The Roles of DNA Topoisomerase IIβ in Transcription

{kind=link}
{kind=link}
Abstract
:1. Discovery of DNA Topoisomerase IIβ
2. Distinctions between Topo IIα and Topo IIβ
3. Genetic Studies Reveal a Role for Topo IIβ in Neural Development
4. Topo IIβ and Transcriptional Regulation of Developmental Genes in Neurons
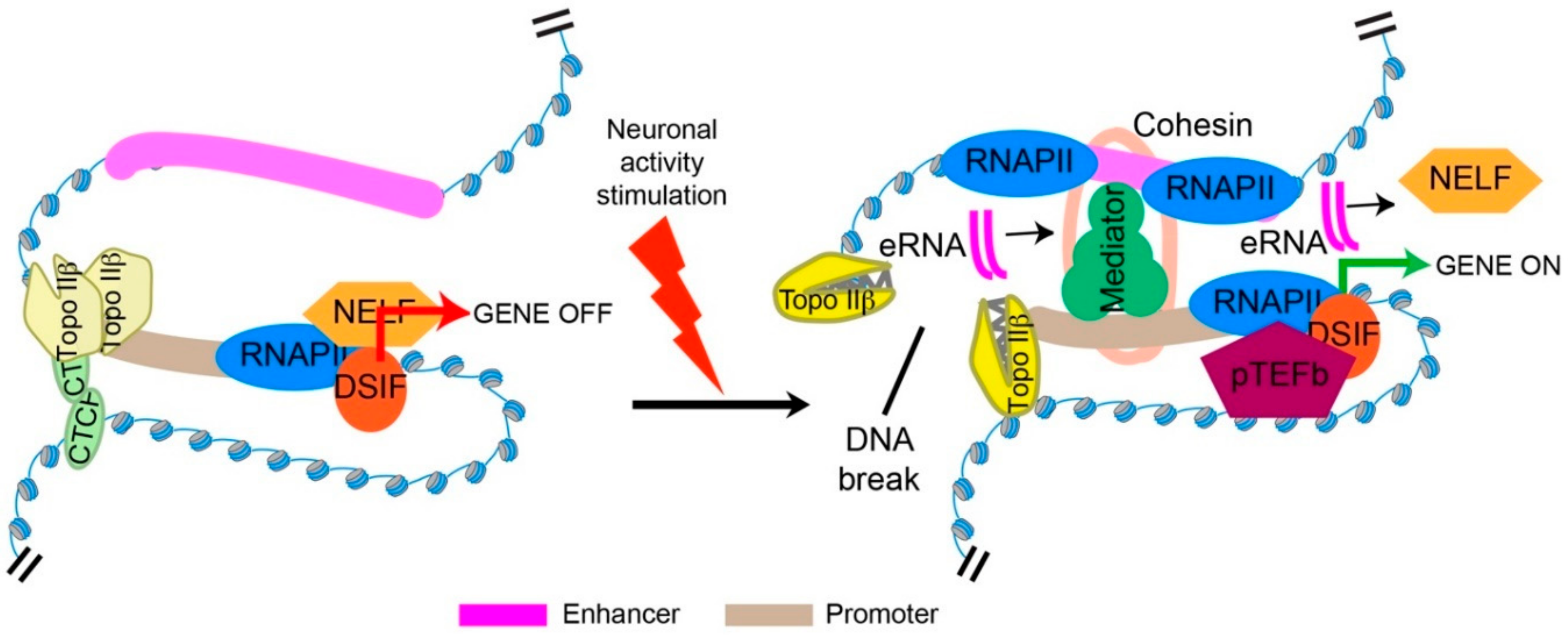
5. Topo IIβ and Transcriptional Regulation through the Formation of Stimulus-Induced DSBs
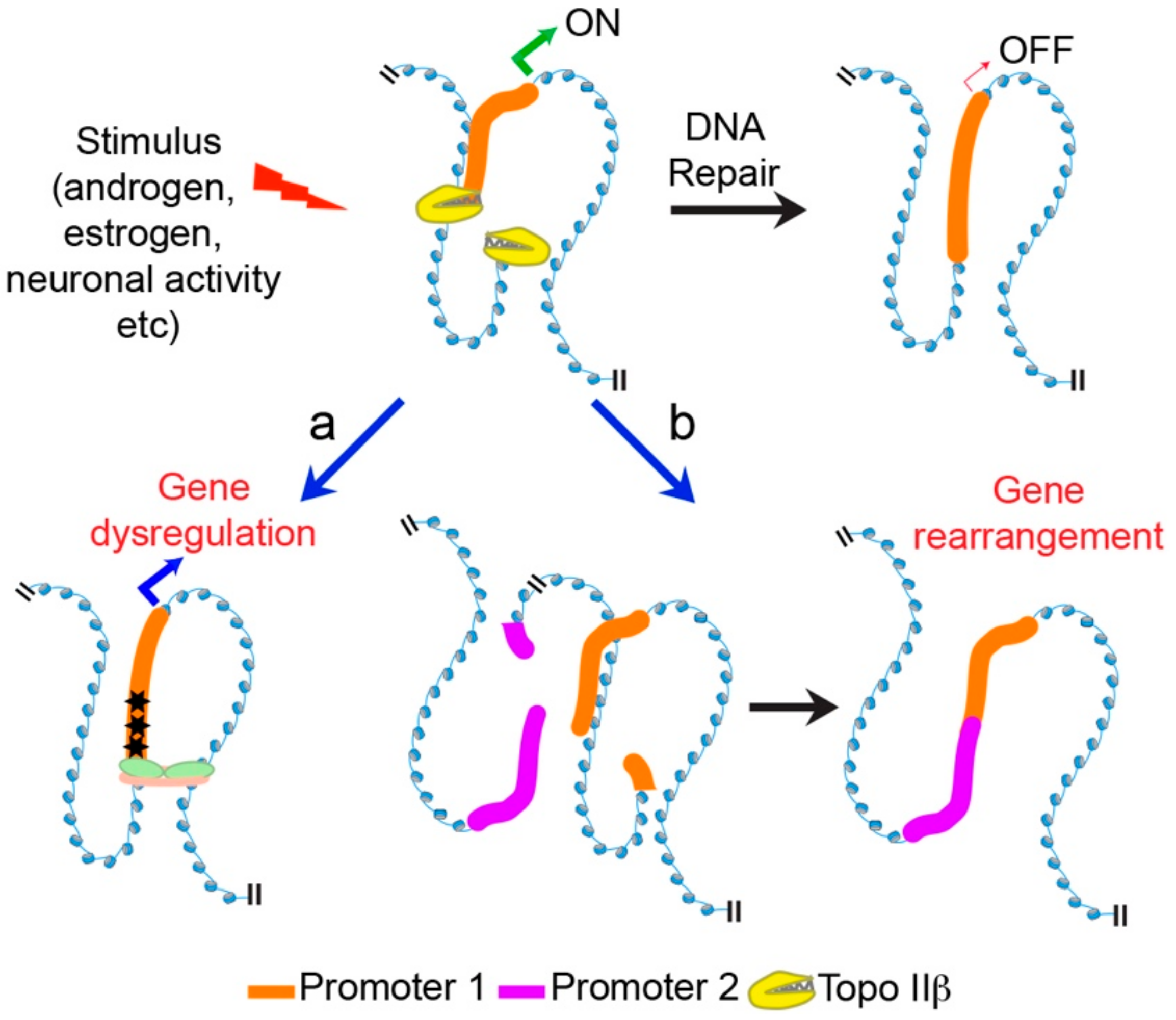
6. Mechanisms Underlying Transcriptional Regulation by Topo IIβ
7. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Drake, F.H.; Zimmerman, J.P.; McCabe, F.L.; Bartus, H.F.; Per, S.R.; Sullivan, D.M.; Ross, W.E.; Mattern, M.R.; Johnson, R.K.; Crooke, S.T.; et al. Purification of topoisomerase II from amsacrine-resistant P388 leukemia cells. Evidence for two forms of the enzyme. J. Biol. Chem. 1987, 262, 16739–16747. [Google Scholar] [PubMed]
- Drake, F.H.; Hofmann, G.A.; Bartus, H.F.; Mattern, M.R.; Crooke, S.T.; Mirabelli, C.K. Biochemical and Pharmacological Properties of p170 and p180 Forms of Topoisomerase II. Biochemistry 1989, 28, 8154–8160. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.D.; Drake, F.H.; Tan, K.B.; Per, S.R.; Crooke, S.T.; Mirabelli, C.K. Characterization and immunological identification of cDNA clones encoding two human DNA topoisomerase II isozymes. Proc. Natl. Acad. Sci. USA 1989, 86, 9431–9435. [Google Scholar] [CrossRef] [PubMed]
- Austin, C.A.; Fisher, L.M. Isolation and characterization of a human cDNA clone encoding a novel DNA topoisomerase II homologue from HeLa cells. FEBS Lett. 1990, 266, 115–117. [Google Scholar] [CrossRef] [Green Version]
- Tsai-Pflugfelder, M.; Liu, L.F.; Liu, A.A.; Tewey, K.M.; Whang-Peng, J.; Knutsen, T.; Huebner, K.; Croce, C.M.; Wang, J.C. Cloning and sequencing of cDNA encoding human DNA topoisomerase II and localization of the gene to chromosome region 17q21-22. Proc. Natl. Acad. Sci. USA 1988, 85, 7177–7181. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.B.; Dorman, T.E.; Falls, K.M.; Chung, T.D.; Mirabelli, C.K.; Crooke, S.T.; Mao, J. Topoisomerase II α and topoisomerase II β genes: Characterization and mapping to human chromosomes 17 and 3, respectively. Cancer Res. 1992, 52, 231–234. [Google Scholar] [PubMed]
- Jenkins, J.R.; Ayton, P.; Jones, T.; Davies, S.L.; Simmons, D.L.; Harris, A.L.; Sheer, D.; Hickson, I.D. Isolation of cDNA clones encoding the β isozyme of human DNA topoisomerase II and localisation of the gene to chromosome 3p24. Nucleic Acids Res. 1992, 20, 5587–5592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, A.J.; Mirski, S.E.L.; Cummings, H.J.; Yu, Q.; Gerlach, J.H.; Cole, S.P.C. Structural organization of the human TOP2A and TOP2B genes. Gene 1998, 221, 255–266. [Google Scholar] [CrossRef]
- Sng, J.-H.; Heaton, V.J.; Bell, M.; Maini, P.; Austin, C.A.; Fisher, L.M. Molecular cloning and characterization of the human topoisomerase IIα and IIβ genes: Evidence for isoform evolution through gene duplication. BBA Gene Struct. Expr. 1999, 1444, 395–406. [Google Scholar] [CrossRef]
- Lynn, R.; Giaever, G.; Swanberg, S.L.; Wang, J.C. Tandem regions of yeast DNA topoisomerase II share homology with different subunits of bacterial gyrase. Science 1986, 233, 647–649. [Google Scholar] [CrossRef] [PubMed]
- Woessner, R.D.; Mattern, M.R.; Mirabelli, C.K.; Johnson, R.K.; Drake, F.H. Proliferation- and cell cycle-dependent differences in expression of the 170 kilodalton and 180 kilodalton forms of topoisomerase II in NIH-3T3 cells. Cell Growth Differ. 1991, 2, 209–214. [Google Scholar] [PubMed]
- Chaly, N.; Chen, X.; Dentry, J.; Brown, D.L. Organization of DNA topoisomerase II isotypes during the cell cycle of human lymphocytes and HeLa cells. Chromosom. Res 1996, 4, 457–466. [Google Scholar] [CrossRef]
- Meyer, K.N.; Kjeldsen, E.; Straub, T.; Knudsen, B.R.; Hickson, I.D.; Kikuchi, A.; Kreipe, H.; Boege, F. Cell cycle-coupled relocation of types I and II topoisomerases and modulation of catalytic enzyme activities. J. Cell Biol. 1997, 136, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Dereuddre, S.; Delaporte, C.; Jacquemin-Sablon, A. Role of topoisomerase II β in the resistance of 9-OH-ellipticine-resistant Chinese hamster fibroblasts to topoisomerase II inhibitors. Cancer Res. 1997, 57, 4301–4308. [Google Scholar] [PubMed]
- Grue, P.; Gräßer, A.; Sehested, M.; Jensen, P.B.; Uhse, A.; Straub, T.; Ness, W.; Boege, F. Essential mitotic functions of DNA topoisomerase IIα are not adopted by topoisomerase IIβ in human H69 cells. J. Biol. Chem. 1998, 273, 33660–33666. [Google Scholar] [CrossRef] [PubMed]
- Capranico, G.; Tinelli, S.; Austin, C.A.; Fisher, M.L.; Zunino, F. Different patterns of gene expression of topoisomerase II isoforms in differentiated tissues during murine development. BBA Gene Struct. Expr. 1992, 1132, 43–48. [Google Scholar] [CrossRef]
- Juenke, J.E.M.; Holden, J.A. The distribution of DNA topoisomerase II isoforms in differentiated adult mouse tissues. BBA Gene Struct. Expr. 1993, 1216, 191–196. [Google Scholar] [CrossRef]
- Watanabe, M.; Tsutsui, K.; Inoue, Y. Differential expressions of the topoisomerase II α and II β mRNAs in developing rat brain. Neurosci. Res. 1994, 19, 51–57. [Google Scholar] [CrossRef]
- Zandvliet, D.W.; Hanby, A.M.; Austin, C.A; Marsh, K.L.; Clark, I.B.; Wright, N.A.; Poulsom, R. Analysis of foetal expression sites of human type II DNA topoisomerase α and β mRNAs by in situ hybridisation. Biochim. Biophys. Acta 1996, 1307, 239–247. [Google Scholar] [CrossRef]
- Turley, H.; Comley, M.; Houlbrook, S.; Nozaki, N.; Kikuchi, A.; Hickson, I.D.; Gatter, K.; Harris, A.L. The distribution and expression of the two isoforms of DNA topoisomerase II in normal and neoplastic human tissues. Br. J. Cancer 1997, 75, 1340–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, C.; Hiasa, H.; Marians, K.J. DNA gyrase and topoisomerase IV: Biochemical activities, physiological roles during chromosome replication, and drug sensitivities. Biochim. Biophys. Acta 1998, 1400, 29–43. [Google Scholar] [CrossRef]
- Wu, C.C.; Li, T.K.; Farh, L.; Lin, L.Y.; Lin, T.S.; Yu, Y.J.; Yen, T.J.; Chiang, C.W.; Chan, N.L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Wendorff, T.J.; Schmidt, B.H.; Heslop, P.; Austin, C.A.; Berger, J.M. The structure of DNA-bound human topoisomerase II α: Conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J. Mol. Biol. 2012, 424, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.; Redwood, C.S.; Jenkins, J.R.; Andersen, A.H.; Hickson, I.D. Human DNA topoisomerases IIα and IIβ can functionally substitute for yeast TOP2 in chromosome segregation and recombination. Mol. Gen. Genet. 1996, 252, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Meczes, E.L.; Marsh, K.L.; Fisher, L.M.; Rogers, M.P.; Austin, C.A. Complementation of temperature-sensitive topoisomerase II mutations in Saccharomyces cerevisiae by a human TOP2β construct allows the study of topoisomerase IIβ inhibitors in yeast. Cancer Chemother. Pharmacol. 1997, 39, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Austin, C.A.; Marsh, K.L. Eukaryotic DNA topoisomerase II β. Bioessays 1998, 20, 215–226. [Google Scholar] [CrossRef]
- Shiozaki, K.; Yanagida, M. A functional 125-kDa core polypeptide of fission yeast DNA topoisomerase II. Mol. Cell. Biol. 1991, 11, 6093–6102. [Google Scholar] [CrossRef] [PubMed]
- Crenshaw, D.G.; Hsieh, T. Function of the hydrophilic carboxyl terminus of type II DNA topoisomerase from Drosophila melanogaster. I. In vitro studies. J. Biol. Chem. 1993, 268, 21328–21334. [Google Scholar] [PubMed]
- Crenshaw, D.G.; Hsieh, T. Function of the hydrophilic carboxyl terminus of type II DNA topoisomerase from Drosophila melanogaster. II. In vivo studies. J. Biol. Chem. 1993, 268, 21335–21343. [Google Scholar] [PubMed]
- Jensen, S.; Andersen, A.H.; Kjeldsen, E.; Biersack, H.; Olsen, E.H.; Andersen, T.B.; Westergaard, O.; Jakobsen, B.K. Analysis of functional domain organization in DNA topoisomerase II from humans and Saccharomyces cerevisiae. Mol. Cell. Biol. 1996, 16, 3866–3877. [Google Scholar] [CrossRef] [PubMed]
- Adachi, N.; Miyaike, M.; Kato, S.; Kanamaru, R.; Koyama, H.; Kikuchi, A. Cellular distribution of mammalian DNA topoisomerase II is determined by its catalytically dispensable C-terminal domain. Nucleic Acids Res. 1997, 25, 3135–3142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickey, J.S.; Osheroff, N. Impact of the C-terminal domain of topoisomerase IIα on the DNA cleavage activity of the human enzyme. Biochemistry 2005, 44, 11546–11554. [Google Scholar] [CrossRef] [PubMed]
- Linka, R.M.; Porter, A.C.G.; Volkov, A.; Mielke, C.; Boege, F.; Christensen, M.O. C-Terminal regions of topoisomerase IIα and IIβ determine isoform-specific functioning of the enzymes in vivo. Nucleic Acids Res. 2007, 35, 3810–3822. [Google Scholar] [CrossRef] [PubMed]
- Meczes, E.L.; Gilroy, K.L.; West, K.L.; Austin, C.A. The impact of the human DNA topoisomerase II C-terminal domain on activity. PLoS ONE 2008, 3, e1754. [Google Scholar] [CrossRef] [PubMed]
- Gilroy, K.L.; Austin, C.A. The impact of the C-Terminal domain on the interaction of human DNA topoisomerase II α and β with DNA. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.J.; Azuma, Y. Non-catalytic roles of the topoisomerase IIα C-terminal domain. Int. J. Mol. Sci. 2017, 18, 2438. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, M.; Grabowski, D.R.; Isaacs, R.J.; Krivacic, K.A.; Rybicki, L.A.; Bukowski, R.M.; Ganapathi, M.K.; Hickson, L.D.; Ganapathi, R. Altered expression and activity of topoisomerases during all-trans retinoic acid-induced differentiation of HL-60 cells. Blood 1998, 92, 2863–2870. [Google Scholar] [PubMed]
- Grabowski, D.R.; Holmes, K.A.; Aoyama, M.; Ye, Y.; Rybicki, L.A.; Bukowski, R.M.; Ganapathi, M.K.; Hickson, I.D.; Ganapathi, R. Altered drug interaction and regulation of topoisomerase IIβ: Potential mechanisms governing sensitivity of HL-60 cells to amsacrine and etoposide. Mol. Pharmacol. 1999, 56, 1340–1345. [Google Scholar] [CrossRef] [PubMed]
- McNamara, S.; Wang, H.; Hanna, N.; Miller, W.H. Topoisomerase IIβ negatively modulates retinoic acid receptor α function: A novel mechanism of retinoic acid resistance. Mol. Cell. Biol. 2008, 28, 2066–2077. [Google Scholar] [CrossRef] [PubMed]
- McNamara, S.; Nichol, J.; Wang, H.; Miller, W.J. Targeting PKCδ mediated topoisomerase ii β overexpression subverts the differentiation block in a retinoic. Leukemia 2010, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, W.; Prescott, E.D.; Burden, S.J.; Wang, J.C. DNA topoisomerase IIβ and neural development. Science 2000, 287, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, K.; Tsutsui, K.; Hosoya, O.; Sano, K.; Tokunaga, A. Immunohistochemical analyses of DNA topoisomerase II isoforms in developing rat cerebellum. J. Comp. Neurol. 2001, 431, 228–239. [Google Scholar] [CrossRef]
- Lyu, Y.L.; Wang, J.C. Aberrant lamination in the cerebral cortex of mouse embryos lacking DNA topoisomerase IIβ. Proc. Natl. Acad. Sci. USA 2003, 100, 7123–7128. [Google Scholar] [CrossRef] [PubMed]
- Kriegstein, A.R.; Noctor, S.C. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004, 27, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Nur-E-Kamal, A.; Meiners, S.; Ahmed, I.; Azarova, A.; Lin, C.P.; Lyu, Y.L.; Liu, L.F. Role of DNA topoisomerase IIβ in neurite outgrowth. Brain Res. 2007, 1154, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Heng, X.; Le, W.-D. The function of DNA topoisomerase IIβ in neuronal development. Neurosci. Bull. 2010, 26, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Heng, X.; Jin, G.; Zhang, X.; Yang, D.; Zhu, M.; Fu, S.; Li, X.; Le, W. Nurr1 regulates Top IIβ and functions in axon genesis of mesencephalic dopaminergic neurons. Mol. Neurodegener. 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Isik, S.; Zaim, M.; Yildiz, M.T.; Negis, Y.; Kunduraci, T.; Karakas, N.; Arikan, G.; Cetin, G. DNA topoisomerase IIβ as a molecular switch in neural differentiation of mesenchymal stem cells. Ann. Hematol. 2015, 94, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Zaim, M.; Isik, S. DNA topoisomerase IIβ stimulates neurite outgrowth in neural differentiated human mesenchymal stem cells through regulation of Rho-GTPases (RhoA/Rock2 pathway) and Nurr1 expression. Stem Cell Res. Ther. 2018, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Nevin, L.M.; Xiao, T.; Staub, W.; Baier, H. Topoisomerase IIβ is required for lamina-specific targeting of retinal ganglion cell axons and dendrites. Development 2011, 138, 2457–2465. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.-wan; Yeung, W.; Law, C. Global developmental delay and intellectual disability associated with a de novo TOP2B mutation. Clin. Chim. Acta 2017, 469, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.F.; Wang, J.C. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 1987, 84, 7024–7027. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Lynch, A.S. Transcription and DNA supercoiling. Curr. Opin. Genet. Dev. 1993, 3, 764–768. [Google Scholar] [CrossRef]
- Salceda, J.; Fernández, X.; Roca, J. Topoisomerase II, not topoisomerase I, is the proficient relaxase of nucleosomal DNA. EMBO J. 2006, 25, 2575–2583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, R.S.; Piña, B.; Roca, J. Topoisomerase II is required for the production of long Pol II gene transcripts in yeast. Nucleic Acids Res. 2012, 40, 7907–7915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, N.; Parvin, J.D. DNA topoisomerase IIα is required for RNA polymerase II transcription on chromatin templates. Nature 2001, 413, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, K.; Sano, K.; Kikuchi, A.; Tokunaga, A. Involvement of DNA topoisomerase IIβ in neuronal differentiation. J. Biol. Chem. 2001, 276, 5769–5778. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Y.L.; Lin, C.-P.; Azarova, A.M.; Cai, L.; Wang, J.C.; Liu, L.F. Role of topoisomerase IIβ in the expression of developmentally regulated genes. Mol. Cell. Biol. 2006, 26, 7929–7941. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.; Miyaji-Yamaguchi, M.; Tsutsui, K.M.; Tsutsui, K. Topoisomerase IIβ activates a subset of neuronal genes that are repressed in AT-rich genomic environment. PLoS ONE 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.K.; Burger, L.; Nikoletopoulou, V.; Deogracias, R.; Thakurela, S.; Wirbelauer, C.; Kaut, J.; Terranova, R.; Hoerner, L.; Mielke, C.; et al. Target genes of Topoisomerase II regulate neuronal survival and are defined by their chromatin state. Proc. Natl. Acad. Sci. USA 2012, 109, E934–E943. [Google Scholar] [CrossRef] [PubMed]
- King, I.F.; Yandava, C.N.; Mabb, A.M.; Hsiao, J.S.; Huang, H.-S.; Pearson, B.L.; Calabrese, J.M.; Starmer, J.; Parker, J.S.; Magnuson, T.; et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature 2013, 501, 58–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, W.; Kawauchi, D.; Körkel-Qu, H.; Deng, H.; Serger, E.; Sieber, L.; Ariel Lieberman, J.; Jimeno-Gonzalez, S.; Lambo, S.; Hanna, B.S.; et al. Chd7 is indispensable for mammalian brain development through activation of a neuronal differentiation programme. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Gao, F.; Pfenning, A.R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T.X.; Yamakawa, H.; Pao, P.C.; et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 2015, 161, 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Uusküla-Reimand, L.; Hou, H.; Samavarchi-Tehrani, P.; Rudan, M.V.; Liang, M.; Medina-Rivera, A.; Mohammed, H.; Schmidt, D.; Schwalie, P.; Young, E.J.; et al. Topoisomerase II β interacts with cohesin and CTCF at topological domain borders. Genome Biol. 2016, 17, 1–22. [Google Scholar] [CrossRef]
- Manville, C.M.; Smith, K.; Sondka, Z.; Rance, H.; Cockell, S.; Cowell, I.G.; Lee, K.C.; Morris, N.J.; Padget, K.; Jackson, G.H.; et al. Genome-wide ChIP-seq analysis of human TOP2B occupancy in MCF7 breast cancer epithelial cells. Biol. Open 2015, 4, 1436–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canela, A.; Maman, Y.; Jung, S.; Wong, N.; Callen, E.; Day, A.; Kieffer-Kwon, K.R.; Pekowska, A.; Zhang, H.; Rao, S.S.P.; et al. Genome organization drives chromosome fragility. Cell 2017, 170, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Kouzine, F.; Gupta, A.; Baranello, L.; Wojtowicz, D.; Ben-Aissa, K.; Liu, J.; Przytycka, T.M.; Levens, D. Transcription-dependent dynamic supercoiling is a short-range genomic force. Nat. Struct. Mol. Biol. 2013, 20, 396–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naughton, C.; Avlonitis, N.; Corless, S.; Prendergast, J.G.; Mati, I.K.; Eijk, P.P.; Cockroft, S.L.; Bradley, M.; Ylstra, B.; Gilbert, N. Transcription forms and remodels supercoiling domains unfolding large-scale chromatin structures. Nat. Struct. Mol. Biol. 2013, 20, 387–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranello, L.; Kouzine, F.; Wojtowicz, D.; Cui, K.; Przytycka, T.M.; Zhao, K.; Levens, D. DNA break mapping reveals topoisomerase II activity genome-wide. Int. J. Mol. Sci. 2014, 15, 13111–13122. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.-G.; Lunyak, V.V.; Perissi, V.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. A topoisomerase IIβ-mediated dsDNA break required for regulated transcription. Science 2006, 312, 1798–1802. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet. 2010, 42, 668–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, R.H.F.; Chang, I.; Hudak, C.S.S.; Hyun, S.; Kwan, H.Y.; Sul, H.S. A Role of DNA-PK for the Metabolic Gene Regulation in Response to Insulin. Cell 2009, 136, 1056–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotter, K.W.; King, H.A.; Archer, T.K. Glucocorticoid receptor transcriptional activation via the BRG1 dependent recruitment of TOP2β and Ku70/86. Mol. Cell. Biol. 2015, 35, 2799–2817. [Google Scholar] [CrossRef] [PubMed]
- Bunch, H.; Lawney, B.P.; Lin, Y.-F.; Asaithamby, A.; Murshid, A.; Wang, Y.E.; Chen, B.P.C.; Calderwood, S.K. Transcriptional elongation requires DNA break-induced signalling. Nat. Commun. 2015, 6, 10191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowe, S.L.; Movsesyan, V.A.; Jorgensen, T.J.; Kondratyev, A. Rapid phosphorylation of histone H2A.X following ionotropic glutamate receptor activation. Eur. J. Neurosci. 2006, 23, 2351–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suberbielle, E.; Sanchez, P.E.; Kravitz, A.V.; Wang, X.; Ho, K.; Eilertson, K.; Devidze, N.; Kreitzer, A.C.; Mucke, L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat. Neurosci. 2013, 16, 613–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.E.; Greenberg, M.E. Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Kim, T.K. Emerging themes in neuronal activity-dependent gene expression. Mol. Cell. Neurosci. 2018, 87, 27–34. [Google Scholar] [CrossRef] [PubMed]
- LeRoy, G.; Loyola, A.; Lane, W.S.; Reinberg, D. Purification and characterization of a human factor that assembles and remodels chromatin. J. Biol. Chem. 2000, 275, 14787–14790. [Google Scholar] [CrossRef] [PubMed]
- Racki, L.R.; Yang, J.G.; Naber, N.; Partensky, P.D.; Acevedo, A.; Purcell, T.J.; Cooke, R.; Cheng, Y.; Narlikar, G.J. The chromatin remodeller ACF acts as a dimeric motor to space nucleosomes. Nature 2009, 462, 1016–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.-K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terzioglu-Usak, S.; Negis, Y.; Karabulut, D.S.; Zaim, M.; Isik, S. Cellular model of Alzheimer’s disease: Aβ1-42 peptide induces amyloid deposition and a decrease in topo isomerase IIβ and nurr1 expression. Curr. Alzheimer Res. 2017, 14, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Cowell, I.G.; Sondka, Z.; Smith, K.; Lee, K.C.; Manville, C.M.; Sidorczuk-Lesthuruge, M.; Rance, H.A.; Padget, K.; Jackson, G.H.; Adachi, N.; et al. Model for MLL translocations in therapy-related leukemia involving topoisomerase II-mediated DNA strand breaks and gene proximity. Proc. Natl. Acad. Sci. USA 2012, 109, 8989–8994. [Google Scholar] [CrossRef] [PubMed]
- Cortes Ledesma, F.; El Khamisy, S.F.; Zuma, M.C.; Osborn, K.; Caldecott, K.W. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 2009, 461, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Herreros, F.; Schuurs-Hoeijmakers, J.H.M.; McCormack, M.; Greally, M.T.; Rulten, S.; Romero-Granados, R.; Counihan, T.J.; Chaila, E.; Conroy, J.; Ennis, S.; et al. TDP2 protects transcription from abortive topoisomerase activity and is required for normal neural function. Nat. Genet. 2014, 46, 516–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madabhushi, R. The Roles of DNA Topoisomerase IIβ in Transcription. Int. J. Mol. Sci. 2018, 19, 1917. https://doi.org/10.3390/ijms19071917
Madabhushi R. The Roles of DNA Topoisomerase IIβ in Transcription. International Journal of Molecular Sciences. 2018; 19(7):1917. https://doi.org/10.3390/ijms19071917
Chicago/Turabian StyleMadabhushi, Ram. 2018. "The Roles of DNA Topoisomerase IIβ in Transcription" International Journal of Molecular Sciences 19, no. 7: 1917. https://doi.org/10.3390/ijms19071917
APA StyleMadabhushi, R. (2018). The Roles of DNA Topoisomerase IIβ in Transcription. International Journal of Molecular Sciences, 19(7), 1917. https://doi.org/10.3390/ijms19071917