Preliminary RNA-Seq Analysis of Long Non-Coding RNAs Expressed in Human Term Placenta

 , , ,
, , ,  , ,
, ,
Abstract
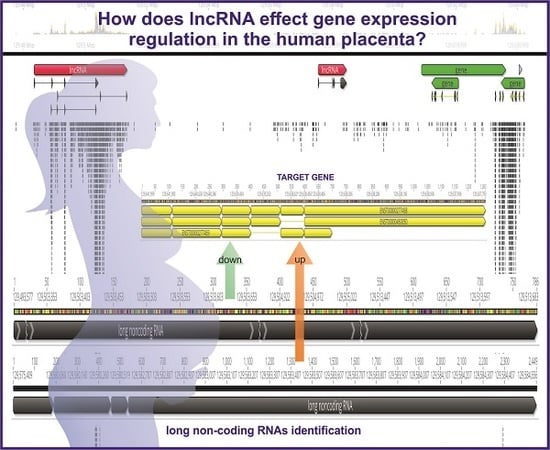
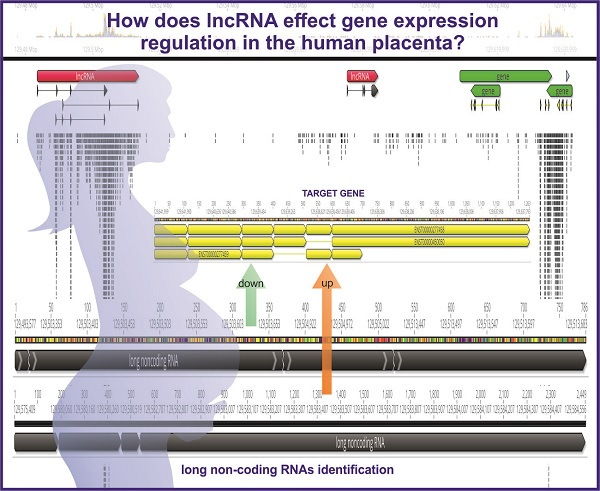
:
1. Introduction
2. Results
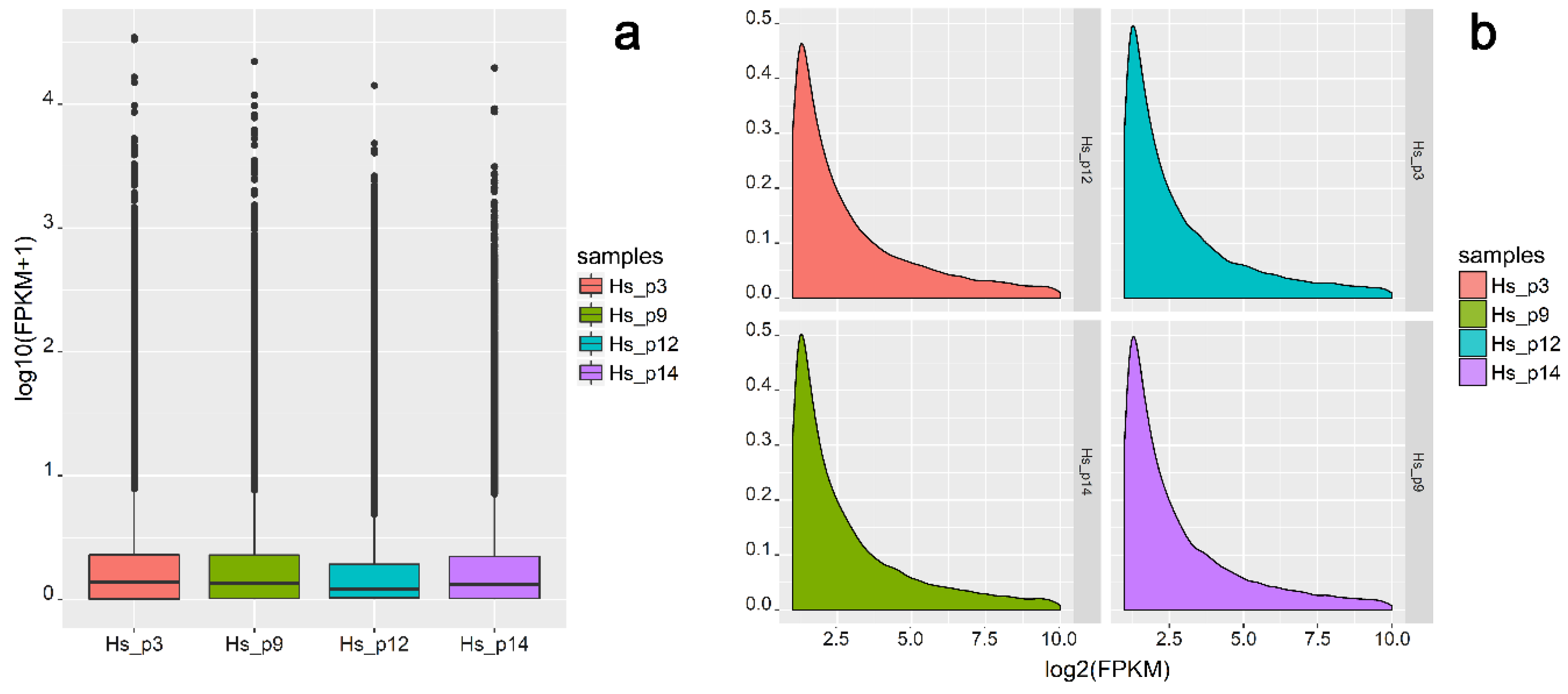
2.1. Characteristics of RNA-Seq Data
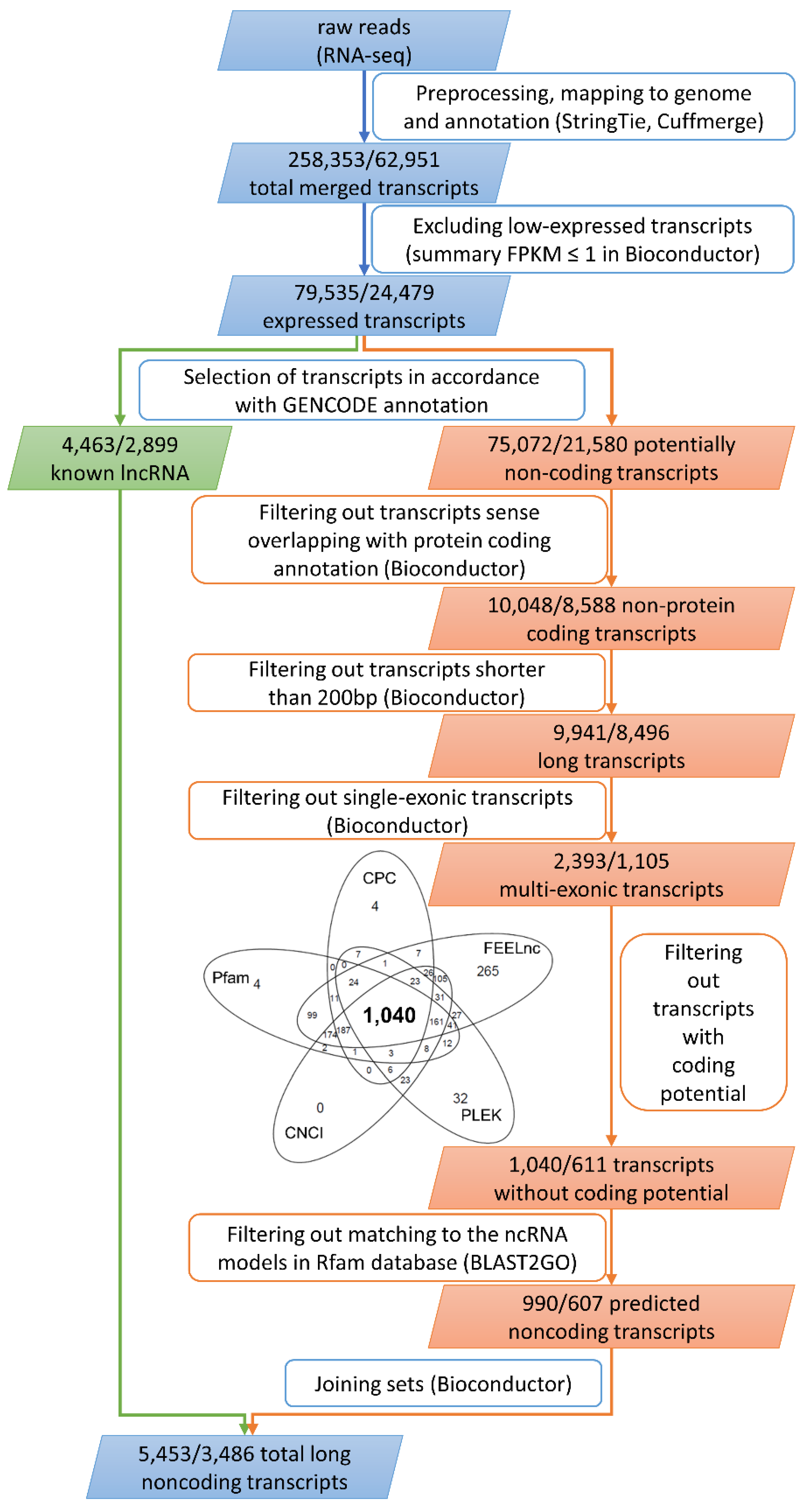
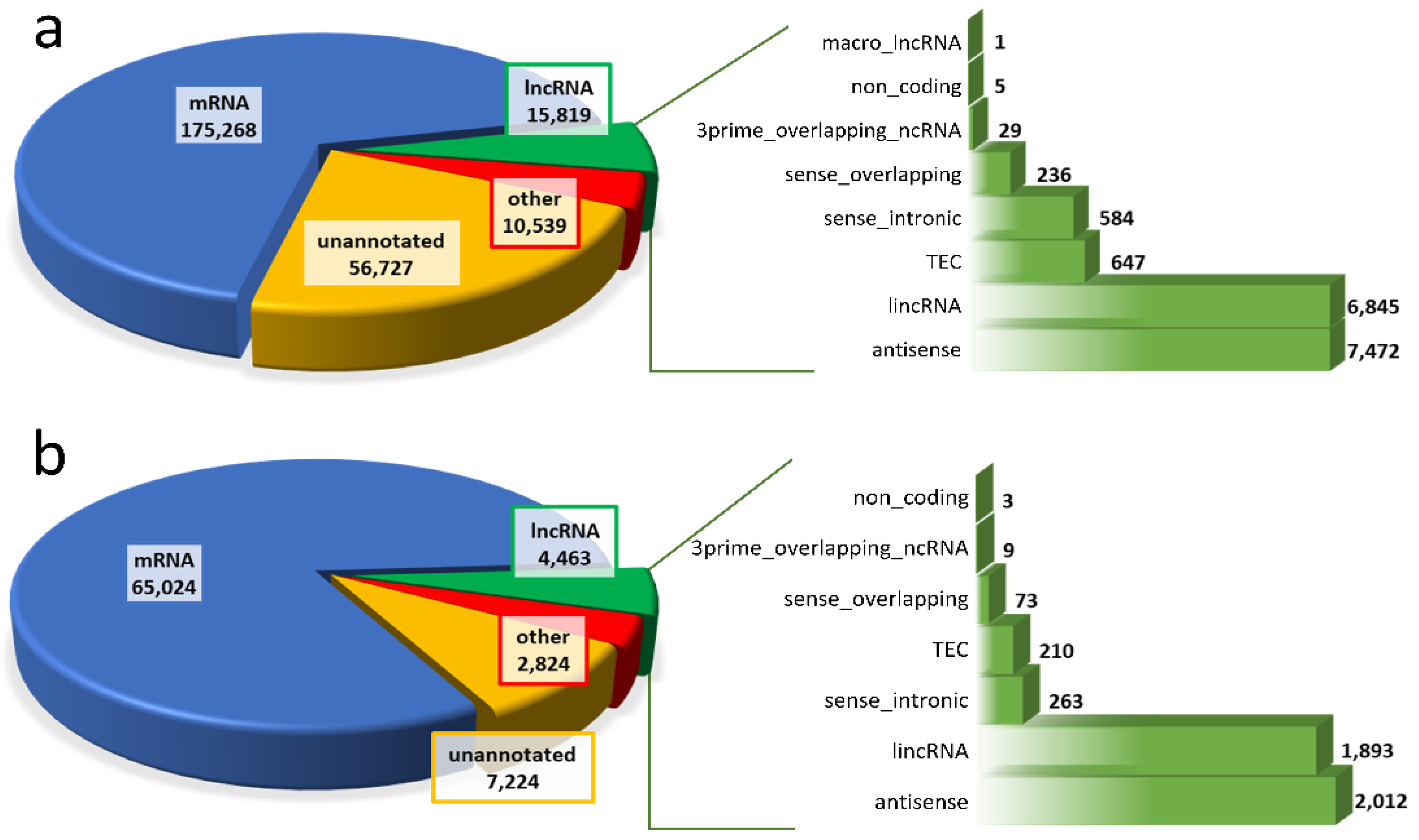
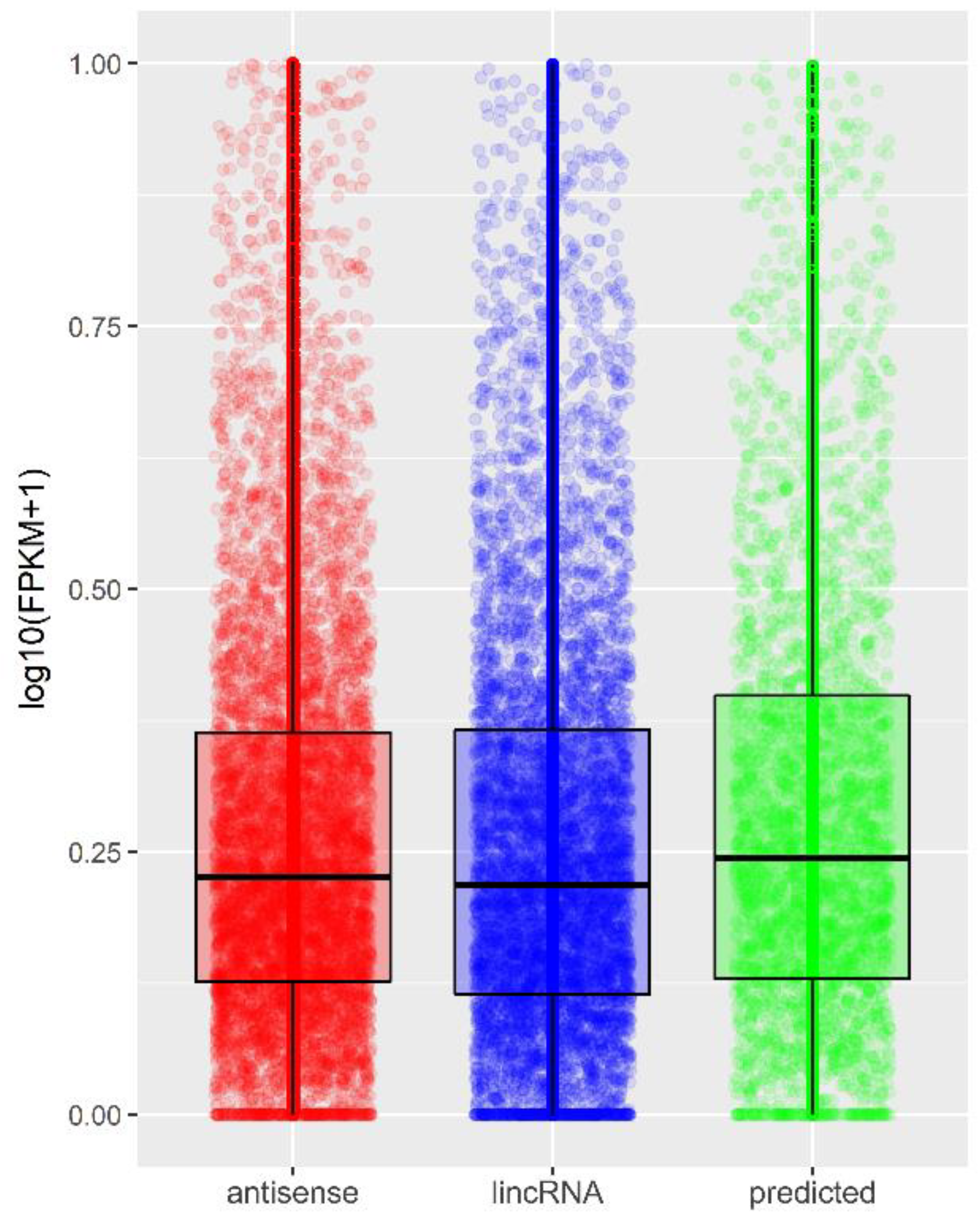
2.2. Identification and Profiling of lncRNAs
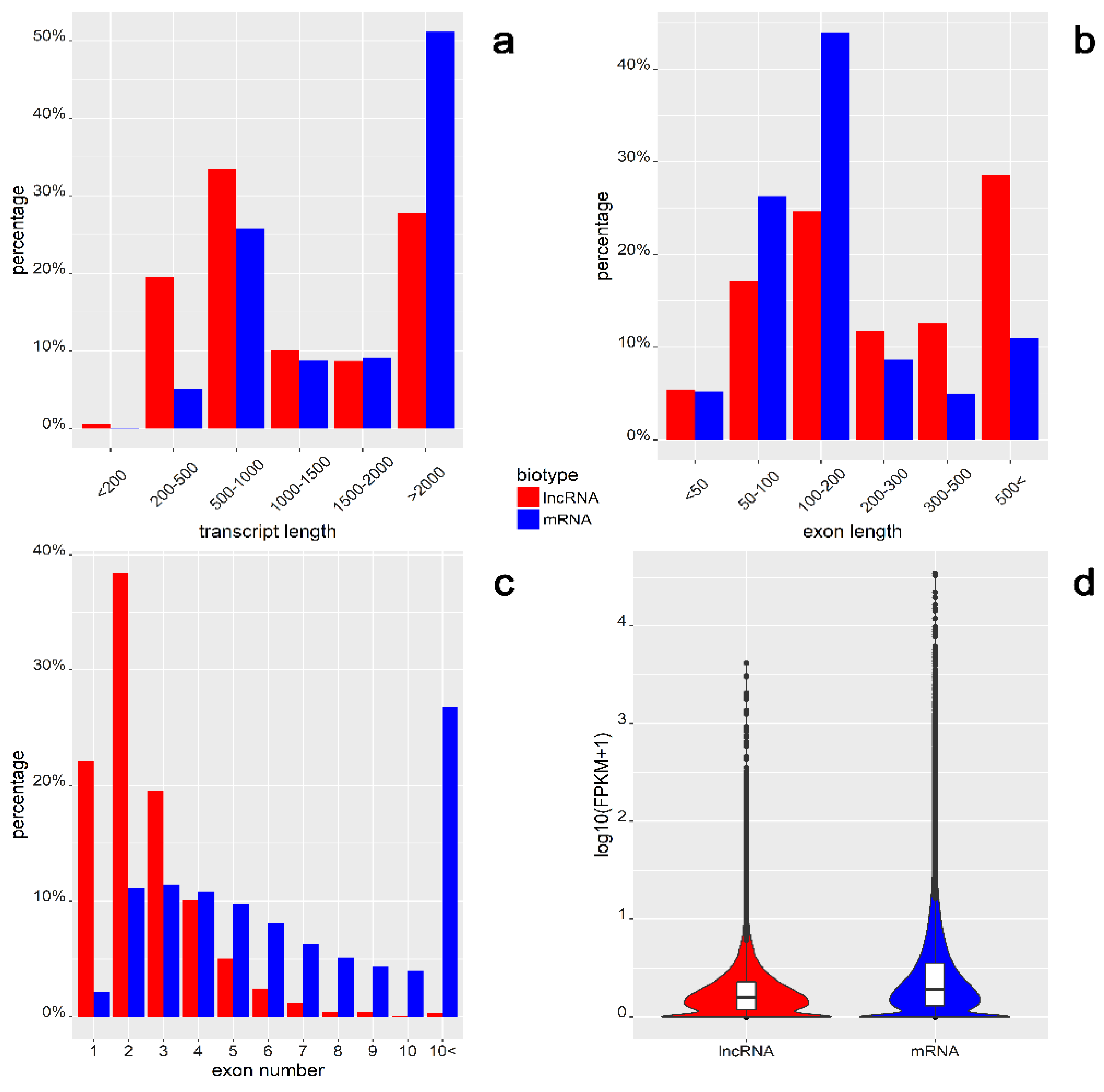
2.3. Feature Comparison of lncRNA and mRNA
2.4. Sex Biases in lncRNA Expression Levels
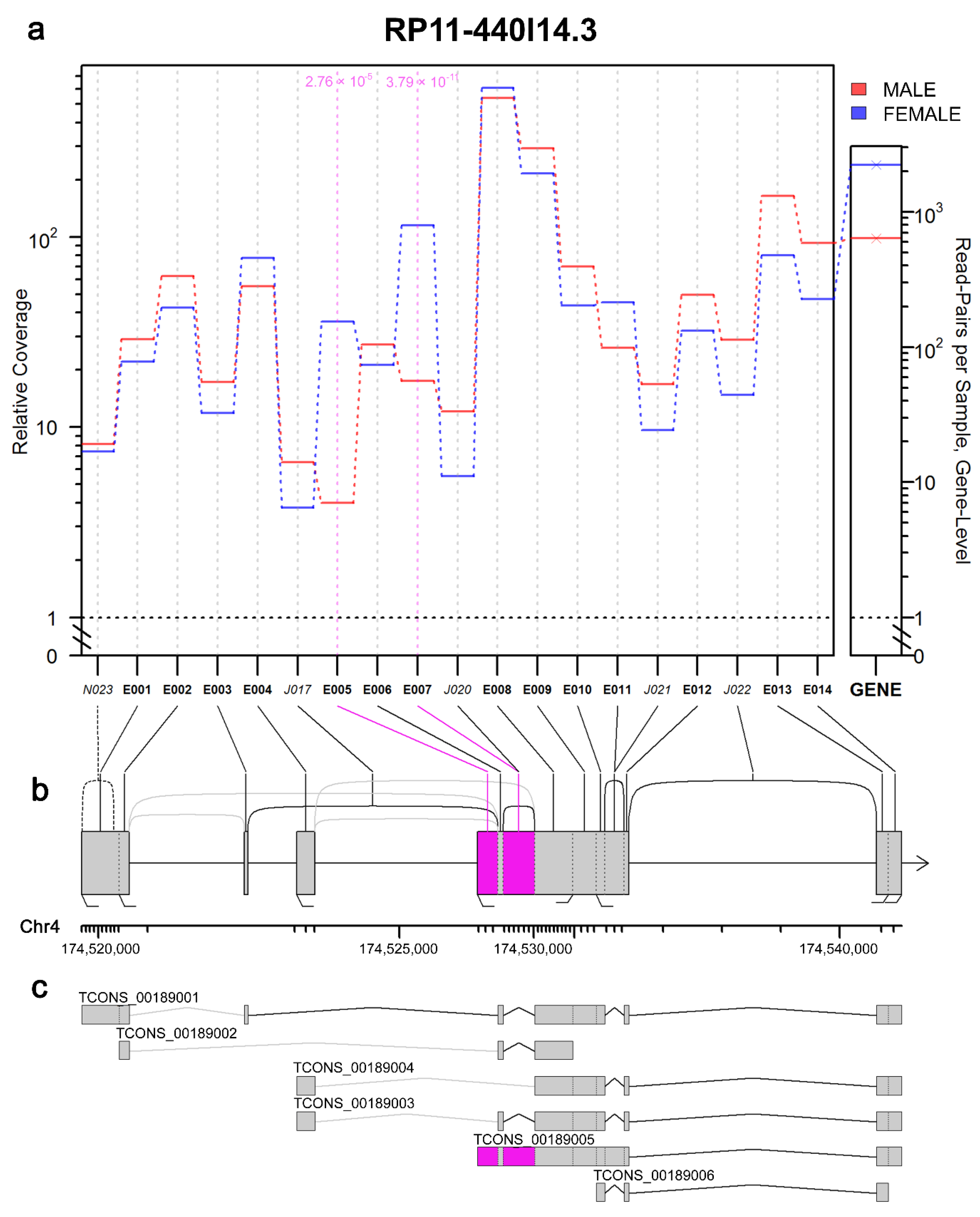
2.5. Splicing Alterations in Placental Transcriptome
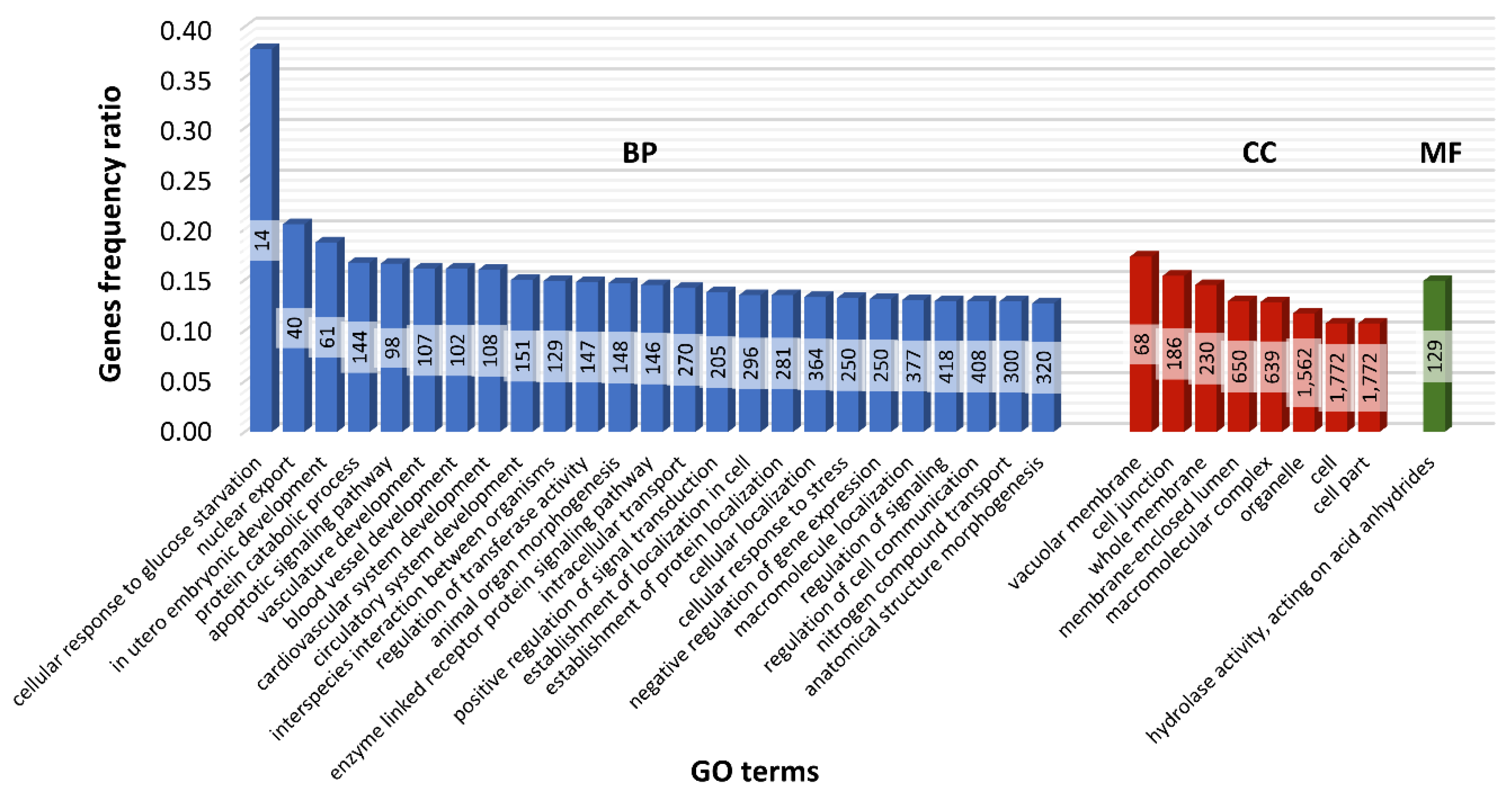
2.6. Functional Analysis of Nearest Neighbor Genes to lncRNAs
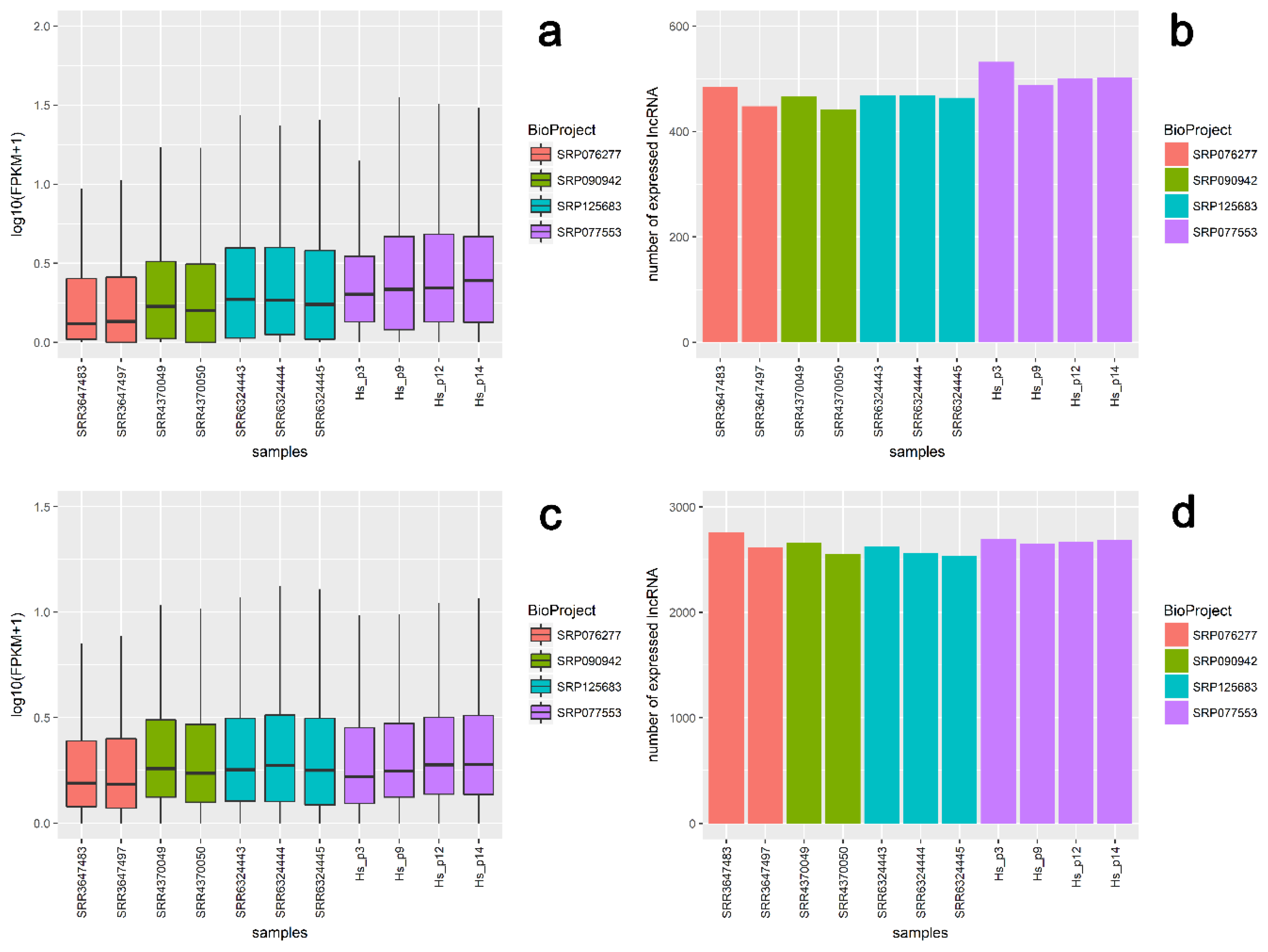
2.7. Validation of RNA-Seq Results Using External Transcriptomic Datasets
3. Discussion
4. Materials and Methods
4.1. Research Material
4.2. Transcriptome Assembly and Identification of Novel Transcripts
4.3. Classification, Characterization, and Validation of lncRNAs
4.4. Different Expression and Splicing Analysis
4.5. LncRNA Target cis Gene Prediction
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Availability of Supporting Data
References
- Murthi, P. Review: Placental homeobox genes and their role in regulating human fetal growth. Placenta 2014, 28, S46–S50. [Google Scholar] [CrossRef] [PubMed]
- Lipka, A.; Paukszto, L.; Majewska, M.; Jastrzebski, J.P.; Myszczynski, K.; Panasiewicz, G.; Szafranska, B. Identification of differentially expressed placental transcripts during multiple gestations in the Eurasian beaver (Castor fiber L.). Reprod. Fertil. Dev. 2017, 29, 2073–2084. [Google Scholar] [CrossRef] [PubMed]
- Majewska, M.; Lipka, A.; Paukszto, L.; Jastrzebski, J.P.; Myszczynski, K.; Gowkielewicz, M.; Jozwik, M.; Majewski, M.K. Transcriptome profile of the human placenta. Funct. Integr. Genom. 2017, 17, 551–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uusküla, L.; Männik, J.; Rull, K.; Minajeva, A.; Kõks, S.; Vaas, P.; Teesalu, P.; Reimand, J.; Laan, M. Mid-gestational gene expression profile in placenta and link to pregnancy complications. PLoS ONE 2012, 7, e49248. [Google Scholar] [CrossRef] [PubMed]
- Buckberry, S.; Bianco-Miotto, T.; Bent, S.J.; Dekker, G.A.; Roberts, C.T. Integrative transcriptome meta-analysis reveals widespread sex-biased gene expression at the human fetal-maternal interface. Mol. Hum. Reprod. 2014, 20, 810–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, D.P.; Salafia, C.M.; Miller, R.K.; Charles, A.K. Non-linear and gender-specific relationships among placental growth measures and the fetoplacental weight ratio. Placenta 2009, 30, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Fox, N.S.; Feinberg, J.; Klauser, C.K.; Rebarber, A. Outcomes in twin pregnancies reduced to singleton pregnancies compared with ongoing twin pregnancies. Am. J. Obstet. Gynecol. 2015, 213, 580.e1–580.e5. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.P.; Scardo, J.A.; Hayes, E.; Abuhamad, A.Z.; Berghella, V. Twins: Prevalence, problems, and preterm births. Am. J. Obstet. Gynecol. 2010, 203, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Ananth, C.V.; Demissie, K.; Smulian, J.C.; Vintzileos, A.M. Relationship among placenta previa, fetal growth restriction, and preterm delivery: A population-based study. Obstet. Gynecol. 2001, 98, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Huppertz, B.; Ghosh, D.; Sengupta, J. An integrative view on the physiology of human early placental villi. Prog. Biophys. Mol. Biol. 2014, 114, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.B.; Jung, S.C.; Lee, J.; Moon, H.S.; Chung, K.W.; Choi, B.O. Dynamic transcriptional events in distal sural nerve revealed by transcriptome analysis. Exp. Neurobiol. 2014, 23, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Sun, J.; Groome, L.J.; Wang, Y. Differential miRNA expression profiles between the first and third trimester human placentas. Am. J. Physiol. Endocrinol. Metab. 2013, 304, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Ding, N.; Li, J.; Jin, X.; Li, L.; Pan, T.; Huo, C.; Li, Y.; Xu, J.; Li, X. Landscape of the long non-coding RNA transcriptome in human heart. Brief. Bioinform. 2018. [Google Scholar] [CrossRef] [PubMed]
- Paralkar, V.R.; Mishra, T.; Luan, J.; Yao, Y.; Kossenkov, A.V.; Anderson, S.M.; Dunagin, M.; Pimkin, M.; Gore, M.; Sun, D.; et al. Lineage and species-specific long noncoding RNAs during erythro-megakaryocytic development. Blood 2014, 123, 1927–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattick, J.S.; Rinn, J.L. Discovery and annotation of long noncoding RNAs. Nat. Struct. Mol. Biol. 2015, 22, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Pauli, A.; Eivind, V.; Lin, M.F.; Garber, M.; Vastenhouw, N.L.; Levin, J.Z.; Fan, L.; Sandelin, A.; Rinn, J.L.; Regev, A.; et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012, 22, 577–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Amaral, P.P.; Mattick, J.S. Noncoding RNA in development. Mamm. Genome 2008, 19, 454–492. [Google Scholar] [CrossRef] [PubMed]
- Pauli, A.; Rinn, J.L.; Schie, A.F. Non-coding RNAs as regulators of embryogenesis. Nat. Rev. Genet. 2011, 12, 136–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouckenheimer, J.; Assou, S.; Riquier, S.; Hou, C.; Philippe, N.; Sansac, C.; Lavabre-Bertrand, T.; Commes, T.; Lemaître, J.M.; Boureux, A.; et al. Long non-coding RNAs in human early embryonic development and their potential in ART. Hum. Reprod. Update 2016, 23, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Mu, Y.; Ma, L.; Wang, C.; Tang, Z.; Yang, S.; Zhou, R.; Hu, X.; Li, M.H.; Li, K. Systematic identification and characterization of long intergenic non-coding RNAs in fetal porcine skeletal muscle development. Sci. Rep. 2015, 5, 8957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleutels, F.; Zwart, R.; Barlow, D.P. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 2002, 415, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Bawa, P.; Zackaria, S.; Verma, M.; Gupta, S.; Srivatsan, R.; Chaudhary, B.; Srinivasan, S. Integrative Analysis of Normal Long Intergenic Non-Coding RNAs in Prostate Cancer. PLoS ONE 2015, 10, e0122143. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wu, K.; Jiang, J.; Huang, J.; Zhang, P.; Zhu, Y.; Hu, G.; Lang, J.; Shi, Y.; Hu, L.; et al. Integrative Analysis Reveals Enhanced Regulatory Effects of Human Long Intergenic Non-Coding RNAs in Lung Adenocarcinoma. J. Genet. Genom. 2015, 42, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Qian, Y.; Zhou, X.; Lin, Y.; Jiang, J.; Chen, J.; Zhao, Z.; Shen, B. Discovery and characterization of long intergenic non-coding RNAs (lincRNA) module biomarkers in prostate cancer: An integrative analysis of RNA-Seq data. BMC Genom. 2015, 16, S3. [Google Scholar] [CrossRef] [PubMed]
- McAninch, D.; Roberts, C.T.; Bianco-Miotto, T. Mechanistic Insight into Long Noncoding RNAs and the Placenta. Int. J. Mol. Sci. 2017, 18, 1371. [Google Scholar] [CrossRef] [PubMed]
- Kaartokallio, T.; Cervera, A.; Kyllönen, A.; Laivuori, K.; Kere, J.; Laivuori, H.; FINNPEC Core Investigator Group. Gene expression profiling of pre-eclamptic placentae by RNA sequencing. Sci. Rep. 2015, 5, 14107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szcześniak, M.W.; Makałowska, I. lncRNA-RNA Interactions across the Human transcriptome. PLoS ONE 2016, 11, e0150353. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, T.L.; Sun, T.; Koeppel, A.F.; Lee, B.; Wang, E.T.; Farber, C.R.; Rich, S.S.; Sundheimer, L.W.; Buttle, R.A.; Chen, Y.I.; et al. Sex differences in the late first trimester human placenta transcriptome. Biol. Sex Differ. 2018, 9, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gormley, M.; Ona, K.; Kapidzic, M.; Garrido-Gomez, T.; Zdravkovic, T.; Fisher, S.J. Preeclampsia: Novel insights from global RNA profiling of trophoblast subpopulations. Am. J. Obstet. Gynecol. 2017, 217, 200.e1–200.e17. [Google Scholar] [CrossRef] [PubMed]
- Long, W.; Rui, C.; Song, X.; Dai, X.; Xue, X.; Lu, Y.; Shen, R.; Li, J.; Li, J.; Ding, H. Distinct expression profiles of lncRNAs between early-onset preeclampsia and preterm controls. Clin. Chim. Acta 2016, 463, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The Landscape of Long Noncoding RNAs in the Human Transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulitsky, I.; Shkumatava, A.; Jan, C.H.; Sive, H.; Bartel, D.P. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 2011, 147, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Huang, K.; Luo, Y.; Li, S. Identification and functional analysis of long non-coding RNAs in mouse cleavage stage embryonic development based on single cell transcriptome data. BMC Genom. 2014, 15, 845. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Xin, L.; Zhu, W.; Li, L.; Li, C.; Wang, Y.; Mu, Y.; Yang, S.; Li, K. Characterization of long non-coding RNA transcriptome in high-energy diet induced nonalcoholic steatohepatitis minipigs. Sci. Rep. 2016, 6, 30709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Wu, Y.; Yang, Y.; Yang, Y.T.; Wang, Z.; Yuan, J.; Yang, Y.; Hua, C.; Fan, X.; Niu, G.; et al. Comprehensive analysis of long non-coding RNAs highlights their spatio-temporal expression patterns and evolutional conservation in Sus scrofa. Sci. Rep. 2017, 7, 43166. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Ballabio, A.; Rupert, J.L.; Lafreniere, R.G.; Grompe, M.; Tonlorenzi, R.; Willard, H.F. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 1991, 349, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Pontier, D.B.; Gribnau, J. Xist regulation and function explored. Hum. Genet. 2011, 130, 223–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wutz, A. Gene silencing in X-chromosome inactivation: Advances in understanding facultative heterochromatin formation. Nat. Rev. Genet. 2011, 12, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Bartolomei, M.S. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell 2013, 152, 1308–1323. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T. Lessons from X-chromosome inactivation: Long ncRNA as guides and tethers to the epigenome. Genes Dev. 2009, 23, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Firulli, A.B. A HANDful of questions: The molecular biology of the heart and neural crest derivatives (HAND)-subclass of basic helix-loop-helix transcription factors. Gene 2003, 312, 27–40. [Google Scholar] [CrossRef]
- Hendershot, T.J.; Liu, H.; Clouthier, D.E.; Shepherd, I.T.; Coppola, E.; Studer, M.; Firulli, A.B.; Pittman, D.L.; Howard, M.J. Conditional deletion of Hand2 reveals critical functions in neurogenesis and cell type-specific gene expression for development of neural crest-derived noradrenergic sympathetic ganglion neurons. Dev. Biol. 2008, 319, 179–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huyen, D.V.; Bany, B.M. Evidence for a conserved function of heart and neural crest derivatives expressed transcript 2 in mouse and human decidualization. Reproduction 2011, 142, 353–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Okada, H.; Tsuzuki, T.; Nishigaki, A.; Yasuda, K.; Kanzaki, H. Progestin-induced heart and neural crest derivatives expressed transcript 2 is associated with fibulin-1 expression in human endometrial stromal cells. Fertil. Steril. 2013, 99, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Tsuzuki, T.; Shindoh, H.; Nishigaki, A.; Yasuda, K.; Kanzaki, H. Regulation of decidualization and angiogenesis in the human endometrium: Mini review. J. Obstet. Gynaecol. Res. 2014, 40, 1180–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgenstern, R.; Zhang, J.; Johansson, K. Microsomal glutathione transferase 1: Mechanism and functional roles. Drug Metab. Rev. 2011, 43, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Gram, A.; Boos, A.; Aslan, S.; Ay, S.S.; Önyay, F.; Kowalewski, M.P. Functional implications of the utero-placental relaxin (RLN) system in the dog throughout pregnancy and at term. Reproduction 2017, 154, 415–431. [Google Scholar] [CrossRef] [PubMed]
- Marzioni, D.; Tamagnone, L.; Capparuccia, L.; Marchini, C.; Amici, A.; Todros, T.; Bischof, P.; Neidhart, S.; Grenningloh, G.; Castellucci, M. Restricted innervation of uterus and placenta during pregnancy: Evidence for a role of the repelling signal Semaphorin 3A. Dev. Dyn. 2004, 231, 839–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartley, S.W.; Mullikin, J.C. Detection and visualization of differential splicing in RNA-Seq data with JunctionSeq. Nucleic Acids. Res. 2016, 44, e127. [Google Scholar] [CrossRef] [PubMed]
- Mourier, T.; Jeffares, D.C. Eukaryotic intron loss. Science 2003, 300, 1393. [Google Scholar] [CrossRef] [PubMed]
- Ha, C.T.; Wu, J.A.; Irmak, S.; Lisboa, F.A.; Dizon, A.M.; Warren, J.W.; Ergun, S.; Dveksler, G.S. Human pregnancy specific beta-1-glycoprotein 1 (PSG1) has a potential role in placental vascular morphogenesis. Biol. Reprod. 2010, 83, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Horne, C.H.; Towler, C.M. Pregnancy-specific beta1-glycoprotein: A review. Obstet. Gynecol. Surv. 1978, 33, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Towler, C.M.; Horne, C.H.; Jandial, V.; Campbell, D.M.; MacGillivray, I. Plasma levels of pregnancy-specific beta 1-glycoprotein in complicated pregnancies. Br. J. Obstet. Gynaecol. 1977, 84, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Silver, R.M.; Heyborne, K.D.; Leslie, K.K. Pregnancy specific beta 1 glycoprotein (SP-1) in maternal serum and amniotic fluid; pre-eclampsia, small for gestational age fetus and fetal distress. Placenta 1993, 14, 583–589. [Google Scholar] [CrossRef]
- Arnold, L.L.; Doherty, T.M.; Flor, A.W.; Simon, J.A.; Chou, J.Y.; Chan, W.Y.; Mansfield, B.C. Pregnancy-specific glycoprotein gene expression in recurrent aborters: A potential correlation to interleukin-10 expression. Am. J. Reprod. Immunol. 1999, 41, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.L.; Semyonov, J.; Cheng, P.J.; Huang, S.Y.; Park, J.I.; Tsai, H.J.; Lin, C.Y.; Grützner, F.; Soong, Y.K.; Cai, J.J.; et al. Widespread divergence of the CEACAM/PSG genes in vertebrates and humans suggests sensitivity to selection. PLoS ONE 2013, 8, e61701. [Google Scholar] [CrossRef] [PubMed]
- De Kreuk, B.J.; Schaefer, A.; Anthony, E.C.; Tol, S.; Fernandez-Borja, M.; Geerts, D.; Pool, J.; Hambach, L.; Goulmy, E.; Hordijk, P.L. The human minor Histocompatibility Antigen1 is a RhoGAP. PLoS ONE 2013, 8, e73962. [Google Scholar] [CrossRef] [PubMed]
- Holland, O.J.; Linscheid, C.; Hodes, H.C.; Nauser, T.L.; Gilliam, M.; Stone, P.; Chamley, L.W.; Petroff, M.G. Minor histocompatibility antigens are expressed in syncytiotrophoblast and trophoblast debris: Implications for maternal alloreactivity to the fetus. Am. J. Pathol. 2012, 180, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Amado-Azevedo, J.; Reinhard, N.R.; van Bezu, J.; van Nieuw Amerongen, G.P.; van Hinsbergh, V.W.M.; Hordijk, P.L. The minor histocompatibility antigen 1 (HMHA1)/ArhGAP45 is a RacGAP and a novel regulator of endothelial integrity. Vascul. Pharmacol. 2018, 101, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Boissel, L.; Houssin, N.; Chikh, A.; Rynditch, A.; Van Hove, L.; Moreau, J. Recruitment of Cdc42 through the GAP domain of RLIP participates in remodeling of the actin cytoskeleton and is involved in Xenopus gastrulation. Dev. Biol. 2007, 312, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Ligeti, E.; Welti, S.; Scheffzek, K. Inhibition and termination of physiological responses by GTPase activating proteins. Physiol. Rev. 2012, 92, 237–272. [Google Scholar] [CrossRef] [PubMed]
- Mishima, M.; Glotzer, M. Cytokinesis: A logical GAP. Curr. Biol. 2003, 13, 589–591. [Google Scholar] [CrossRef]
- Linscheid, C.; Heitmann, E.; Singh, P.; Wickstrom, E.; Qiu, L.; Hodes, H.; Nauser, T.; Petroff, M.G. Trophoblast expression of the minor histocompatibility antigen HA-1 is regulated by oxygen and is increased in placentas from preeclamptic women. Placenta 2015, 36, 832–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assou, S.; Boumela, I.; Haouzi, D.; Monzo, C.; Dechaud, H.; Kadoch, I.J.; Hamamah, S. Transcriptome analysis during human trophectoderm specification suggests new roles of metabolic and epigenetic genes. PLoS ONE 2012, 7, e39306. [Google Scholar] [CrossRef] [PubMed]
- Blakeley, P.; Fogarty, N.M.; del Valle, I.; Wamaitha, S.E.; Hu, T.X.; Elder, K.; Snell, P.; Christie, L.; Robson, P.; Niakan, K.K. Defining the three cell lineages of the human blastocyst by single-cell RNA-seq. Development 2015, 142, 3151–3165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Home, P.; Kumar, R.P.; Ganguly, A.; Saha, B.; Milano-Foster, J.; Bhattacharya, B.; Ray, S.; Gunewardena, S.; Paul, A.; Camper, S.A.; et al. Genetic redundancy of GATA factors in the extraembryonic trophoblast lineage ensures the progression of preimplantation and postimplantation mammalian development. Development 2017, 144, 876–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keniry, A.; Oxley, D.; Monnier, P.; Kyba, M.; Dandolo, L.; Smits, G.; Reik, W. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat. Cell Biol. 2012, 14, 659–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saben, J.; Zhong, Y.; McKelvey, S.; Dajani, N.K.; Andres, A.; Badger, T.M.; Gomez-Acevedo, H.; Shankar, K. A comprehensive analysis of the human placenta transcriptome. Placenta 2014, 35, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, D.H.; Chu, E.T.; Spektor, R.; Soloway, P.D. Long Non-Coding RNA Regulation of Reproduction and Development. Mol. Reprod. Dev. 2015, 82, 932–956. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.L.; Liu, M.; Yang, Y.; Yang, H.; Liao, Q.; Bai, Y.; Li, Y.X.; Li, D.; Peng, C.; Wang, Y.L. The imprinted H19 gene regulates human placental trophoblast cell proliferation via encoding miR-675 that targets Nodal Modulator 1 (NOMO1). RNA Biol. 2012, 9, 1002–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Luo, X.; Gao, Q.; Wang, Y.; Gao, Q.; Long, W. Dysregulation of LncRNAs in Placenta and Pathogenesis of Preeclampsia. Curr. Drug Targets 2017, 10, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, M.; Zhao, D.; Yi, P.; Lu, L.; Han, J.; Zheng, X.; Zhou, Y.; Li, L. The H19 gene imprinting in normal pregnancy and pre-eclampsia. Placenta 2009, 30, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Giabicani, É.; Brioude, F.; Le Bouc, Y.; Netchine, I. Imprinted disorders and growth. Ann. Endocrinol. (Paris) 2017, 78, 112–113. [Google Scholar] [CrossRef] [PubMed]
- Zuckerwise, L.; Li, J.; Lu, L.; Men, Y.; Geng, T.; Buhimschi, C.S.; Buhimschi, I.A.; Bukowski, R.; Guller, S.; Paidas, M.; et al. H19 long noncoding RNA alters trophoblast cell migration and invasion by regulating TβR3 in placentae with fetal growth restriction. Oncotarget 2016, 7, 38398–38407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, W.; Jingli, F.; Wei, S.W.; Li, W.L. Genomic imprinting status of IGF-II and H19 in placentas of fetal growth restriction patients. J. Genet. 2010, 89, 213–216. [Google Scholar] [PubMed]
- Li, X.; Ma, C.; Zhang, L.; Li, N.; Zhang, X.; He, J.; He, R.; Shao, M.; Wang, J.; Kang, L.; et al. LncRNAAC132217.4, a KLF8-regulated long non-coding RNA, facilitates oral squamous cell carcinoma metastasis by upregulating IGF2 expression. Cancer Lett. 2017, 407, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Aisemberg, J.; Bariani, M.V.; Vercelli, C.A.; Wolfson, M.L.; Franchi, A.M. Lipopolysaccharide-induced murine embryonic resorption involves nitric oxide-mediatedinhibition of the NAD+-dependent 15-hydroxyprostaglandin dehydrogenase. Reproduction 2012, 144, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Girard, A.; Sachidanandam, R.; Hannon, G.J.; Carmell, M.A. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 2006, 442, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Aravin, A.; Gaidatzis, D.; Pfeffer, S.; Lagos-Quintana, M.; Landgraf, P.; Iovino, N.; Morris, P.; Brownstein, M.J.; Kuramochi-Miyagawa, S.; Nakano, T.; et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature 2006, 442, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.D. Non-coding RNA: A new frontier in regulatory biology. Natl. Sci. Rev. 2014, 1, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.Q.; Nolasco, S.; Soares, H. Non-coding RNAs: Multi-tasking molecules in the cell. Int. J. Mol. Sci. 2013, 14, 16010–16039. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Bateman, A.; Finn, R.D. Predicting active site residue annotations in the Pfam database. BMC Bioinform. 2007, 8, 298. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Mao, X.; Cai, T.; Luo, J.; Wei, L. KOBAS server: A web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006, 34, 720–724. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class-Code | Description | Isoform (TCONS) | Locus (XLOC) |
|---|---|---|---|
| “-” | unknown, intergenic region | 395 | 344 |
| “o” | overlapped with existed gene with a dramatic difference in gene structures | 208 | 170 |
| “x” | overlapped with existed gene in an opposite direction | 160 | 150 |
| “i” | located in introns | 2 | 2 |
| “=” | complete match (of known lncRNA) | 3747 | 2698 |
| “j” | potentially novel isoform (of known lncRNA) | 941 | 579 |
| Gene_ID | lncRNAVariant ID | Ensembl Gene ID | HGNC Symbol | Gene Name | Biotype | Locus | Samples | Expression Level [FPKM] | log2fc | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Male | Female | Male | Female | ||||||||
| XLOC_042918 | 11 | ENSG00000237125 | HAND2-AS1 | HAND2-AS1 | antisense | 4:173524692-173591465 | Hs_p3, Hs_p9 | Hs_p12, Hs_p14 | 2.416 | 23.984 | 3.312 |
| XLOC_050164 | 1 | ENSG00000227012 | LINC02527 | RP1-97J1.2 | lincRNA | 6:111900309-111909386 | Hs_p3, Hs_p9 | Hs_p12, Hs_p14 | 2.095 | 0.000 | “-Inf” |
| XLOC_061548 | 9 | ENSG00000229807 | XIST | XIST | lincRNA | X:73792204-73852753 | Hs_p3, Hs_p9 | Hs_p12, Hs_p14 | 1.185 | 90.740 | 6.259 |
| XLOC_062450 | 1 | ENSG00000229308 | NA | AC010084.1 | lincRNA | Y:4036485-4106081 | Hs_p3, Hs_p9 | Hs_p12, Hs_p14 | 1.444 | 0.000 | “-Inf” |
| XLOC_062528 | 3 | ENSG00000233864 | TTTY15 | TTTY15 | lincRNA | Y:12662333-12692233 | Hs_p3, Hs_p9 | Hs_p12, Hs_p14 | 5.355 | 0.000 | “-Inf” |
| SRR3647483 | SRR3647497 | SRR4370049 | SRR4370050 | SRR6324443 | SRR6324444 | SRR6324445 | Hs_p3 | Hs_p9 | Hs_p12 | Hs_p14 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Novel | Min. | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
| 1st Qu. | 0.0200 | 0.0000 | 0.0272 | 0.0000 | 0.0343 | 0.0527 | 0.0288 | 0.1313 | 0.0843 | 0.1331 | 0.1333 | |
| Median | 0.1178 | 0.1345 | 0.2295 | 0.2124 | 0.2744 | 0.2801 | 0.2483 | 0.3100 | 0.3456 | 0.3519 | 0.3937 | |
| Mean | 0.3073 | 0.3158 | 0.3851 | 0.3723 | 0.4278 | 0.4400 | 0.4262 | 0.4341 | 0.4693 | 0.4878 | 0.5055 | |
| 3rd Qu. | 0.4167 | 0.4262 | 0.5216 | 0.5149 | 0.6155 | 0.6120 | 0.6010 | 0.5563 | 0.6927 | 0.7024 | 0.7068 | |
| Max. | 3.5141 | 3.5084 | 3.1126 | 3.0138 | 2.8940 | 2.8243 | 2.7285 | 3.5662 | 3.0406 | 3.6352 | 2.8402 | |
| Known | Min. | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
| 1st Qu. | 0.0810 | 0.0732 | 0.1281 | 0.1034 | 0.1093 | 0.1085 | 0.0909 | 0.0972 | 0.1283 | 0.1410 | 0.1412 | |
| Median | 0.1932 | 0.1882 | 0.2657 | 0.2461 | 0.2649 | 0.2865 | 0.2619 | 0.2273 | 0.2574 | 0.2870 | 0.2867 | |
| Mean | 0.3159 | 0.3127 | 0.3951 | 0.3755 | 0.4016 | 0.4227 | 0.4067 | 0.3698 | 0.3945 | 0.4150 | 0.4228 | |
| 3rd Qu. | 0.4016 | 0.4150 | 0.5165 | 0.4978 | 0.5357 | 0.5665 | 0.5464 | 0.4826 | 0.5086 | 0.5361 | 0.5451 | |
| Max. | 3.4224 | 3.3734 | 3.4381 | 3.3992 | 4.5228 | 5.1047 | 5.1712 | 3.7855 | 3.5651 | 3.3986 | 3.5616 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majewska, M.; Lipka, A.; Paukszto, L.; Jastrzebski, J.P.; Gowkielewicz, M.; Jozwik, M.; Majewski, M.K. Preliminary RNA-Seq Analysis of Long Non-Coding RNAs Expressed in Human Term Placenta. Int. J. Mol. Sci. 2018, 19, 1894. https://doi.org/10.3390/ijms19071894
Majewska M, Lipka A, Paukszto L, Jastrzebski JP, Gowkielewicz M, Jozwik M, Majewski MK. Preliminary RNA-Seq Analysis of Long Non-Coding RNAs Expressed in Human Term Placenta. International Journal of Molecular Sciences. 2018; 19(7):1894. https://doi.org/10.3390/ijms19071894
Chicago/Turabian StyleMajewska, Marta, Aleksandra Lipka, Lukasz Paukszto, Jan Pawel Jastrzebski, Marek Gowkielewicz, Marcin Jozwik, and Mariusz Krzysztof Majewski. 2018. "Preliminary RNA-Seq Analysis of Long Non-Coding RNAs Expressed in Human Term Placenta" International Journal of Molecular Sciences 19, no. 7: 1894. https://doi.org/10.3390/ijms19071894
APA StyleMajewska, M., Lipka, A., Paukszto, L., Jastrzebski, J. P., Gowkielewicz, M., Jozwik, M., & Majewski, M. K. (2018). Preliminary RNA-Seq Analysis of Long Non-Coding RNAs Expressed in Human Term Placenta. International Journal of Molecular Sciences, 19(7), 1894. https://doi.org/10.3390/ijms19071894