1. Introduction
The cancerous transformation of cells and the subsequent stages of tumorigenesis involve a metabolic and physiological reprogramming to support constitutive growth. Such derangements include enhanced glucose uptake and aerobic glycolytic metabolism (“the Warburg effect”), as well as accelerated glutamine transport and glutaminolysis, characterized as “glutamine addiction” [
1,
2,
3,
4]. While these physiological aspects of cancer have been recognized for decades, there is currently a renewed interest in their molecular underpinnings in the hope of finding novel therapeutic strategies.
Cellular metabolism within a tumor microenvironment is subject to fluctuating nutrient conditions as well as active communication and metabolite exchange between tumor and stromal cells [
5]. Likewise, nutrient transporters play an important role in fueling the dynamic metabolic economies within tumors. The advancement of genome and transcriptome sequencing tools have led to the creation of vastly informative databases such as the Cancer Genome Anatomy Project (CGAP), the Cancer Genome Atlas (TCGA), and the Cancer Gene Expression Database (CGED), among others, and these data assemblies have been critical to the investigation of potential therapeutic targets in tumor metabolism [
6,
7,
8,
9]. In 2005, the human expressed sequence tag (EST) database from the CGAP was used to identify a coordinately enhanced expression of two amino acid transporters, alanine-serine-cysteine transporter 2 (ASCT2) and large neutral amino acid transporter 1 (LAT1), across a variety of human cancers relative to normal tissue [
10]. At the time, this finding was consistent with enhanced functions of both transporters in cancerous vs. normal cells [
11,
12].
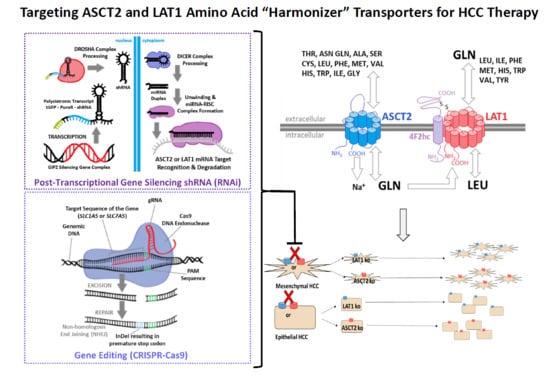
The ASCT2 and LAT1 amino acid transporters function in a cooperative, tertiary antiporter mechanism (
Supplemental Figure S1). ASCT2 is a broad specificity Na
+-dependent antiporter (exchanger) which exploits the electrochemical gradient of sodium across the plasma membrane to exchange one small, neutral amino acid from the cytoplasmic side for one small, neutral amino acid and a sodium ion on the extracellular side. LAT1 also functions as an antiporter, but its mechanism is Na
+-independent. In the tertiary active transport model, LAT1 utilizes the intracellular pool of amino acids in part generated by ASCT2 activity to exchange one small, neutral amino acid from the cytoplasmic side for one large, essential amino acid from the extracellular side. This cooperative mechanism was suggested to provide a rationale for the coordinate upregulation of both transporters in cancer [
10]. The model was reinforced by the concurrent discovery that CD147 and CD98hc nucleated a “metabolic activation-related complex” with LAT1, ASCT2, and monocarboxylate transporters (MCT) in the plasma membrane of cancer cells [
13]. As ASCT2 and LAT1 ostensibly equilibrate intracellular amino acid pools through exchange mechanisms that are responsive to metabolic demands, such transporters have recently been designated as “amino acid harmonizers” by Bröer et al. [
14].
Why the ASCT2 and LAT1 partnership? In the tertiary active transport/harmonizer model, the complementary transport activities of ASCT2 and LAT1 sustain an intracellular pool of essential amino acids that ostensibly stimulates the activity of mammalian (now, mechanistic) target-of-rapamycin (mTOR) growth signaling complex 1 (mTORC1), a serine threonine kinase which is a critical activator of cap-dependent protein translation and also acts to inhibit autophagy (
Supplemental Figure S2). More than a decade ago, studies from our lab [
15] and others [
16] initially implicated ASCT2 in mTORC1 signaling, which helped prompt more recent investigations into the role of these two transporters in cancer. Subsequent studies showed that ASCT2 and LAT1 also function in, myc-driven transcriptional programs, mitochondrial glutamine metabolism, and glycolytic diversion [
15,
16,
17,
18,
19]. Thus, their potential as therapeutic targets has been well founded to-date.
ASCT2 has been targeted with shRNA and small molecular inhibitors in a broad spectrum of cancers including melanoma [
20], prostate cancer [
21], human head and neck squamous cell carcinoma [
22], and breast cancer [
23], among others. These studies generally found that suppression of ASCT2 led to detrimental effects on glutamine transport, mTORC1 signaling, proliferation, and cell cycle progression. LAT1 has also been targeted with shRNA and small molecular inhibitors in a broad spectrum of cancers including esophageal squamous cell carcinoma [
24], gastric cancer [
25], cholangiocarcinoma [
26], breast cancer [
27], ovarian cancer [
28], and endometrial carcinoma [
29], among others. Like ASCT2, these studies generally found that the suppression of LAT1 led to detrimental effects on leucine transport, mTORC1 signaling, proliferation, and cell cycle progression. More recent studies with ASCT2 or LAT1 knockout cancer cell lines suggest a more complex role for these transporters in tumorigenesis [
14,
30].
Previous work from our lab with an inducible long antisense RNA system clearly implicated ASCT2 as a viable therapeutic target in human HCC cells [
31]. Here, those studies were extended using viral shRNA encoding vectors and the CRISPR-Cas9 gene editing system because they offer more specific and tractable modalities for in vivo applications. Both approaches were pursued and reported here because recent studies have yielded contradictory results with genes implicated in cancer targeted by RNA interference or CRISPR-Cas9 mediated gene editing [
32]. Given the enhanced expression of ASCT2 and LAT1 in HCC, the studies presented here were designed to assess the therapeutic potential of targeting each transporter, and to determine whether primary (epithelial) or metastatic (mesenchymal) phenotype human HCC cells are differentially reliant on these transporters for growth and viability.
Contrary to expectations, the results from both approaches contradicted earlier studies with inducible long-antisense RNA [
31]. While shRNA and particularly CRISPR-Cas9 profoundly suppressed glutamine and leucine initial-rate transport, the results indicated that neither suppression nor knockout of ASCT2 or LAT1 is alone sufficient to inhibit the in vitro growth of epithelial or mesenchymal human HCC cells, or to suppress mTORC1 signaling. Potential reasons for the conflicting results with long antisense RNA and suggestions for a path forward in this research area are discussed in light of our findings and recent results from ASCT2 and LAT1 knockout studies in other types of cancer cells.
3. Discussion
The studies presented here represent a culmination of several years of research identifying and pursuing amino acid transporters as potential targets for cancer therapy. ASCT2 and LAT1 are both transporters that have been implicated by our lab and others in driving the growth of cancerous cells. Prior studies from our lab implicated ASCT2 as vital for HCC cell survival [
31] and mTORC1 signaling [
15], and also, implicated LAT1 as a “partner in crime” in driving the growth of many human cancers [
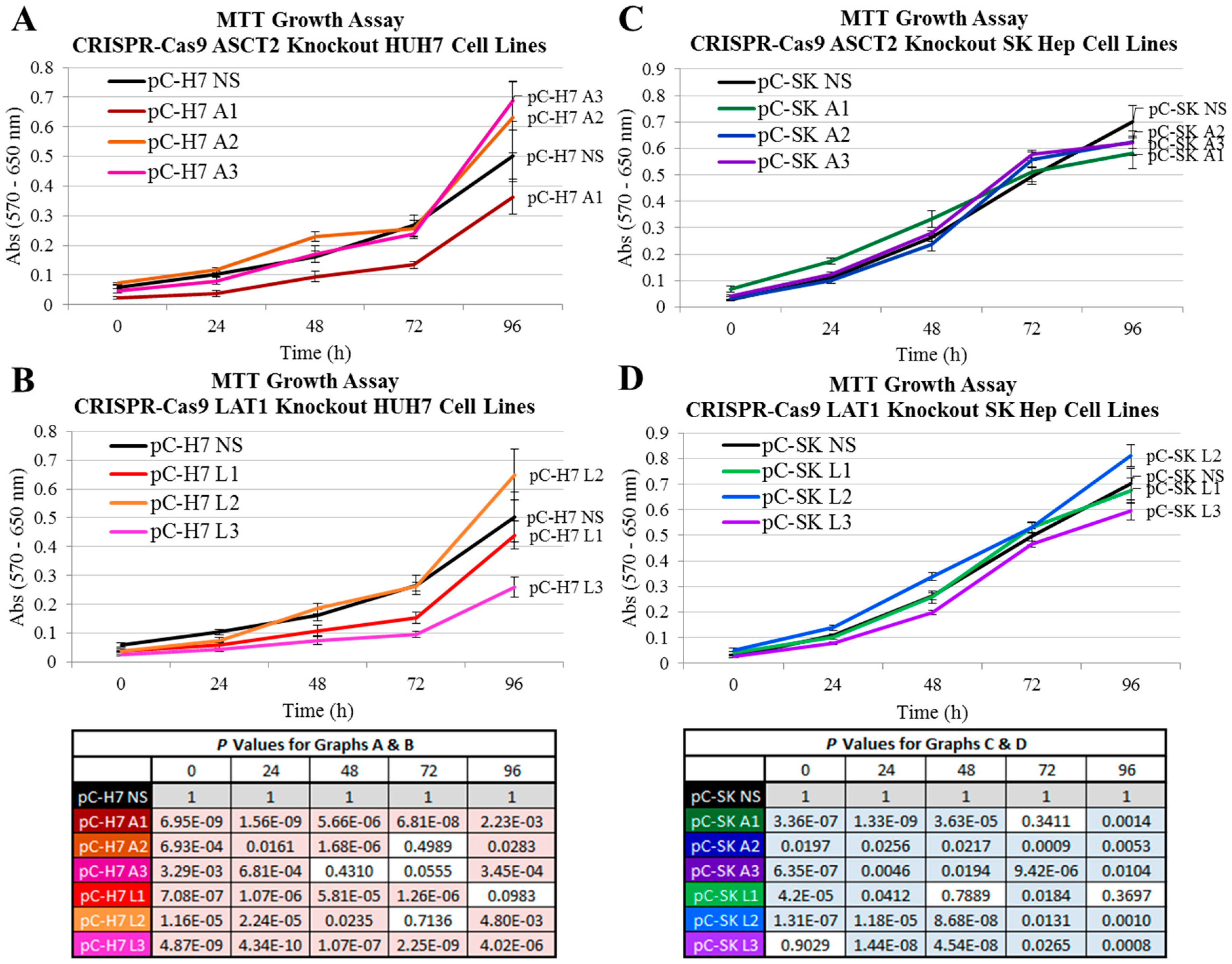
10]. The overarching question has been “can ASCT2 or LAT1 be effective targets for cancer therapy?” Likewise, the studies here were designed to assess whether either transporter might play a particularly important role in driving the biology of epithelial (modeling primary) and mesenchymal (modeling invasive/metastatic) cancer cells. The results suggest that alone, neither transporter is necessary for sustained in vitro growth of epithelial or mesenchymal human liver cancer cells.
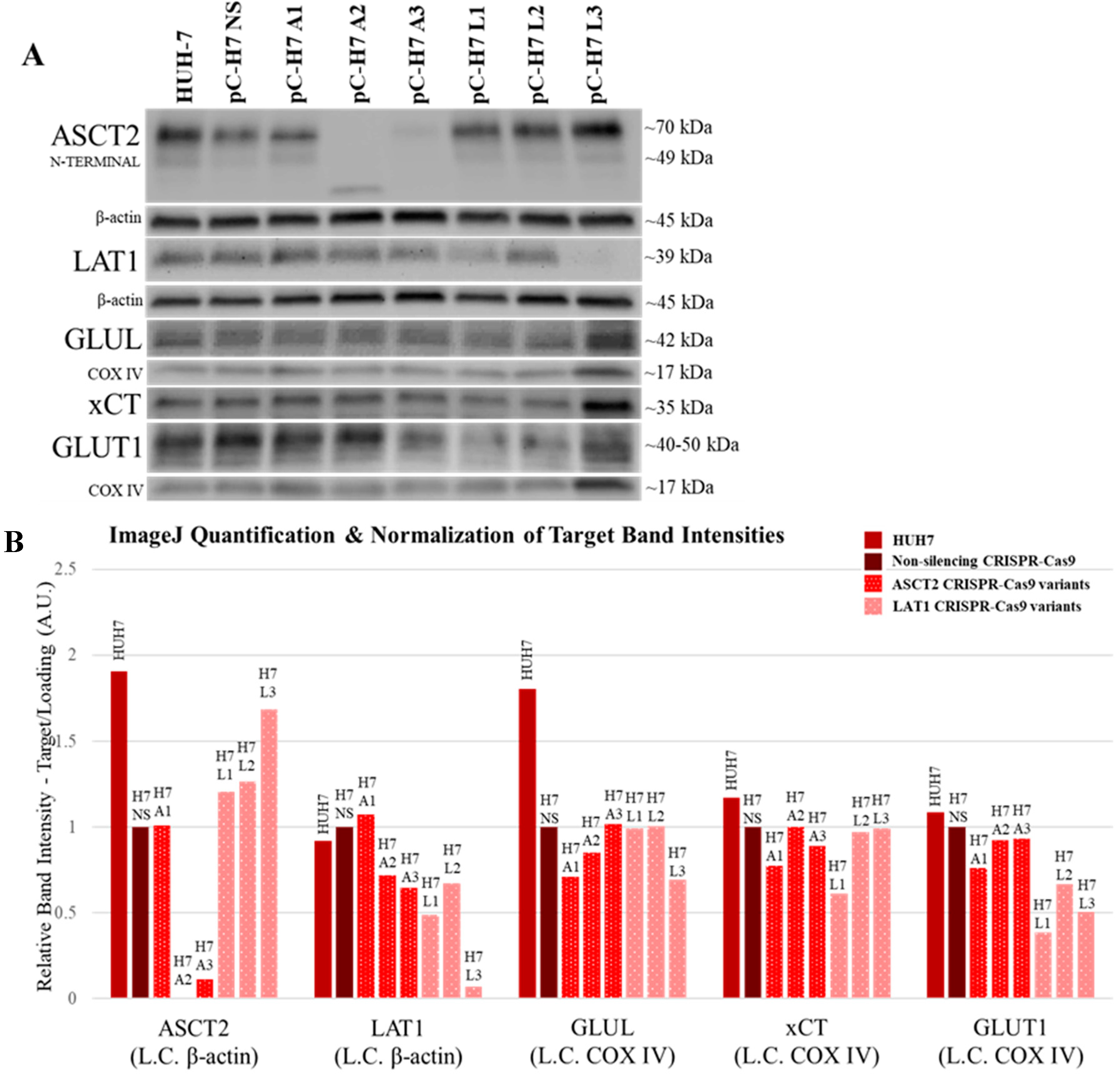
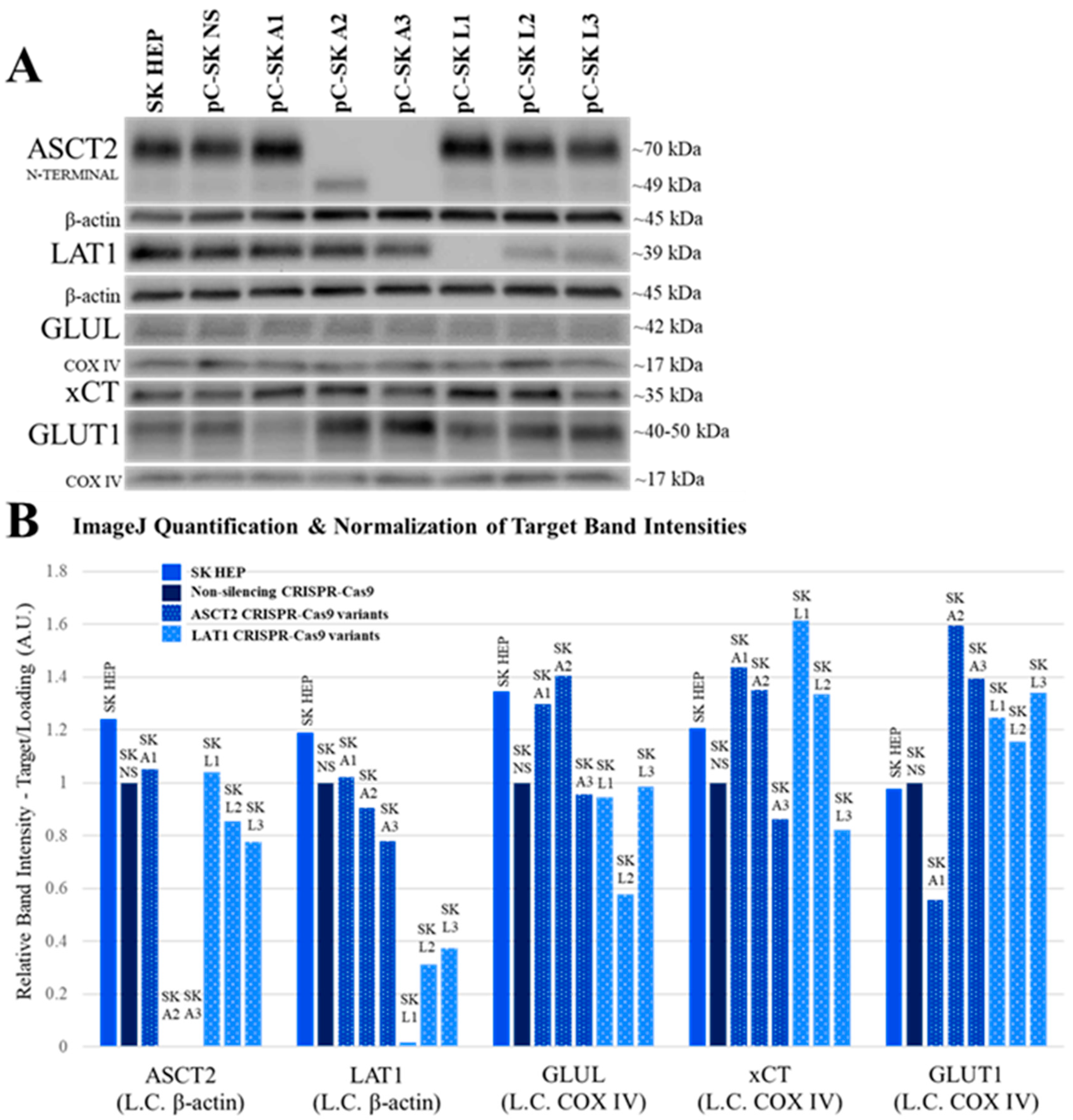
Using more refined and contemporary approaches (shRNA and CRISPR-Cas9 systems) in the present study relative to our prior studies with inducible ASCT2 antisense RNA, both ASCT2 and LAT1 were successfully repressed or eliminated below detectable levels. The results suggest that cancer cells adapt to profound transporter suppression—likely far beyond what could be reasonably obtained in vivo. In spite of effective ASCT2 or LAT1 knockout (
Figure 1 and
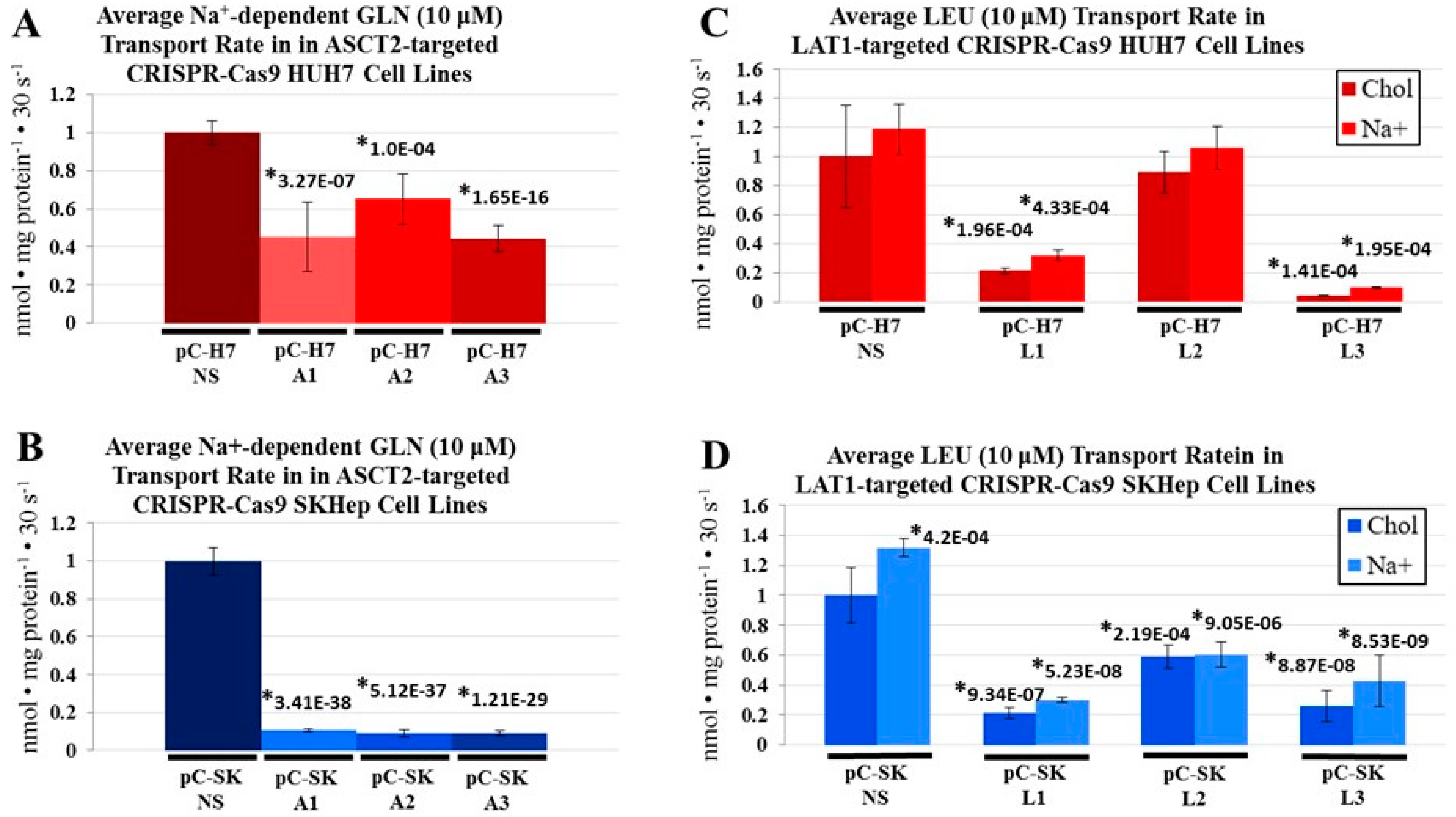
Figure 2), and linked reductions in glutamine and leucine transport rates (
Figure 3 and
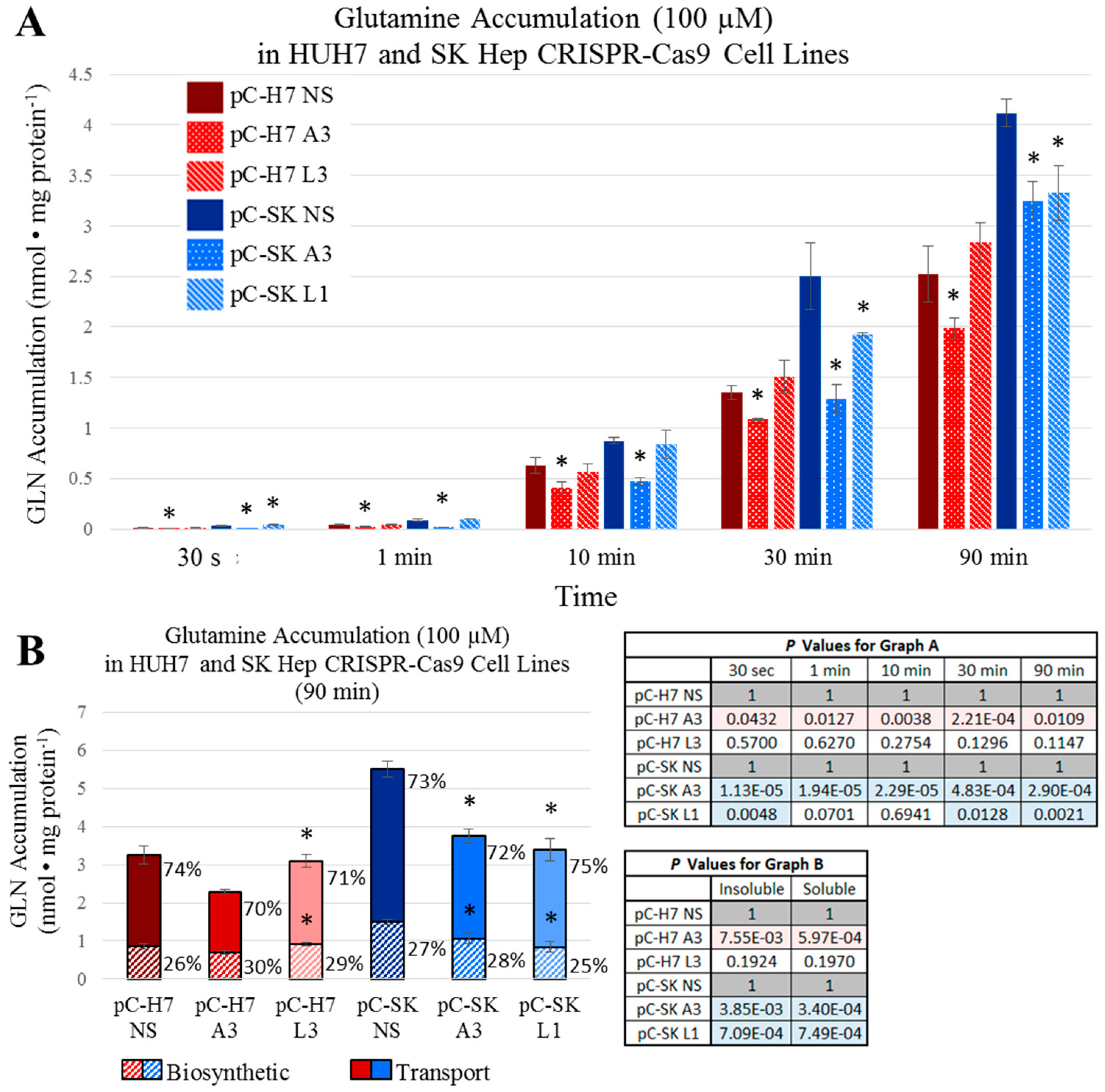
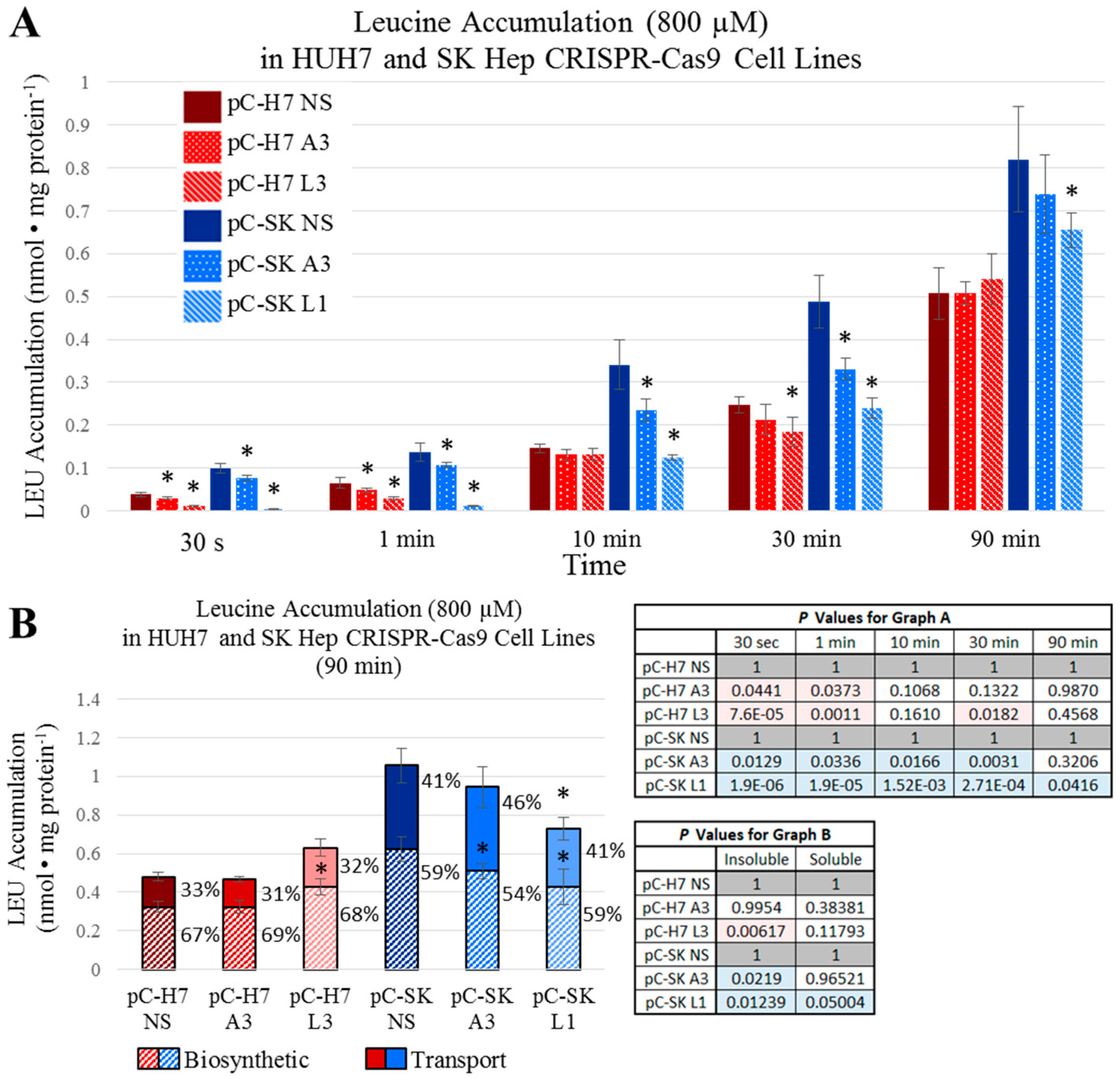
Figure 4), both epithelial and mesenchymal human HCC cells continued to grow at rates equal to, or only moderately less than cognate controls (Figures 5). These results were further reinforced by the observation that neither LAT1, nor ASCT2 CRISPR-Cas9 knockout cells exhibited sustained repression of intracellular glutamine or leucine accumulation, incorporation into protein and other macromolecules (
Figure 6 and
Figure 7), or reductions in long-term or acute mTORC1 signaling (
Supplemental Figures S22 and S23). Collectively, all results were consistent—human liver cancer cells adapt to profoundly diminished ASCT2 or LAT1 activity.
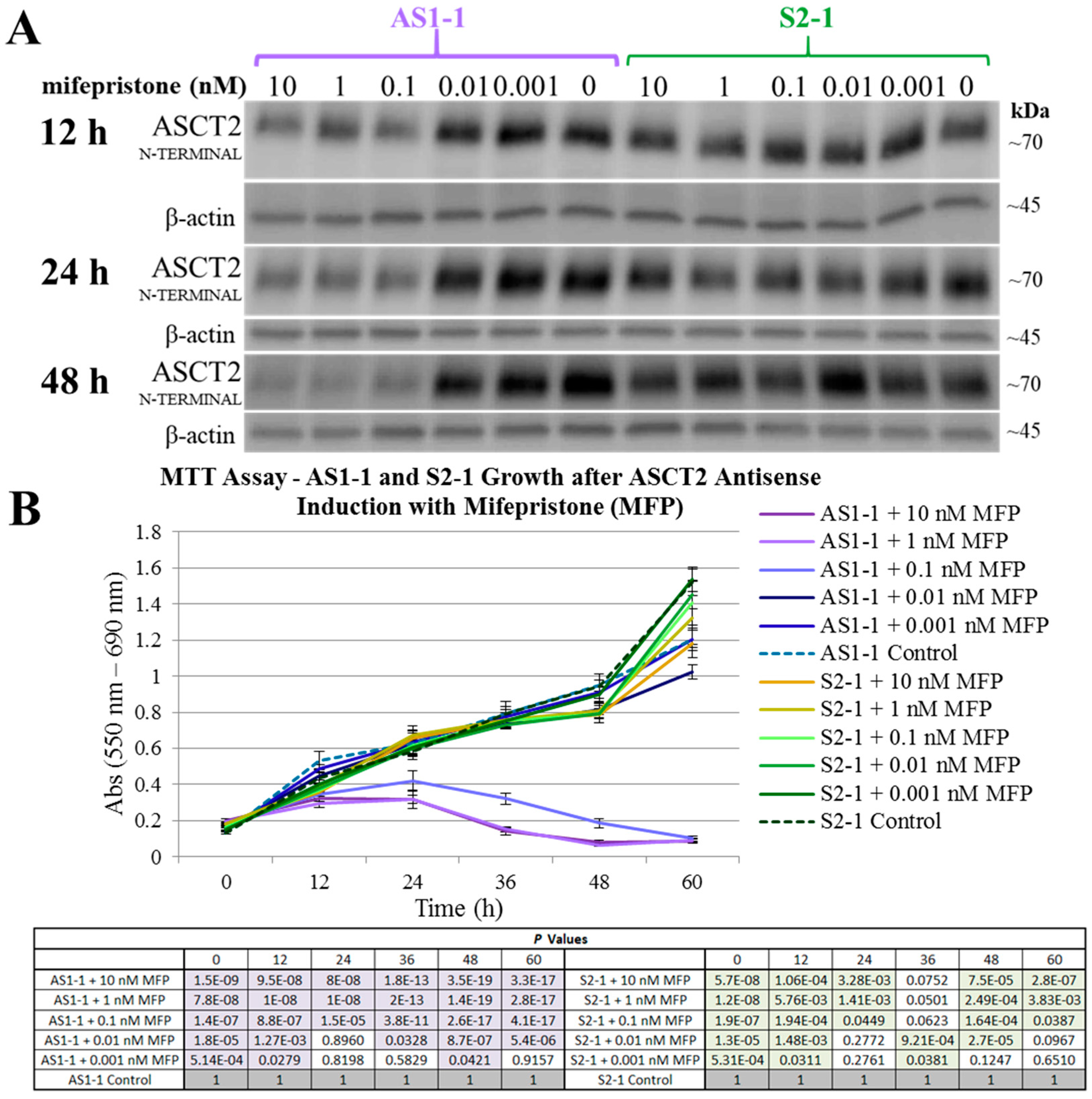
The results in this study regarding ASCT2 also contradict earlier work from our laboratory using an inducible ASCT2 antisense RNA, which showed that ASCT2 silencing led to apoptotic cell death and early depression of mTORC1 signaling [
15,
31]. The reason for the disparity is likely grounded in off-target effects of the long ASCT2 antisense RNA deployed in that study [
34]. Using CRISPR-Cas9, ASCT2 expression levels in HUH7 (
Figure 1) and SKHep (
Figure 2) were repressed to levels that far exceed those achieved with the antisense RNA targeting methodology (
Figure 8). Therefore, the cause of cell death in prior work was likely not alone attributable to ASCT2 suppression. The induced antisense transcript was large, roughly ~1.3 kb in length, complementary to the endogenous ASCT2 mRNA transcript, but a Protein Kinase R (PKR)-induced stress response was ruled out as an off-target effect in those studies [
31]. However, it is possible that once the antisense transcript formed a double-stranded complex with the endogenous ASCT2 mRNA transcript and is subsequently cleaved by Dicer, the resulting array of small microRNA (miRNA) duplexes could (1) allow the RNA-induced silencing complex (RISC) to target more than just the endogenous ASCT2 mRNA transcript, or (2) overwhelm the cellular miRNA processing machinery leading to cell death [
35]. Therefore, ASCT2 may not have as critical of a role in hepatoma cell survival in vitro as previously thought, when evaluated in the light of results presented here.
Is all hope lost in targeting these transporters for cancer therapy based on these results? In a word, no. If anything, the results presented here further inform our approach to targeting metabolic derangements, including the so-called “glutamine addiction” of cancer cells [
17]. Old models are constantly refined by often unexpected experimental results, and subsequent paradigm shifts. In this case, the old model that native levels of ASCT2 or LAT1 activity are necessary for cancer cell growth and mTORC1 signaling were not supported by our data. Recent studies have shown that tumorigenic potential is markedly diminished in ASCT2 knockout cancer cells [
14,
36]; likewise, the same was true in LAT1 knockout cells [
30]. The ability to support in vitro growth and the attributes required for effective tumorigenesis (stromal cell recruitment, angiogenesis, establishment of tumor metabolic economies, etc.), while related, are likewise quite distinct.
As cancer is now widely recognized as a disease of tissues, not just cells, stromal-carcinoma metabolic cycles clearly must be established in order for a tumor to form, grow, and progress. Recent work showed despite moderate to no effects on in vitro growth, ASCT2 knockout cells generate smaller tumors in mouse xenograft models [
14,
36]. We hypothesize that the disparity in lack of an in vitro growth phenotype and diminished tumorigenic potential resides in the compromised ability of the carcinoma cells to engage in metabolic cycling, which is crucial in tumors [
37]. In other words, while loss of a rapid exchange mechanism (ASCT2 or LAT1) has modest effects if any, on in vitro growth and long-term amino acid accumulation (
Figure 5,
Figure 6 and
Figure 7) in a nutrient-rich environment, in a tissue context where resources are more austere, the inability to sufficiently fuel metabolic cycles through export likely compromises the tissue-generating stromal recruitment and support component of tumorigenesis.
A new model has emerged for the role of amino acid transporters in cancer: Both ASCT2 and LAT1 are obligate amino acid exchangers—a role characterized as cellular “harmonizers” by Bröer et al. [
14], as they equilibrate intracellular amino acid levels to align with metabolic demands. Other Na
+-dependent transporters known as “loaders” mediate net unidirectional delivery of amino acids, such as members of the SLC38 System N and A transporters [
38]. While slower than harmonizers, they provide substrates that sustain mTORC1 activity either directly or indirectly, which is likely why we saw no effect of ASCT2 or LAT1 knockout on basal or amino acid-induced mTORC1 signaling, as reported and suggested by Bröer et al. [
14]. To fulfill their physiological role, the harmonizers ASCT2 and LAT1 necessarily must have higher innate activities than loaders, and indeed this is precisely why they have been regarded as “dominant” glutamine transporters, especially when initial-rate amino acid transport is measured as an assessment of carrier contributions [
11]. As harmonizers, ASCT2 and LAT1 provide substrates via exchange to surrounding cells (e.g., stromal cells), whose metabolic economies are likely complimentary to the carcinoma cells [
37]. One cell’s byproduct is another cell’s fuel. Their export role in a heterotypic tumor is likely why the emerging studies to-date have indicated modest in vitro growth effects of LAT1 or ASCT2 knockout in different human cancer cell types, but more profound impacts on tumorigenesis [
14,
36]. Studies are currently underway to test whether the same holds true in the ASCT2 and LAT1 knockout SKHep and HUH7 liver cancer cell cohorts reported here.
Another facet of targeting amino acid transporters in cancer that will come to bear is the integrated (amino acid) stress response (ISR) [
39,
40]. In the ISR, amino acid starvation, as might be induced in vivo by transporter knockout, induces a general control nonderepressible 2 (GCN2) kinase-dependent phosphorylation of the translational regulator eukaryotic initiation factor 2 alpha (eIF2α) and arrest of cap-dependent translation, while activation of transcription factor ATF4 leads to expression of a third functional class of amino acid transporter—“rescue”—such as SNAT2 [
39]. In recent work, Bröer et al. reported that upon ASCT2 knockout in HeLa and osteosarcoma cells, an ISR upregulated the “loader” SNAT1 to functionally replace ASCT2 in a GCN2-dependent manner [
14]. In contrast, ASCT2 knockout in colon and lung carcinoma cells did not induce an ISR, whereas LAT1 knockout did so [
14,
36]. While the ISR was not investigated extensively in the studies reported here, no evidence of compensatory SNAT1 or SNAT2 upregulation was observed, but ongoing studies are assessing these links more deeply. Clearly the ISR (amino acid—GCN2/eIF2α/ATF4 stress response) will be a key consideration in targeting amino acid transporters in oncology, especially as it relates to mechanisms of developed resistance.
One last possibility exists in the targeting of ASCT2 and LAT1 in cancer therapy: double-knockout. No one has yet reported the double knockout of ASCT2 and LAT1, which may be lethal to the cells. Attempts by our lab to generate double-knockouts were unsuccessful, which suggests, but certainly does not prove, that an ASCT2−/−/LAT1−/− genotype might be lethal to cancer cells. We are currently exploring this possibility using different approaches.
In conclusion, the studies presented here indicate that ASCT2 and LAT1 can each be successfully targeted and eliminated, but neither knockout alone is sufficient to prevent cancer cell growth in vitro, contrary to earlier RNAi-based studies that suggested critical roles for each. Such disparities are not uncommon and continue to emerge in oncology [
32]. Recent studies demonstrating that ASCT2 or LAT1 knockout diminishes human cancer cell tumorigenesis in mouse xenograft models likewise indicates that their role as amino acid exchangers is likely more essential in vivo than in vitro. The field of amino acid transporters as therapeutic targets still holds promise in this regard, informed now by recent research results and linked paradigms for experimental design.
4. Materials and Methods
4.1. Cell Culture
The human hepatoma cell lines used herein were Hep3B, HUH7, and SKHep cells (American Type Culture Collection, Manassas, VA, USA). Two additional cell lines, AS1-1 and S2-1, were used that were previously established by transfecting SKHep with an inducible, dual vector system which produces an antisense RNA against the ASCT2 mRNA transcript, as previously described [
31]. All cell lines were maintained at 37 °C in a humidified atmosphere of 5% CO
2-95% air in Dulbecco’s Modified Eagle Medium (DMEM, 4.5 mg/mL
d-glucose) (Gibco, Waltham, MA, USA) supplemented with 10% triple 0.1 μm filtered fetal bovine serum (FBS) (Atlanta Biologicals, Flowery Branch, GA, USA), 2 mM
l-glutamine (Thermo Scientific, Waltham, MA, USA), 1% antibiotic/anti-mycotic solution (100 U/mL penicillin G, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin; Thermo Scientific). All cell lines were authenticated by short tandem repeat (STR) analysis by the Johns Hopkins Genetics Resource Core Facility (Baltimore, MD, USA). Cell count measurements were performed using an Invitrogen™ Tali™ image-based cytometer (Thermo Scientific) and a hemocytometer (Hausser Scientific Co., Horsham, PA, USA).
4.2. RNAi—shRNAmir Transfection and Antibiotic Selection
pGIPZ plasmid vectors were obtained from Open Biosystems as
Escherichia coli (
E. coli) stocks (60% glycerol) and stored at −80 °C [
41]; pGIPZ plasmid constituents are depicted in
Supplemental Figures S3 and S4, respectively. Five shRNAmir sequences were used to target the alanine-serine-cysteine transporter 2 (ASCT2) mRNA transcript (clone IDs: V3LHS_393625, V3LHS_393624, V3LHS_393623, and V3LHS_393622, and V2LHS_56725), and these shRNAmir designs are referred to herein as A1, A2, A3, A4, and A5, respectively. Four shRNAmir sequences selected to target the large neutral amino acid transporter 1 (LAT1) mRNA transcript (clone IDs: V2LHS_17487, V3LHS_353675, V3LHS_353676, and V3LHS_353679), and these shRNAmir designs are referred to herein as L1, L2, L3, and L4, respectively. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) shRNA transcript used as a positive GIPZ system control is V3LHS_382663. The scrambled shRNAmir sequence that served as a nonsilencing (NS) negative control is RHS4346. The binding sites for each shRNA variant on the target ASCT2 and LAT1 mRNA sequences (accession numbers NM_005628 and NM_003486, respectively) were visualized using Sfold Software [
42,
43] (
Supplemental Figures S5 and S6). The shRNAmir sense and anti-sense sequences are listed in
Supplemental Table S1. All pGIPZ shRNAmir plasmid vectors.
PureYield™ Promega plasmid midiprep kit (Promega, Madison, WI, USA) was used to extract and purify plasmid stocks according to the manufacturer protocol. Plasmid concentrations were measured with a Nanodrop 2000 Micro-Volume UV-Vis spectrophotometer (Thermo Scientific), and each plasmid stock was stored at −20 °C. Transfections were performed by electroporation into suspended HCC cells (5 × 10
6 cells/mL) using the Neon
® Transfection System (Thermo Scientific). All stably transfected hepatoma cells were maintained under selective pressure in growth media supplemented with 0.5 μg/mL puromycin (Gibco) for SKHep and 3.0 μg/mL for Hep3B; antibiotic concentrations were established by preliminary survival dosing assays. Expression of the GIPZ vector’s visual reporter, turboGFP (tGFP), was imaged using an EVOS
® FLoid
® fluorescent microscope (Invitrogen, Carlsbad, CA, USA) with an 482/18 nm excitation preset (
Supplemental Figures S7 and S8).
4.3. CRISPR-Cas9 Transfection and Antibiotic Selection
pCLIP-All-EFS-Puro CRISPR-Cas9 all-in-one plasmid vectors were obtained from transOMIC technologies as PrimePlus
E. coli stocks (60% glycerol) and stored at −80 °C [
44]. A comprehensive map of the pCLIP-All-EFS-Puro vector plasmid vector is depicted in
Supplemental Figure S21. Three CRISPR-Cas9 sequences were selected to target the
SLC1A5 gene (clone IDs: TEVH-1126600, TEVH-1193742, and TEVH-120884); these sequences are referred to herein as A1, A2, and A3, respectively. Three CRISPR-Cas9 sequences were selected to target
SLC7A5 gene (clone IDs: TEVH-1112259, TEVH-1179401, and TEVH-1246543), and these sequences referred to herein as L1, L2, and L3, respectively. The clone ID of the non-silencing CRISPR-Cas9 sequence that served as a CRISPR-Cas9 silencing system control is TELA1011, referred to herein as NS. Each unique guide RNA (gRNA) sequence is listed in
Supplemental Table S2, and the targeted regions in each gene were determined using the following accession numbers: NC_000019.10 and NC_000016.10 for
SLC1A5 and
SLC7A5, respectively. pCLIP-All-EFS-Puro plasmid preparation, quantification, electroporation, and selection procedures were the same as described earlier for the pGIPZ shRNA plasmid vectors, with the exception that the HUH7 cell line was used instead of Hep3B. Stably transfected cells were maintained under selective pressure in growth media supplemented with 0.5 μg/mL puromycin (Gibco) for SKHep and 3.0 μg/mL for HUH7.
4.4. RNA Isolation and RT-qPCR
All cell lines were cultured in 150 mm culture dish; when the cells reached 80–90% confluence, an RNA isolation was performed using TRIzol
® reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer instructions, and this was followed by an alcohol precipitation to concentrate the samples. RNA integrity was assessed by visualization of 28S and 18S band quality after electrophoresis on a 1% agarose gel. A Nanodrop 2000 Micro-Volume UV-Vis spectrophotometer was used to quantify RNA concentrations. All concentrated RNA samples were stored at −80 °C until use. One µg of total RNA was used from each sample for reverse transcription (RT) reactions, which contained reverse transcriptase, oligo(dT), 5× reaction buffer, MgCl
2, RNasin, dNTP (all from Promega). RT-qPCR was performed using proprietary TaqMan
® Gene Expression Assay primers exclusive to human ASCT2, LAT1, alanine-serine-cysteine transporter 1 (ASCT1), sodium-dependent neutral amino acid transporter 2 (SNAT2), large neutral amino acid transporter 2 (LAT2), glutamine synthetase (GLUL), and cystine-glutamate transporter (xCT) mRNA transcripts, and all measurements were normalized to two housekeeping genes (HKG): TATA-binding protein (TBP) and hydroxymethylbilane synthase (HMBS) (all TaqMan probes were from Applied Biosystems, Foster City, CA, USA). All primer sets selected for this study spanned intragenic regions (introns) within each gene. The 2
−ΔΔCt method was used to calculate the fold difference in expression relative to nonsilencing (NS) controls generated from each parent cell line. Greater than a two-fold increase in expression was considered significant. All RT-qPCR reactions were carried out using Eppendorf Mastercycler
® ep Realplex instrumentation (Eppendorf, Hauppauge, NY, USA) and analytical software (Eppendorf Realplex software, Version 2.2). Each run was performed a minimum of three times, and within each run, each sample was run in triplicate for each target transcript. These experiments complied with MIQE (Minimum Information about Quantitative Real-Time PCR Experiments) guidelines [
45].
4.5. Western Blot Analysis
For Western blot analysis of ASCT2, LAT1, GAPDH, GLUL, and glucose transporter 1 (GLUT1), total cellular protein lysates were prepared using 2× Laemmli lysis buffer (2% SDS (Sigma, St. Louis, MO, USA), 62.5 mM Tris-HCl (pH 6.8), and 1% protease inhibitor cocktail (Thermo Scientific)). Lysis was performed in 6-well plates when the cell monolayers reached confluence. Total cellular protein concentration was measured with a Nanodrop 2000 Micro-Volume UV-Vis spectrophotometer, and equal protein from each sample was prepared for vertical electrophoresis in 2% SDS, 10% glycerol, 62.5 mM Tris-HCl (pH 6.8), 0.05% bromophenol blue, and 50 mM dithiothreitol (DTT) (Fisher Scientific). For Western blot analysis of mTORC1 phosphorylated targets (4EBP1 and p70S6K), total cellular protein lysates were prepared using a lysis buffer which contained phosphatase inhibitors (10 mM Tris (pH 6.8), 1 mM EDTA, 5 mM EGTA, 0.5% NP40 (US Biological, Salem, MA, USA), 0.1% TritonX 100, 1 mM sodium orthovanadate (Sigma), 50 mM β-glycerophosphate (Sigma), and 1% protease inhibitor cocktail (Thermo Scientific)). This lysis buffer formulation was used for the analysis of total and phosphorylated eukaryotic initiation factor 4E-binding protein 1 (4E-BP1TOTAL and 4E-BP1T37/46) and 70 kDa ribosomal protein S6 kinase (p70S6KTOTAL and p70S6KT389). Total cellular protein concentration was measured with a bicinchoninic acid (BCA) chemical assay (Pierce Chemical, Rockford, IL, USA) and an Epoch microplate spectrophotometer. Equal protein from each sample was prepared for vertical electrophoresis in the same buffer that was used for lysis, supplemented with 10% glycerol, 0.05% bromophenol blue, and 50 mM dithiothreitol (DTT). All of the total cellular protein lysates analyzed in 4E-BP1 and p70S6K Western blots were run in parallel with a positive control cell lysate from MCF7 cells treated with insulin (Cell Signaling Technologies, Danvers, MA, USA).
For all Western blots, vertical electrophoresis was run on precast 4–20% Mini-PROTEAN
® TGX™ polyacrylamide gels (Bio-Rad, Hercules, CA, USA) at 150 V for 45 min, and protein samples were transferred to Immobilon-P polyvinylidene difluoride (PVDF) membranes (Millipore) at 75 V for 90 min. After 1 h of incubation in blocking buffer (5% bovine serum albumin (BSA) in TBST (0.1% Tween-20, 150 mM NaCl, 20 mM Tris (pH 7.5)) at room temperature, the PVDF membranes were incubated overnight at 4 °C with the appropriate primary antibody in blocking buffer. After four washes with TBST), membranes were incubated with secondary antibody in blocking buffer for 1 h at room temperature, and immunoreactive bands were visualized by adding 10 mL of chemiluminescent substrate (LumiGLO
® and peroxide both from Cell Signaling Technologies) and detected with a G:Box chemiluminescence imager (Syngene, Frederick, MD, USA) and GeneSnap image acquisition software (Syngene, version 7.09). The molecular size of the chemiluminescent bands was determined by comparison to a biotinylated ladder (Cell Signaling Technologies), and band intensities were quantified using ImageJ software (Available online:
https://imagej.nih.gov/ij/download.html). Target band intensity was normalized to the band intensities of the loading controls measured on the same blot. For Western blots that measured total and phosphorylated 4E-BP1, a change in kDa weight of the 4E-BP1 protein due to phosphorylation was the principal focus, and banding patterns for 4E-BP1 Western blots were qualitatively evaluated.
Primary polyclonal rabbit IgG antibodies against LAT1 were obtained from Abcam (Cambridge, MA, USA). Primary IgG antibodies against GLUT1 and xCT were obtained from Thermo Scientific. All other primary monoclonal rabbit IgG antibodies as well as secondary HRP-conjugated goat, anti-rabbit IgG antibodies were obtained from Cell Signaling Technologies. Manufacturer-recommended antibody concentrations were applied to each blot during all hybridization steps.
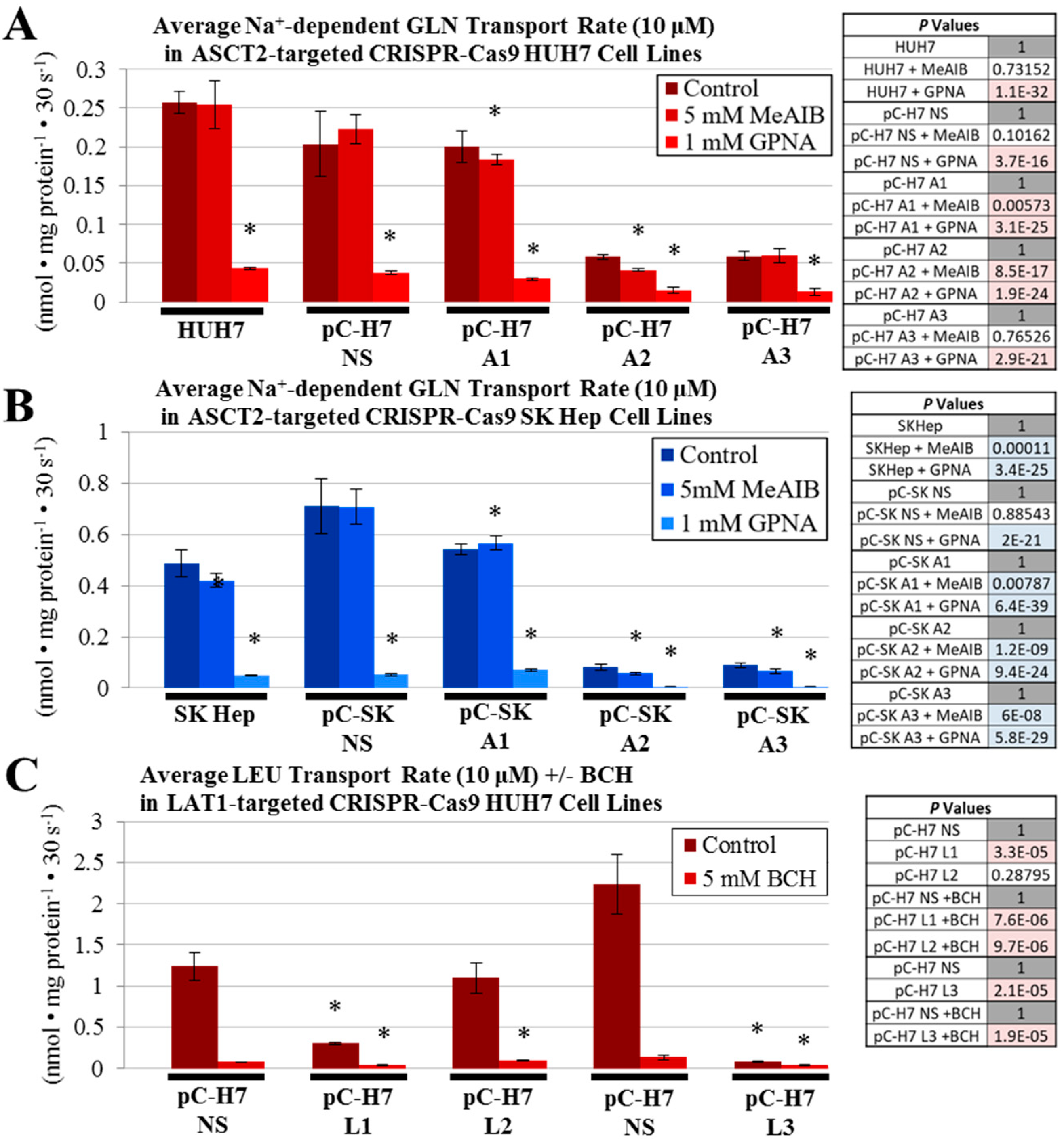
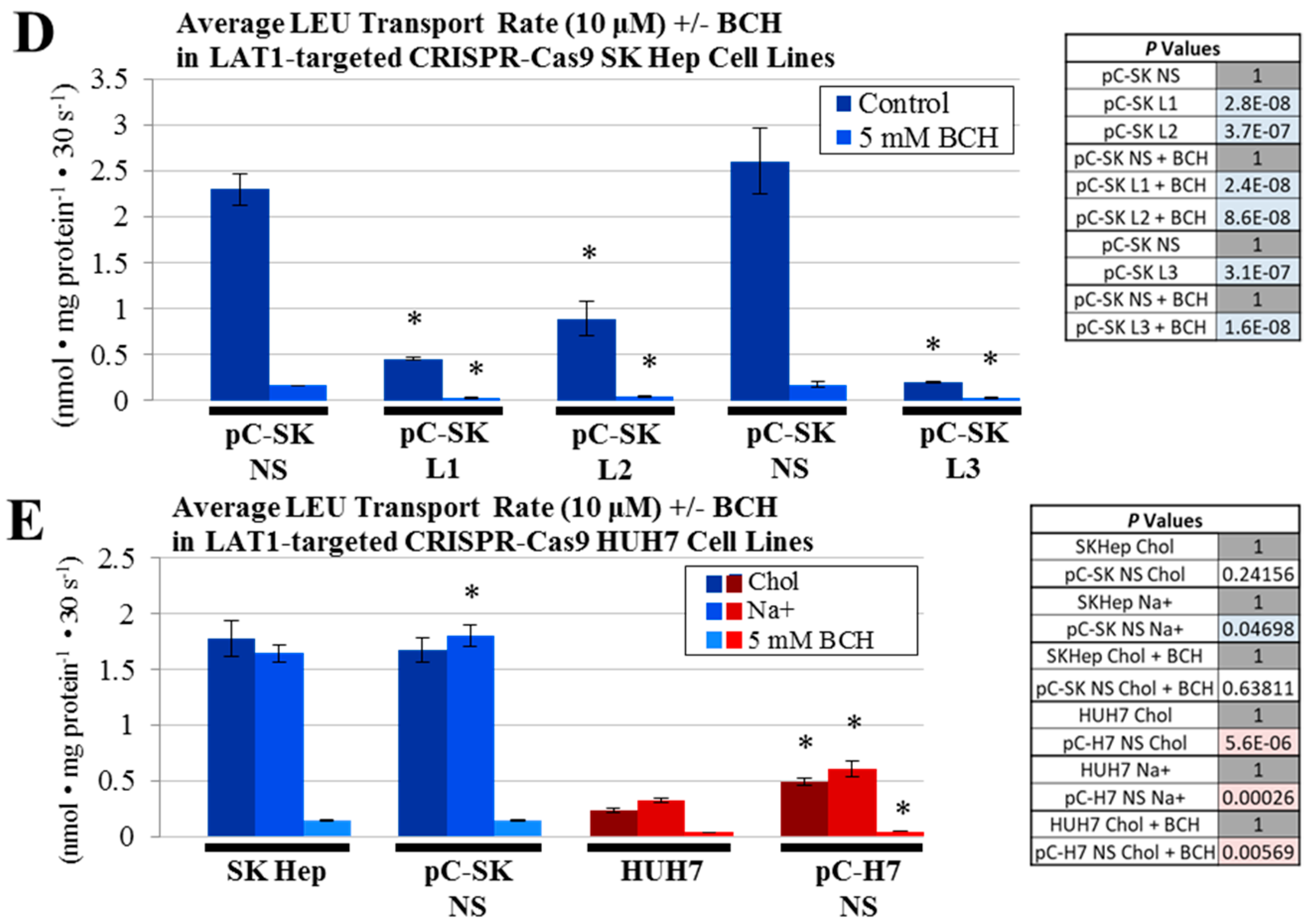
4.6. Glutamine and Leucine Transport Analysis
All cell lines were seeded at a density of 1 × 10
5 cells/well in 24-well culture plates and allowed to grow to 80–90% confluence, normally one or two days later. Radiolabeled glutamine (
l-[
3H]Gln) and leucine (
l-[
3H]Leu) (Perkin-Elmer, Waltham, MA, USA), each at a 3 μCi/mL were used to assess amino acid transport as described previously [
11,
31]. Initial-rate transport measurements were performed in Na
+-containing (NaKRP) or Na
+-free (cholKRP) Krebs–Ringer Phosphate Buffers in the presence of unlabeled
l-Gln or
l-Leu, each at 10 μM—herein referred to as “uptake mix”. All transport measurements were carried out at 37 °C and were terminated after 30 s by three rapid washes with an ice-cold phosphate-buffered saline (PBS) solution. Transported amino acids were extracted with lysis buffer (0.2 mL/well of 0.2% SDS and 0.2 N NaOH) for 30 min, and 0.1 mL of the lysate was neutralized with 10 μL 2 N HCl and analyzed by scintillation spectrophotometry in a Tri-Carb B2910TR liquid scintillation analyzer and associated QuantaSmart™ software (version 4.0, PerkinElmer). The remaining lysate was used to measure the cellular protein concentration of each sample by a BCA assay using an Epoch microplate spectrophotometer. Initial rate uptake of glutamine and leucine were calculated from the counts per min (cpm) per sample and the specific activity of each uptake mix (in cpm/nmol), and these measurements were normalized to cellular protein content in a Microsoft Excel spreadsheet. For glutamine uptake studies, transport values obtained in the absence of extracellular Na
+ (diffusion and Na
+-independent uptake) were subtracted from those in the presence of Na
+ (total uptake) to yield Na
+-dependent rates that are reported in units of nmol·mg
−1 protein·30 s
−1. In some experiments, a system A glutamine transport inhibitor, 2-methylaminoisobutyric acid (5 mM, MeAIB; Sigma) and a pan-specific glutamine transport inhibitor, glutaminyl-para-nitroanilide (1 mM, GPNA; MP Biomedicals, Santa Ana, CA, USA) were included in
l-[
3H] Glutamine uptake mixtures to differentiate between system A and system ASC-mediated glutamine transport. Likewise, a LAT1 inhibitor, 2-amino-2-norbornanecarboxlic acid (5 mM, BCH; Sigma) was included in some
l-[
3H] Leucine uptake mixtures to characterize leucine transport mediated by the System L (including LAT1) transporters. All transport values depicted are the average ± SD of four separate determinations. When applicable, to compare transport values between separate cluster plates, non-silencing (NS) controls from each parent cell line were set to a value of 1 and all other transport values were mathematically adjusted to reflect this change.
4.7. Glutamine and Leucine Accumulation Analyses
For longer-term accumulation of amino acids, l-[3H]Gln or l-[3H]Leu were at 3 μCi/mL were added to DMEM (4.5 mg/mL d-glucose) supplemented with 10% triple 0.1 μm filtered dialyzed fetal bovine serum (dFBS) and 100 μM unlabeled l-glutamine. All measurements were carried out at 37 °C and were terminated at specific times (30 s, 1 min, 10 min, 30 min, 1 h, and 1.5 h) by three rapid washes with an ice-cold phosphate-buffered saline (PBS) solution. Free and biosynthetically-incorporated fractions of accumulated radiolabeled amino acid were assessed by acid precipitation. Cells were treated (0.2 mL/well) with 10% (w/v) trichloroacetic acid (TCA) (Acros Organics, Morris, NJ, USA) and incubated on ice; after 30 min, 0.1 mL of the supernatant, representing the free (soluble) fraction of radiolabeled amino acid, was removed analyzed by scintillation spectrophotometry. The remaining acid-precipitated cellular macromolecules in each well, representative of the incorporated fraction of radiolabeled amino acid, were washed three times with ice-cold PBS (1 mL/well/wash), solubilized by adding a standard lysis buffer (0.2 mL/well of 0.2% SDS and 0.2 N NaOH), neutralized, and analyzed by scintillation spectrophotometry as previously described. Free radiolabeled amino acid values (nmol AA·mg−1 protein) were calculated as the counts per min (cpm) of the supernatant divided by the specific activity of the relevant amino acid uptake mix (glutamine or leucine; cpm/nmol) and normalized to the protein concentration (mg) of each well as determined by a bicinchoninic acid (BCA) assay. Incorporated radiolabeled amino acid values (incorporated nmol AA·mg−1 protein) were likewise calculated from the solubilized acid-precipitated fraction. All transport values depicted are the average ± SD of four separate determinations.
4.8. Cell Proliferation Assays—MTT Analysis
For methylthiazol tetrazolium (MTT) analysis, SKHep and its derivative cell lines were plated at an initial density of 2.0 × 103 cells/well, and Hep3B, HUH7, and their derivative cell lines were plated at an initial density of 8.0 × 103 cells/well, all in 48-well plates. After cell cultures reached 10–20% confluence (T0) before the plates were subjected to MTT analysis at the following time points: T0, 24, 48, 72, 96, 120, 144, and 168 h. Cells were washed with PBS and incubated for three hours in a 0.5 mg/mL solution of MTT in phenol red-free DMEM (Gibco) at 37 °C. After incubation, formazan crystals in each well were solubilized with an acidic solution of 0.04 M HCl in absolute isopropanol, and absorbance measured with an Epoch microplate spectrophotometer. Background absorbance was subtracted from each measurement (550–690 nm), and values were calculated as the average ± SD of at least four separate determinations.
4.9. mTORC1 Analysis
To evaluate the influence of distinct extracellular signals on mammalian target of rapamycin complex 1 (mTORC1) activity, cells were treated with culture medium starvation and repletion steps, part of a modified protocol based on a procedure described in Nicklin et al., 2009 [
16].
Supplemental Figure S16 depicts the order and formulation of each treatment in this analysis. All cell lines were plated at a density of 5.0 × 10
5 cells/well in 6-well plates, and when this population was 80–90% confluent, the cells were deprived of FBS (serum-free DMEM) for 18–22 h in order to remove growth factor signaling. The cells were next incubated in FBS-free, amino acid-free sodium Krebs-Ringer bicarbonate buffer (NaKRB) (MgSO
4, KCl, KH
2PO
4, NaCl, glucose, NaHCO
3, phenol red, and CaCl
2) for 3 h to remove extracellular amino acid signaling. Cells were next differentially treated with: (1) NaKRB + 2 mM
l-Gln + essential amino acids (EAA: histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine, all at normal media concentrations); or NaKRB + GLN + EAA plus one of the following four additives: (2) 1 mM GPNA, an ASCT2 inhibitor; (3) 5 mM BCH (Sigma), a LAT1 inhibitor; (4) 20 nM rapamycin (LC Laboratories, Woburn, MA, USA), an mTORC1 inhibitor; or (5) EtOH vehicle control, followed by an incubation time of 1 h at 37 °C. A cellular lysate was prepared from each treatment condition (indicated by the microfuge tube symbol in
Supplemental Figure S16) according to the previously described parameters for phosphorylation-specific Western blots in the ‘Western blot Analysis’ section of Materials and Methods.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








