimmer, A.; Muller, U.; Samson, E.; Quintanilla-Martinez, L.; et al. Patched target igf2 is indispensable for the formation of medulloblastoma and rhabdomyosarcoma. J. Biol. Chem. 2000, 275, 28341–28344. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, T.J.; Aguilera, D.; Castellino, R.C. The rationale for targeted therapies in medulloblastoma. Neuro-Oncology 2014, 16, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Frasson, C.; Rampazzo, E.; Accordi, B.; Beggio, G.; Pistollato, F.; Basso, G.; Persano, L. Inhibition of pi3k signalling selectively affects medulloblastoma cancer stem cells. Biomed. Res. Int. 2015, 2015, 973912. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.H.; Xu, Q.F.; Cui, Y.H.; Li, N.; Bian, X.W.; Lv, S.Q. Medulloblastoma stem cells: Promising targets in medulloblastoma therapy. Cancer Sci. 2016, 107, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, Y.; Zhang, Y.; Zhang, Z.; Peng, J.; Li, Z.; Han, L.; You, Q.; Chen, X.; Rao, X.; et al. Downregulation of cancer stem cell properties via mtor signaling pathway inhibition by rapamycin in nasopharyngeal carcinoma. Int. J. Oncol. 2015, 47, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Becher, O.J.; Rosenblum, M.K.; Pandolfi, P.P.; Manova-Todorova, K.; Holland, E.C. Pi3k pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008, 22, 436–448. [Google Scholar] [CrossRef] [PubMed]
- Kenney, A.M.; Widlund, H.R.; Rowitch, D.H. Hedgehog and pi-3 kinase signaling converge on nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development 2004, 131, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Merchant, A.A.; Matsui, W. Targeting hedgehog—A cancer stem cell pathway. Clin. Cancer Res. 2010, 16, 3130–3140. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog signaling and bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef] [PubMed]
- Ahlfeld, J.; Favaro, R.; Pagella, P.; Kretzschmar, H.A.; Nicolis, S.; Schuller, U. Sox2 requirement in sonic hedgehog-associated medulloblastoma. Cancer Res. 2013, 73, 3796–3807. [Google Scholar] [CrossRef] [PubMed]
- Salaroli, R.; Ronchi, A.; Buttarelli, F.R.; Cortesi, F.; Marchese, V.; Della Bella, E.; Renna, C.; Baldi, C.; Giangaspero, F.; Cenacchi, G. Wnt activation affects proliferation, invasiveness and radiosensitivity in medulloblastoma. J. Neurooncol. 2015, 121, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, F.; Scoppettuolo, M.N.; Carotenuto, M.; De Antonellis, P.; Dato, V.D.; De Vita, G.; Zollo, M. Norcantharidin impairs medulloblastoma growth by inhibition of wnt/beta-catenin signaling. J. Neurooncol. 2012, 106, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Zinke, J.; Schneider, F.T.; Harter, P.N.; Thom, S.; Ziegler, N.; Toftgard, R.; Plate, K.H.; Liebner, S. Beta-catenin-gli1 interaction regulates proliferation and tumor growth in medulloblastoma. Mol. Cancer 2015, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.B.; Oppenheimer, J.J.; Weinberger, M.; Rubin, B.K.; Irwin, R.S. Children with chronic wet or productive cough—Treatment and investigations: A systematic review. Chest 2016, 149, 120–142. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: Results from phase ii pediatric brain tumor consortium studies pbtc-025b and pbtc-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Jones, D.T.; Jager, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome sequencing of shh medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Dijkgraaf, G.J.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of hedgehog pathway transcriptional output through bet bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.C.; Peaceman, D.; Ozdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking hedgehog/gli pathway. J. Clin. Investig. 2011, 121, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Morfouace, M.; Shelat, A.; Jacus, M.; Freeman, B.B., 3rd; Turner, D.; Robinson, S.; Zindy, F.; Wang, Y.D.; Finkelstein, D.; Ayrault, O.; et al. Pemetrexed and gemcitabine as combination therapy for the treatment of group3 medulloblastoma. Cancer Cell 2014, 25, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Jones, D.T.; Kool, M.; Robinson, G.W.; Gilbertson, R.J.; Cho, Y.J.; Pomeroy, S.L.; Korshunov, A.; Lichter, P.; Taylor, M.D.; et al. Medulloblastomics: The end of the beginning. Nat. Rev. Cancer 2012, 12, 818–834. [Google Scholar] [CrossRef] [PubMed]
- Conciatori, F.; Ciuffreda, L.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M. Mtor cross-talk in cancer and potential for combination therapy. Cancers 2018, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef] [PubMed]
- Hutson, T.E.; Escudier, B.; Esteban, E.; Bjarnason, G.A.; Lim, H.Y.; Pittman, K.B.; Senico, P.; Niethammer, A.; Lu, D.R.; Hariharan, S.; et al. Randomized phase iii trial of temsirolimus versus sorafenib as second-line therapy after sunitinib in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2014, 32, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Bellmunt, J.; Clancy, J.; Wang, K.; Niethammer, A.G.; Hariharan, S.; Escudier, B. Randomized phase iii trial of temsirolimus and bevacizumab versus interferon alfa and bevacizumab in metastatic renal cell carcinoma: Intoract trial. J. Clin. Oncol. 2014, 32, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Hess, G.; Barlev, A.; Chung, K.; Hill, J.W.; Fonseca, E. Cost of palliative radiation to the bone for patients with bone metastases secondary to breast or prostate cancer. Radiat. Oncol. 2012, 7, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grunwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase iii trial. Lancet 2008, 372, 449–456. [Google Scholar] [CrossRef]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; de Vries, E.G.; et al. Everolimus for advanced pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.E.; Hainsworth, J.D.; Baudin, E.; Peeters, M.; Horsch, D.; Winkler, R.E.; Klimovsky, J.; Lebwohl, D.; Jehl, V.; Wolin, E.M.; et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (radiant-2): A randomised, placebo-controlled, phase 3 study. Lancet 2011, 378, 2005–2012. [Google Scholar] [CrossRef]
- Ohtsu, A.; Ajani, J.A.; Bai, Y.X.; Bang, Y.J.; Chung, H.C.; Pan, H.M.; Sahmoud, T.; Shen, L.; Yeh, K.H.; Chin, K.; et al. Everolimus for previously treated advanced gastric cancer: Results of the randomized, double-blind, phase iii granite-1 study. J. Clin. Oncol. 2013, 31, 3935–3943. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., 3rd; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; O’Regan, R.; Ozguroglu, M.; Toi, M.; Xu, B.; Jerusalem, G.; Masuda, N.; Wilks, S.; Arena, F.; Isaacs, C.; et al. Everolimus for women with trastuzumab-resistant, her2-positive, advanced breast cancer (bolero-3): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014, 15, 580–591. [Google Scholar] [CrossRef]
- Wolff, A.C.; Lazar, A.A.; Bondarenko, I.; Garin, A.M.; Brincat, S.; Chow, L.; Sun, Y.; Neskovic-Konstantinovic, Z.; Guimaraes, R.C.; Fumoleau, P.; et al. Randomized phase iii placebo-controlled trial of letrozole plus oral temsirolimus as first-line endocrine therapy in postmenopausal women with locally advanced or metastatic breast cancer. J. Clin. Oncol. 2013, 31, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B.; et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef]
- Demetri, G.D.; Chawla, S.P.; Ray-Coquard, I.; Le Cesne, A.; Staddon, A.P.; Milhem, M.M.; Penel, N.; Riedel, R.F.; Bui-Nguyen, B.; Cranmer, L.D.; et al. Results of an international randomized phase iii trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J. Clin. Oncol. 2013, 31, 2485–2492. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Kurkjian, C.; Infante, J.R.; Bauer, T.M.; Burris, H.A., 3rd; Greco, F.A.; Shih, K.C.; Thompson, D.S.; Lane, C.M.; Finney, L.H.; et al. A phase 1 study of the sachet formulation of the oral dual pi3k/mtor inhibitor bez235 given twice daily (bid) in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.; Aggarwal, R.; Hong, D.; Schellens, J.H.; van der Noll, R.; Specht, J.; Witteveen, P.O.; Werner, T.L.; Dees, E.C.; Bergsland, E.; et al. First-in-human phase i study of gsk2126458, an oral pan-class i phosphatidylinositol-3-kinase inhibitor, in patients with advanced solid tumor malignancies. Clin. Cancer Res. 2016, 22, 1932–1939. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; Bell-McGuinn, K.M.; Molina, J.R.; Bendell, J.; Spicer, J.; Kwak, E.L.; Pandya, S.S.; Millham, R.; Borzillo, G.; Pierce, K.J.; et al. First-in-human study of pf-05212384 (pki-587), a small-molecule, intravenous, dual inhibitor of pi3k and mtor in patients with advanced cancer. Clin. Cancer Res. 2015, 21, 1888–1895. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Aksoy, O.; Wong, R.A.; Ilkhanizadeh, S.; Novotny, C.J.; Gustafson, W.C.; Truong, A.Y.; Cayanan, G.; Simonds, E.F.; Haas-Kogan, D.; et al. A kinase inhibitor targeted to mtorc1 drives regression in glioblastoma. Cancer Cell 2017, 31, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Fazio, N.; Singh, S.; Buzzoni, R.; Carnaghi, C.; Wolin, E.; Tomasek, J.; Raderer, M.; Lahner, H.; Voi, M.; et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (radiant-4): A randomised, placebo-controlled, phase 3 study. Lancet 2016, 387, 968–977. [Google Scholar] [CrossRef]
- Dolly, S.O.; Wagner, A.J.; Bendell, J.C.; Kindler, H.L.; Krug, L.M.; Seiwert, T.Y.; Zauderer, M.G.; Lolkema, M.P.; Apt, D.; Yeh, R.F.; et al. Phase i study of apitolisib (gdc-0980), dual phosphatidylinositol-3-kinase and mammalian target of rapamycin kinase inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2016, 22, 2874–2884. [Google Scholar] [CrossRef] [PubMed]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the pi3k pathway in medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medulloblastoma Classification System | Clinical Features | WNT | SHH | Group 3 | Group 4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Taylor Classification | Histology | Classic, Rarely LCA | Desmoplastic/Nodular, Classic, LCA | Classic, LCA | Classic, LCA | ||||||||
| Prognosis | Very good | Infants Good; Other Intermidiate | Poor | Intermidiate | |||||||||
| Cavalli classification | α | β | α | β | γ | δ | α | β | γ | α | β | γ | |
| Metastasis | 9% | 21% | 20% | 33% | 9% | 9% | 43% | 20% | 40% | 40% | 41% | 39% | |
| Genetic alterations | Monosomy 6 | TP53 mutations | PTEN loss | TERT promoter mut | 8q loss | GFI1 and GFI1B ↑, OTX2 amp, DDX31 loss | MYC amp | MYCN and CDK amp, 8p loss, 7q gain | SNCAIP dup, i17q | CDK amp, 8p loss, 7q gain | |||
| Age | Child; Ado | Ado; Adult | Child; Ado | Infant | Infant | Adult | Infant; Child | Child; Ado | Infant; Children | Child; Ado | Child; Ado | Child; Ado | |
| Subtype histoloogy | LCA, desmoplastic | Desmoplastic | MBEN, desmoplastic | Desmoplastic | |||||||||
| Survival | 97% | 100% | 70% | 67% | 88% | 89% | 66% | 56% | 42% | 67% | 75% | 83% | |
| mTOR | mTORC1 activation | PI3K/AKT/mTOR activation | |||||||||||
| Types of mTOR Inhibitors | Name | Target | Disease | Trial Phase |
|---|---|---|---|---|
| Rapalogs | Temsirolimus | mTOR | RCC and MCL | Completed phase III |
| Everolimus | mTOR | RCC, PNET, Lung, GEP, NET, Gastric, BC, mRCC | Completed phase III | |
| Ridaforolimus | mTOR | Sarcoma | Completed phase III | |
| Second-generation mTOR inhibitors | BEZ235 | PI3K/mTOR | BC, RCC, Endometrial, PNET | Discontinued |
| GSK2126458 | PI3K/mTOR | Colon/Rectum, RCC, BC, Endometrial, Melanoma, Ovary/Primary Peritoneal, Pancreas, Prostate | Phase I | |
| Gedatolisib (PF-04691502; PKI-587) | PI3K/mTOR | SCLC, Ovarian, Endometrial, Renal, Colorectal, Glioblastoma | Phase I | |
| Apitolisib (GDC-0980) | PI3K/mTOR | MPM, Colorectal, GIST, Sarcoma, BC | Phase I | |
| Third-generation mTOR inhibitors | Rapalink-1 | mTOR (mutant forms too) | Glioblastoma | No clinical data |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldaregia, J.; Odriozola, A.; Matheu, A.; Garcia, I. Targeting mTOR as a Therapeutic Approach in Medulloblastoma. Int. J. Mol. Sci. 2018, 19, 1838. https://doi.org/10.3390/ijms19071838
Aldaregia J, Odriozola A, Matheu A, Garcia I. Targeting mTOR as a Therapeutic Approach in Medulloblastoma. International Journal of Molecular Sciences. 2018; 19(7):1838. https://doi.org/10.3390/ijms19071838
Chicago/Turabian StyleAldaregia, Juncal, Ainitze Odriozola, Ander Matheu, and Idoia Garcia. 2018. "Targeting mTOR as a Therapeutic Approach in Medulloblastoma" International Journal of Molecular Sciences 19, no. 7: 1838. https://doi.org/10.3390/ijms19071838
APA StyleAldaregia, J., Odriozola, A., Matheu, A., & Garcia, I. (2018). Targeting mTOR as a Therapeutic Approach in Medulloblastoma. International Journal of Molecular Sciences, 19(7), 1838. https://doi.org/10.3390/ijms19071838