Computational Approaches to Prioritize Cancer Driver Missense Mutations

Abstract
:1. Introduction
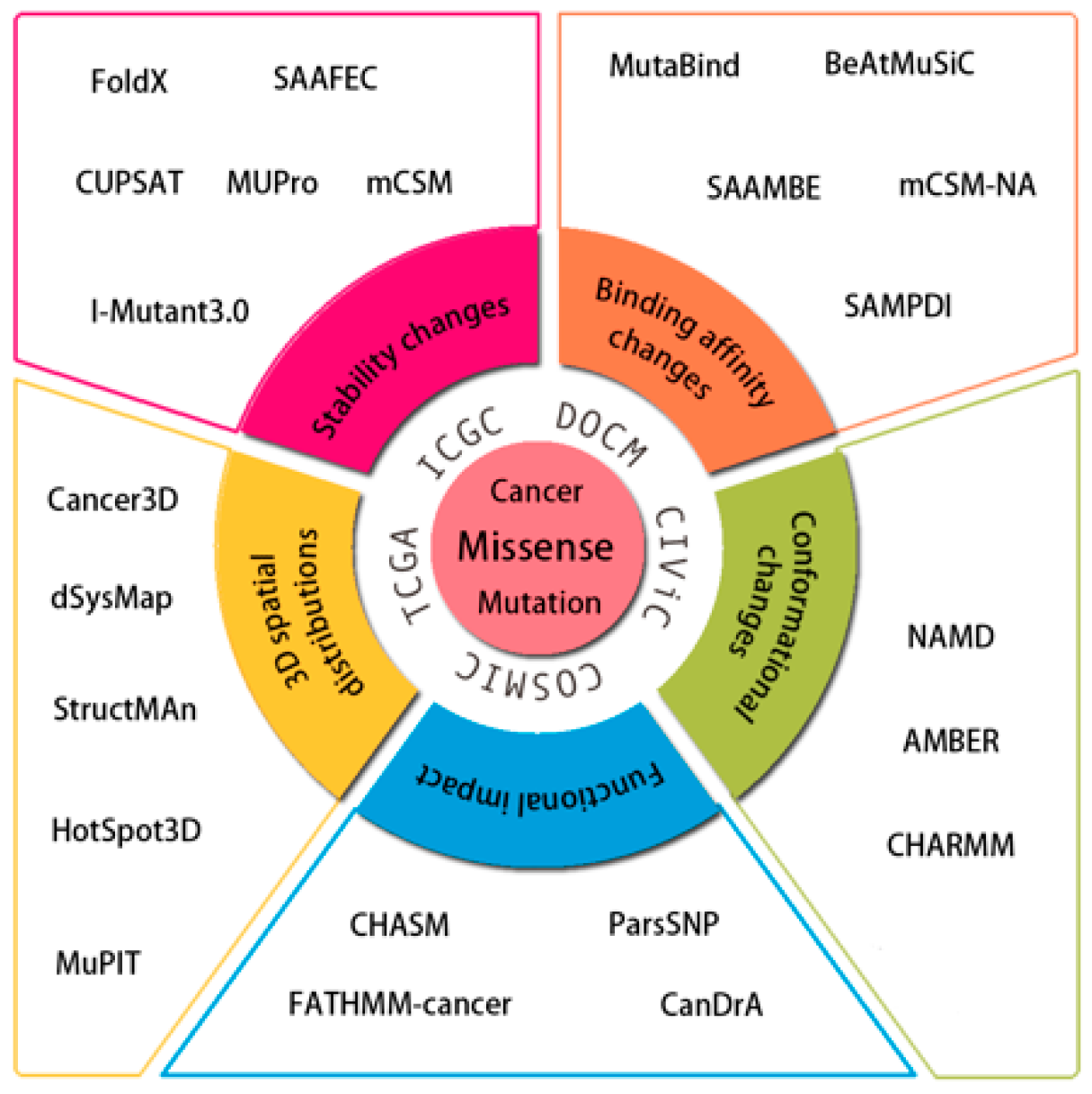
2. Data Resources for Cancer Missense Mutations
3. Computational Methods and Web Tools
3.1. 3D Spatial Distributions of Cancer Missense Mutations
3.2. Assessing Changes in Protein Conformation induced by Mutations
3.3. Estimating the Effects of Mutations on Protein Stability
3.4. Estimating Quantitative Effects of Mutations on Protein–Protein or Protein–Nucleic Acid Interactions
3.5. Assessing Driver Status of Cancer Mutations
Acknowledgments
Conflicts of Interest
References
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M.; Cancer Genome Atlas Research Network. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [PubMed] [Green Version]
- Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; Gerhard, D.S.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, E.R.; Vogelstein, B. A Genetic Model for Colorectal Tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Tabin, C.J.; Bradley, S.M.; Bargmann, C.I.; Weinberg, R.A.; Papageorge, A.G.; Scolnick, E.M.; Dhar, R.; Lowy, D.R.; Chang, E.H. Mechanism of Activation of a Human Oncogene. Nature 1982, 300, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O'Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.X.; Zhao, J.F.; Zhao, Z.M. Advances in computational approaches for prioritizing driver mutations and significantly mutated genes in cancer genomes. Brief. Bioinform. 2016, 17, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Porta-Pardo, E.; Kamburov, A.; Tamborero, D.; Pons, T.; Grases, D.; Valencia, A.; Lopez-Bigas, N.; Getz, G.; Godzik, A. Comparison of algorithms for the detection of cancer drivers at subgene resolution. Nat. Methods 2017, 14, 782. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J. ‘Latent drivers’ expand the cancer mutational landscape. Curr. Opin. Struct. Biol. 2015, 32, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Leedham, S.; Tomlinson, I. The Continuum Model of Selection in Human Tumors: General Paradigm or Niche Product? Cancer Res. 2012, 72, 3131–3134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krutovskikh, V.; Mazzoleni, G.; Mironov, N.; Omori, Y.; Aguelon, A.M.; Mesnil, M.; Berger, F.; Partensky, C.; Yamasaki, H. Altered homologous and heterologous gap-junctional intercellular communication in primary human liver tumors associated with aberrant protein localization but not gene mutation of connexin 32. Int. J. Cancer 1994, 56, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, C.F.; Riley, D.J.; Allred, D.C.; Chen, P.L.; Von Hoff, D.; Osborne, C.K.; Lee, W.H. Aberrant subcellular localization of BRCA1 in breast cancer. Science 1995, 270, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.C.; Link, W. Protein localization in disease and therapy. J. Cell Sci. 2011, 124, 3381–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, S.; Mak, M.W.; Kung, S.Y. R3P-Loc: A compact multi-label predictor using ridge regression and random projection for protein subcellular localization. J. Theor. Biol. 2014, 360, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Mak, M.W.; Kung, S.Y. HybridGO-Loc: Mining hybrid features on gene ontology for predicting subcellular localization of multi-location proteins. PLoS ONE 2014, 9, e89545. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011, 39, D945–D950. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Goncearenco, A.; Panchenko, A.R. Annotating Mutational Effects on Proteins and Protein Interactions: Designing Novel and Revisiting Existing Protocols. Methods Mol. Biol. 2017, 1550, 235–260. [Google Scholar] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tamborero, D.; Rubio-Perez, C.; Deu-Pons, J.; Schroeder, M.P.; Vivancos, A.; Rovira, A.; Tusquets, I.; Albanell, J.; Rodon, J.; Tabernero, J.; et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018, 10, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonetti, F.L.; Tornador, C.; Nabau-Moreto, N.; Molina-Vila, M.A.; Marino-Buslje, C. Kin-Driver: A database of driver mutations in protein kinases. Database 2014, 2014, bau104. [Google Scholar] [CrossRef] [PubMed]
- MacConaill, L.E.; Garcia, E.; Shivdasani, P.; Ducar, M.; Adusumilli, R.; Breneiser, M.; Byrne, M.; Chung, L.; Conneely, J.; Crosby, L.; et al. Prospective Enterprise-Level Molecular Genotyping of a Cohort of Cancer Patients. J. Mol. Diagn. 2014, 16, 660–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ainscough, B.J.; Griffith, M.; Coffman, A.C.; Wagner, A.H.; Kunisaki, J.; Choudhary, M.N.; McMichael, J.F.; Fulton, R.S.; Wilson, R.K.; Griffith, O.L.; et al. DoCM: A database of curated mutations in cancer. Nat. Methods 2016, 13, 806–807. [Google Scholar] [CrossRef] [PubMed]
- Griffith, M.; Spies, N.C.; Krysiak, K.; McMichael, J.F.; Coffman, A.C.; Danos, A.M.; Ainscough, B.J.; Ramirez, C.A.; Rieke, D.T.; Kujan, L.; et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet. 2017, 49, 170–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.D.S.; Bava, K.A.; Gromiha, M.M.; Prabakaran, P.; Kitajima, K.; Uedaira, H.; Sarai, A. ProTherm and ProNIT: Thermodynamic databases for proteins and protein-nucleic acid interactions. Nucleic Acids Res. 2006, 34, D204–D206. [Google Scholar] [CrossRef] [PubMed]
- Moal, I.H.; Fernandez-Recio, J. SKEMPI: A Structural Kinetic and Energetic database of Mutant Protein Interactions and its use in empirical models. Bioinformatics 2012, 28, 2600–2607. [Google Scholar] [CrossRef] [PubMed]
- Porta-Pardo, E.; Hrabe, T.; Godzik, A. Cancer3D: Understanding cancer mutations through protein structures. Nucleic Acids Res. 2015, 43, D968–D973. [Google Scholar] [CrossRef] [PubMed]
- Mosca, R.; Tenorio-Laranga, J.; Olivella, R.; Alcalde, V.; Ceol, A.; Soler-Lopez, M.; Aloy, P. dSysMap: Exploring the edgetic role of disease mutations. Nat. Methods 2015, 12, 167–168. [Google Scholar] [CrossRef] [PubMed]
- Gress, A.; Ramensky, V.; Buch, J.; Keller, A.; Kalinina, O.V. StructMAn: Annotation of single-nucleotide polymorphisms in the structural context. Nucleic Acids Res. 2016, 44, W463–W468. [Google Scholar] [CrossRef] [PubMed]
- Harper, K. Modeling Cancer Mutations in 3-D. Cancer Discov. 2017, 7, 787–788. [Google Scholar]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.C.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niknafs, N.; Kim, D.; Kim, R.; Diekhans, M.; Ryan, M.; Stenson, P.D.; Cooper, D.N.; Karchin, R. MuPIT interactive: Webserver for mapping variant positions to annotated, interactive 3D structures. Hum. Genet. 2013, 132, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Ryslik, G.A.; Cheng, Y.W.; Cheung, K.H.; Bjornson, R.D.; Zelterman, D.; Modis, Y.; Zhao, H.Y. A spatial simulation approach to account for protein structure when identifying non-random somatic mutations. BMC Bioinform. 2014, 15, 231. [Google Scholar] [CrossRef] [PubMed]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Getov, I.; Petukh, M.; Alexov, E. SAAFEC: Predicting the Effect of Single Point Mutations on Protein Folding Free Energy Using a Knowledge-Modified MM/PBSA Approach. Int. J. Mol. Sci. 2016, 17, 512. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Ascher, D.B. Blundell, T.L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Parthiban, V.; Gromiha, M.M.; Schomburg, D. CUPSAT: Prediction of protein stability upon point mutations. Nucleic Acids Res. 2006, 34, W239–W242. [Google Scholar] [CrossRef] [PubMed]
- Masso, M.; Vaisman, I.I. AUTO-MUTE: Web-based tools for predicting stability changes in proteins due to single amino acid replacements. Protein Eng. Des. Sel. 2010, 23, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Giollo, M.; Martin, A.J.M.; Walsh, I.; Ferrari, C.; Tosatto, S.C.E. NeEMO: A method using residue interaction networks to improve prediction of protein stability upon mutation. BMC Genom. 2014, 15, S7. [Google Scholar] [CrossRef] [PubMed]
- Laimer, J.; Hofer, H.; Fritz, M.; Wegenkittl, S.; Lackner, P. MAESTRO—Multi agent stability prediction upon point mutations. BMC Bioinform. 2015, 16, 116. [Google Scholar] [CrossRef] [PubMed]
- Wainreb, G.; Wolf, L.; Ashkenazy, H.; Dehouck, Y.; Ben-Tal, N. Protein stability: A single recorded mutation aids in predicting the effects of other mutations in the same amino acid site. Bioinformatics 2011, 27, 3286–3292. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Rossi, I.; Casadio, R. A three-state prediction of single point mutations on protein stability changes. Bmc Bioinformatics 2008, 9 (Suppl. 2), S6. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.L.; Randall, A.; Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins Struct. Funct. Bioinform. 2006, 62, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Lin, J.; Chu, Y.W. iStable: Off-the-shelf predictor integration for predicting protein stability changes. BMC Bioinform. 2013, 14, S5. [Google Scholar] [CrossRef]
- Li, M.; Simonetti, F.L.; Goncearenco, A.; Panchenko, A.R. MutaBind estimates and interprets the effects of sequence variants on protein-protein interactions. Nucleic Acids Res. 2016, 44, W494–W501. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, M. BeAtMuSiC: Prediction of changes in protein-protein binding affinity on mutations. Nucleic Acids Res. 2013, 41, W333–W339. [Google Scholar] [CrossRef] [PubMed]
- Petukh, M.; Li, M.; Alexov, E. Predicting Binding Free Energy Change Caused by Point Mutations with Knowledge-Modified MM/PBSA Method. PLoS Comput. Biol. 2015, 11, e1004276. [Google Scholar] [CrossRef] [PubMed]
- Petukh, M.; Dai, L.; Alexov, E. SAAMBE: Webserver to Predict the Charge of Binding Free Energy Caused by Amino Acids Mutations. Int. J. Mol. Sci. 2016, 17, 547. [Google Scholar] [CrossRef] [PubMed]
- Brender, J.R.; Zhang, Y. Predicting the Effect of Mutations on Protein-Protein Binding Interactions through Structure-Based Interface Profiles. PLoS Comput. Biol. 2015, 11, e1004494. [Google Scholar] [CrossRef] [PubMed]
- Kruger, D.M.; Gohlke, H. DrugScorePPI webserver: Fast and accurate in silico alanine scanning for scoring protein-protein interactions. Nucleic Acids Res. 2010, 38, W480–W486. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Han, J.G.; Shyu, C.R.; Korkin, D. Determining Effects of Non-synonymous SNPs on Protein-Protein Interactions using Supervised and Semi-supervised Learning. PLoS Comput. Biol. 2014, 10, e1003592. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Ascher, D.B. mCSM-NA: Predicting the effects of mutations on protein-nucleic acids interactions. Nucleic Acids Res. 2017, 45, W241–W246. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.H.; Sun, L.X.; Jia, Z.; Li, L.; Alexov, E. Predicting protein-DNA binding free energy change upon missense mutations using modified MM/PBSA approach: SAMPDI webserver. Bioinformatics 2018, 34, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Kamburov, A.; Lawrence, M.S.; Polak, P.; Leshchiner, I.; Lage, K.; Golub, T.R.; Lander, E.S.; Getz, G. Comprehensive assessment of cancer missense mutation clustering in protein structures. Proc. Natl. Acad. Sci. USA 2015, 112, E5486–E5495. [Google Scholar] [CrossRef] [PubMed]
- Tokheim, C.; Bhattacharya, R.; Niknafs, N.; Gygax, D.M.; Kim, R.; Ryan, M.; Masica, D.L.; Karchin, R. Exome-Scale Discovery of Hotspot Mutation Regions in Human Cancer Using 3D. Protein Struct. Cancer Res. 2016, 76, 3719–3731. [Google Scholar]
- Davis, M.J.; Ha, B.H.; Holman, E.C.; Halaban, R.; Schlessinger, J.; Boggon, T.J. RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc. Natl. Acad. Sci. USA 2013, 110, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.; Scott, A.D.; Sengupta, S.; Bailey, M.H.; Batra, P.; Ning, J.; Wyczalkowski, M.A.; Liang, W.W.; Zhang, Q.; McLellan, M.D.; et al. Protein-structure-guided discovery of functional mutations across 19 cancer types. Nat. Genet. 2016, 48, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.; Boye, K.; Flatmark, K. Molecular modelling and simulations in cancer research. Biochim. Biophys. Acta Rev. Cancer 2013, 1836, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, K.; Liu, D.; Tarpey, P.; Gallant, E.; Lam, A.; Witham, S.; Alexov, E.; Chaubey, A.; Stevenson, R.E.; Schwartz, C.E.; et al. An X-linked channelopathy with cardiomegaly due to a CLIC2 mutation enhancing ryanodine receptor channel activity. Hum. Mol. Genet. 2012, 21, 4497–4507. [Google Scholar] [CrossRef] [PubMed]
- Witham, S.; Takano, K.; Schwartz, C.; Alexov, E. A missense mutation in CLIC2 associated with intellectual disability is predicted by in silico modeling to affect protein stability and dynamics. Proteins Struct. Funct. Bioinform. 2011, 79, 2444–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukamoto, H.; Farrens, D.L. A Constitutively Activating Mutation Alters the Dynamics and Energetics of a Key Conformational Change in a Ligand-free G Protein-coupled Receptor. J. Biol. Chem. 2013, 288, 28207–28216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.W.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.B.; Eastwood, M.P.; Zhang, X.W.; Kim, E.T.; Arkhipov, A.; Dror, R.O.; Jumper, J.; Kuriyan, J.; Shaw, D.E. Oncogenic Mutations Counteract Intrinsic Disorder in the EGFR Kinase and Promote Receptor Dimerization. Cell 2012, 149, 860–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, K.; Rogozin, I.B.; Panchenko, A.R. Oncogenic potential is related to activating effect of cancer single and double somatic mutations in receptor tyrosine kinases. Hum. Mutat. 2012, 33, 1566–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutto, L.; Gervasio, F.L. Effects of oncogenic mutations on the conformational free-energy landscape of EGFR kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 10616–10621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salsbury, F.R. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr. Opin. Pharmacol. 2010, 10, 738–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwier, M.C.; Chong, L.T. Reaching biological timescales with all-atom molecular dynamics simulations. Curr. Opin. Pharmacol. 2010, 10, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Scheraga, H.A.; Khalili, M.; Liwo, A. Protein-folding dynamics: Overview of molecular simulation techniques. Annu. Rev. Phys. Chem. 2007, 58, 57–83. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Zheng, W.J. All-Atom Molecular Dynamics Simulations of Actin-Myosin Interactions: A Comparative Study of Cardiac alpha Myosin, beta Myosin, and Fast Skeletal Muscle Myosin. Biochemistry 2013, 52, 8393–8405. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Zheng, W.J. All-Atom Structural Investigation of Kinesin-Microtubule Complex Constrained by High-Quality Cryo-Electron-Microscopy Maps. Biochemistry 2012, 51, 5022–5032. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Zheng, W.J. Probing the Structural and Energetic Basis of Kinesin-Microtubule Binding Using Computational Alanine-Scanning Mutagenesis. Biochemistry 2011, 50, 8645–8655. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Luo, Q.A.; Xue, X.G.; Li, Z.S. Molecular dynamics studies of the 3D structure and planar ligand binding of a quadruplex dimer. J. Mol. Model. 2011, 17, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Luo, Q.; Li, Z.S. Molecular Dynamics Study on the Interactions of Porphyrin with Two Antiparallel Human Telomeric Quadruplexes. J. Phys. Chem. B 2010, 114, 6216–6224. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Zhou, Y.H.; Luo, Q.; Li, Z.S. The 3D structures of G-Quadruplexes of HIV-1 integrase inhibitors: Molecular dynamics simulations in aqueous solution and in the gas phase. J. Mol. Model. 2010, 16, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. Charmm—A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.H.; Shoemaker, B.A.; Thangudu, R.R.; Ferraris, J.D.; Burg, M.B.; Panchenko, A.R. Mutations in DNA-Binding Loop of NFAT5 Transcription Factor Produce Unique Outcomes on Protein-DNA Binding and Dynamics. J. Phys. Chem. B 2013, 117, 13226–13234. [Google Scholar] [CrossRef] [PubMed]
- Demir, O.; Baronio, R.; Salehi, F.; Wassman, C.D.; Hall, L.; Hatfield, G.W.; Chamberlin, R.; Kaiser, P.; Lathrop, R.H.; Amaro, R.E. Ensemble-Based Computational Approach Discriminates Functional Activity of p53 Cancer and Rescue Mutants. PLoS Comput. Biol. 2011, 7, e1002238. [Google Scholar] [CrossRef] [PubMed]
- Stehr, H.; Jang, S.H.J.; Duarte, J.M.; Wierling, C.; Lehrach, H.; Lappe, M.; Lange, B.M.H. The structural impact of cancer-associated missense mutations in oncogenes and tumor suppressors. Mol. Cancer 2011, 10, 54. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.H.; Norris, J.; Schwartz, C.; Alexov, E. Revealing the Effects of Missense Mutations Causing Snyder-Robinson Syndrome on the Stability and Dimerization of Spermine Synthase. Int. J. Mol. Sci. 2016, 17, 77. [Google Scholar] [CrossRef] [PubMed]
- Kales, S.C.; Ryan, P.E.; Nau, M.M.; Lipkowitz, S. Cbl and Human Myeloid Neoplasms: The Cbl Oncogene Comes of Age. Cancer Res. 2010, 70, 4789–4794. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.N.; Thacker, S.; Jaini, R.; Eng, C. Dynamics and structural stability effects of germline PTEN mutations associated with cancer versus autism phenotypes. J. Biomol. Struct. Dyn. 2018, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.H.; Grauffel, C.; Wu, L.S.; Kuo, P.H.; Doudeva, L.G.; Lim, C.; Shen, C.K.; Yuan, H.S. Structural analysis of disease-related TDP-43 D169G mutation: Linking enhanced stability and caspase cleavage efficiency to protein accumulation. Sci. Rep. 2016, 6, 21581. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Rahman, S.; Choudhry, H.; Zamzami, M.A.; Sarwar Jamal, M.; Islam, A.; Ahmad, F.; Hassan, M.I. Computing disease-linked SOD1 mutations: Deciphering protein stability and patient-phenotype relations. Sci. Rep. 2017, 7, 4678. [Google Scholar] [CrossRef] [PubMed]
- Kepp, K.P. Towards a “Golden Standard” for computing globin stability: Stability and structure sensitivity of myoglobin mutants. Biochim. Biophys. Acta 2015, 1854, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Kepp, K.P. Computing stability effects of mutations in human superoxide dismutase 1. J. Phys. Chem. B 2014, 118, 1799–1812. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Vihinen, M. Performance of protein stability predictors. Hum. Mutat. 2010, 31, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potapov, V.; Cohen, M.; Schreiber, G. Assessing computational methods for predicting protein stability upon mutation: Good on average but not in the details. Protein Eng. Des. Sel. 2009, 22, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Nishi, H.; Tyagi, M.; Teng, S.L.; Shoemaker, B.A.; Hashimoto, K.; Alexov, E.; Wuchty, S.; Panchenko, A.R. Cancer Missense Mutations Alter Binding Properties of Proteins and Their Interaction Networks. PLoS ONE 2013, 8, e66273. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.L.; Madej, T.; Panchenko, A.; Alexov, E. Modeling effects of human single nucleotide polymorphisms on protein-protein interactions. Biophys. J. 2009, 96, 2178–2188. [Google Scholar] [CrossRef] [PubMed]
- Ghersi, D.; Singh, M. Interaction-based discovery of functionally important genes in cancers. Nucleic Acids Res. 2014, 42, e18. [Google Scholar] [CrossRef] [PubMed]
- Domankevich, V.; Opatowsky, Y.; Malik, A.; Korol, A.B.; Frenkel, Z.; Manov, I.; Avivi, A.; Shams, I. Adaptive patterns in the p53 protein sequence of the hypoxia- and cancer-tolerant blind mole rat Spalax. BMC Evol. Biol. 2016, 16, 177. [Google Scholar] [CrossRef] [PubMed]
- Goncearenco, A.; Shoemaker, B.A.; Zhang, D.C.; Sarychey, A.; Panchenko, A.R. Coverage of protein domain families with structural protein-protein interactions: Current progress and future trends. Prog. Biophys. Mol. Biol. 2014, 116, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, B.A.; Zhang, D.C.; Tyagi, M.; Thangudu, R.R.; Fong, J.H.; Marchler-Bauer, A.; Bryant, S.H.; Madej, T.; Panchenko, A.R. IBIS (Inferred Biomolecular Interaction Server) reports, predicts and integrates multiple types of conserved interactions for proteins. Nucleic Acids Res. 2012, 40, D834–D840. [Google Scholar] [CrossRef] [PubMed]
- Goncearenco, A.; Li, M.; Simonetti, F.L.; Shoemaker, B.A.; Panchenko, A.R. Exploring Protein-Protein Interactions as Drug Targets for Anti-cancer Therapy with In Silico Workflows. Methods Mol. Biol. 2017, 1647, 221–236. [Google Scholar] [PubMed]
- Acuner-Ozbabacan, E.S.; Engin, B.H.; Guven-Maiorov, E.; Kuzu, G.; Muratcioglu, S.; Baspinar, A.; Chen, Z.; Van Waes, C.; Gursoy, A.; Keskin, O.; et al. The structural network of Interleukin-10 and its implications in inflammation and cancer. BMC Genom. 2014, 15, S2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Petukh, M.; Alexov, E.; Panchenko, A.R. Predicting the Impact of Missense Mutations on Protein-Protein Binding Affinity. J. Chem. Theory Comput. 2014, 10, 1770–1780. [Google Scholar] [CrossRef] [PubMed]
- David, A.; Sternberg, M.J.E. The Contribution of Missense Mutations in Core and Rim Residues of Protein-Protein Interfaces to Human Disease. J. Mol. Biol. 2015, 427, 2886–2898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Norris, J.; Kalscheuer, V.; Wood, T.; Wang, L.; Schwartz, C.; Alexov, E.; Van Esch, H. A Y328C missense mutation in spermine synthase causes a mild form of Snyder-Robinson syndrome. Hum. Mol. Genet. 2013, 22, 3789–3797. [Google Scholar] [CrossRef] [PubMed]
- Kucukkal, T.G.; Alexov, E. Structural, Dynamical, and Energetical Consequences of Rett Syndrome Mutation R133C in MeCP2. Comput. Math. Methods Med. 2015, 2015, 746157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Witham, S.; Petukh, M.; Moroy, G.; Miteva, M.; Ikeguchi, Y.; Alexov, E. A rational free energy-based approach to understanding and targeting disease-causing missense mutations. J. Am. Med. Inform. Assoc. 2013, 20, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, L.; Gao, Y.; Zhang, J.; Zhenirovskyy, M.; Alexov, E. Predicting folding free energy changes upon single point mutations. Bioinformatics 2012, 28, 664–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petukh, M.; Kucukkal, T.G.; Alexov, E. On Human Disease-Causing Amino Acid Variants: Statistical Study of Sequence and Structural Patterns. Hum. Mutat. 2015, 36, 524–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolzhanskaya, N.; Gonzalez, M.A.; Sperziani, F.; Stefl, S.; Messing, J.; Wen, G.Y.; Alexov, E.; Zuchner, S.; Velinov, M. A Novel p.Leu(381)Phe Mutation in Presenilin 1 is Associated with Very Early Onset and Unusually Fast Progressing Dementia as well as Lysosomal Inclusions Typically Seen in Kufs Disease. J. Alzheimers Dis. 2014, 39, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Boccuto, L.; Aoki, K.; Flanagan-Steet, H.; Chen, C.F.; Fan, X.; Bartel, F.; Petukh, M.; Pittman, A.; Saul, R.; Chaubey, A.; et al. A mutation in a ganglioside biosynthetic enzyme, ST3GAL5, results in salt & pepper syndrome, a neurocutaneous disorder with altered glycolipid and glycoprotein glycosylation. Hum. Mol. Genet. 2014, 23, 418–433. [Google Scholar] [PubMed]
- Peng, Y.H.; Alexov, E. Investigating the linkage between disease-causing amino acid variants and their effect on protein stability and binding. Proteins Struct. Funct. Bioinform. 2016, 84, 232–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilis, D.; McLennan, H.R.; Dehouck, Y.; Cabrita, L.D.; Rooman, M.; Bottomley, S.P. In vitro and in silico design of alpha1-antitrypsin mutants with different conformational stabilities. J. Mol. Biol. 2003, 325, 581–589. [Google Scholar] [CrossRef]
- Zhang, Z.; Norris, J.; Schwartz, C.; Alexov, E. In Silico and In Vitro Investigations of the Mutability of Disease-Causing Missense Mutation Sites in Spermine Synthase. PLoS ONE 2011, 6, e20373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.B.; Wu, Z.L. Identification of amino acid residues responsible for increased thermostability of feruloyl esterase A from Aspergillus niger using the PoPMuSiC algorithm. Bioresour. Technol. 2011, 102, 2093–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Jia, Z.; Peng, Y.H.; Godar, S.; Getov, I.; Teng, S.L.; Alper, J.; Alexov, E. Forces and Disease: Electrostatic force differences caused by mutations in kinesin motor domains can distinguish between disease-causing and non-disease-causing mutations. Sci. Rep. 2017, 7, 8237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zheng, Y.L.; Petukh, M.; Pegg, A.; Ikeguchi, Y.; Alexov, E. Enhancing Human Spermine Synthase Activity by Engineered Mutations. PLoS Comput. Biol. 2013, 9, e1002924. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chakravorty, A.; Alexov, E. DelPhiForce, a Tool for Electrostatic Force Calculations: Applications to Macromolecular Binding. J. Comput. Chem. 2017, 38, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.H.; Alexov, E. Computational investigation of proton transfer, pKa shifts and pH-optimum of protein-DNA and protein-RNA complexes. Proteins Struct. Funct. Bioinform. 2017, 85, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Teng, S.L. Wang, L.J.; Schwartz, C.E.; Alexov, E.; Computational Analysis of Missense Mutations Causing Snyder-Robinson Syndrome. Hum. Mutat. 2010, 31, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Kales, S.C.; Ma, K.; Shoemaker, B.A.; Crespo-Barreto, J.; Cangelosi, A.L.; Lipkowitz, S.; Panchenko, A.R. Balancing Protein Stability and Activity in Cancer: A New Approach for Identifying Driver Mutations Affecting CBL Ubiquitin Ligase Activation. Cancer Res. 2016, 76, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Naramura, M.; Nadeau, S.; Mohapatra, B.; Ahmad, G.; Mukhopadhyay, C.; Sattler, M.; Raja, S.M.; Natarajan, A.; Band, V.; Band, H. Mutant Cbl proteins as oncogenic drivers in myeloproliferative disorders. Oncotarget 2011, 2, 245–250. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Mustonen, V.; Reva, B.; Ritchie, G.R.S.; Creixell, P.; Karchin, R.; Vazquez, M.; Fink, J.L.; Kassahn, K.S.; Pearson, J.V.; et al. Computational approaches to identify functional genetic variants in cancer genomes. Nat. Methods 2013, 10, 723–729. [Google Scholar] [PubMed] [Green Version]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Day, I.N.M.; Gaunt, T.R. Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics 2013, 29, 1504–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Duncan, D.T.; Zhang, B. CanProVar: A human cancer proteome variation database. Hum. Mutat. 2010, 31, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.; Chen, S.N.; Isik, L.; Tyekucheva, S.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Cancer-specific high-throughput annotation of somatic mutations: Computational prediction of driver missense mutations. Cancer Res. 2009, 69, 6660–6667. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Amit, Y.; Geman, D. Shape quantization and recognition with randomized trees. Neural Comput. 1997, 9, 1545–1588. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.S.; Parsons, D.W.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.S.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Sjoblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Chen, H.; Liang, H.; Meric-Bernstam, F.; Mills, G.B.; Chen, K. CanDrA: Cancer-specific driver missense mutation annotation with optimized features. PLoS ONE 2013, 8, e77945. [Google Scholar] [CrossRef] [PubMed]
- Martelotto, L.G.; Ng, C.K.Y.; De Filippo, M.R.; Zhang, Y.; Piscuoglio, S.; Lim, R.S.; Shen, R.L.; Norton, L.; Reis-Filho, J.S.; Weigelt, B. Benchmarking mutation effect prediction algorithms using functionally validated cancer-related missense mutations. Genome Biol. 2014, 15, 484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.D.; Swamidass, S.J.; Bose, R. Unsupervised detection of cancer driver mutations with parsimony-guided learning. Nat. Genet. 2016, 48, 1288–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncearenco, A.; Rager, S.L.; Li, M.H.; Sang, Q.X.; Rogozin, I.B.; Panchenko, A.R. Exploring background mutational processes to decipher cancer genetic heterogeneity. Nucleic Acids Res. 2017, 45, W514–W522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Brown, A.-L.; Goncearenco, A.; Panchenko, A.R. Nucleotide and codon background mutability shape cancer mutational spectrum and advance driver mutation identification. bioRxiv 2018, 354506. [Google Scholar] [CrossRef]


{kind=link}
| Name | Description | Web Site | Ref. |
|---|---|---|---|
| Databases of cancer somatic mutations | |||
| COSMIC | Somatic mutations in cancer | http://cancer.sanger.ac.uk/cosmic | [18] |
| TCGA | Cancer Genome Atlas | http://cancergenome.nih.gov/ | [1] |
| ICGC | International Cancer Genome Consortium | https://icgc.org | [2] |
| DOCM | A highly curated database of somatic mutations with characterized functional or clinical significance in cancer. | http://docm.genome.wustl.edu | [25] |
| CIViC | Provide supported clinical interpretations of cancer-related mutations | https://civic.genome.wustl.edu/home | [26] |
| Databases of thermodynamic parameters | |||
| Protherm | Changes in thermodynamic parameters upon mutation for protein stability | http://gibk26.bse.kyutech.ac.jp/jouhou/Protherm/protherm.html | [27] |
| SKEMPI | Changes in thermodynamic parameters and kinetic rate constants upon mutation for protein-protein interactions | https://life.bsc.es/pid/mutation_database/ | [28] |
| ProNIT | Changes in thermodynamic parameters upon mutation for protein-nucleic acid interactions | http://gibk26.bse.kyutech.ac.jp/jouhou/pronit/pronit.html | [27] |
| Name | Description | Web Site | Ref. |
|---|---|---|---|
| Analyzing 3D spatial distributions of cancer missense mutations | |||
| Cancer3D | Mapping somatic missense mutations from human proteins to protein structure from Protein Data Bank (PDB) | http://www.cancer3d.org | [29] |
| COSMIC-3D | Understanding cancer mutations in the context of 3D protein structure | https://cancer.sanger.ac.uk/cosmic3d/ | [32] |
| cBioPortal | Visualization and analysis of large cancer studies. It is based on TCGA and incorporates the overlapping data from COSMIC | http://cbioportal.org/ | [33,34] |
| dSysMap | The systematic mapping of disease-related missense mutations on the structurally annotated binary human interactome | https://dsysmap.irbbarcelona.org | [30] |
| MuPIT | Mapping the genomic coordinates of SNVs onto the 3D protein structures | http://mupit.icm.jhu.edu/MuPIT_Interactive/ | [35] |
| StructMAn | Annotating nsSNVs in the context of the structural neighborhood of the resulting variations in the protein | http://structman.mpi-inf.mpg.de | [31] |
| SpacePAC | Identification of mutational clusters while considering protein tertiary structure | https://www.bioconductor.org/packages/release/bioc/html/SpacePAC.html | [36] |
| Predicting protein stability changes upon mutations | |||
| FoldX | ΔΔG using empirical force fields | http://fold-x.embl-heidelberg.de | [37] |
| SAAFEC | ΔΔG using multiple linear regression | http://compbio.clemson.edu/SAAFEC/ | [38] |
| mCSM | ΔΔG using graph-based signatures | http://biosig.unimelb.edu.au/mcsm/ | [39] |
| CUPSAT | ΔΔG using mean force atom pair and torsion angle potentials | http://cupsat.tu-bs.de/ | [40] |
| AUTO-MUTE | ΔΔG using knowledge-based potentials | http://proteins.gmu.edu/automute | [41] |
| NeEMO | ΔΔG using residue interaction networks | http://protein.bio.unipd.it/neemo/ | [42] |
| MAESTRO | ΔΔG using multi agent stability prediction | http://biwww.che.sbg.ac.at/MAESTRO | [43] |
| ProMaya | ΔΔG using random forests regression | http://bental.tau.ac.il/ProMaya/ | [44] |
| I-Mutant3.0 * | ΔΔG using SVMs | http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi | [45] |
| MUPro * | Predicts qualitative decrease/increase of stability using SVM | http://mupro.proteomics.ics.uci.edu/ | [46] |
| iStable * | ΔΔG using SVM | http://predictor.nchu.edu.tw/iStable | [47] |
| Predicting protein-protein binding affinity changes upon mutations | |||
| MutaBind | ΔΔG using molecular mechanics force fields, statistical potentials and fast side-chain optimization algorithms built via multiple linear regression and random forest | https://www.ncbi.nlm.nih.gov/research/mutabind/ | [48] |
| BeAtMuSiC | ΔΔG using a set of statistical potentials | http://babylone.ulb.ac.be/beatmusic | [49] |
| SAAMBE | ΔΔG using modified MM-PBSA based energy terms and a set of statistical terms built via multiple linear regression | http://compbio.clemson.edu/saambe_webserver/ | [50,51] |
| BindProf | ΔΔG using structure-based interface profiles | https://zhanglab.ccmb.med.umich.edu/BindProf/ | [52] |
| DrugScorePPI | ΔΔG for alanine-scanning mutations located on interface using knowledge-based scoring functions | http://cpclab.uni-duesseldorf.de/dsppi/ | [53] |
| SNP-IN | A classifier of effects on protein-protein interactions using supervised and semi-supervised learning | http://korkinlab.org/snpintool/ | [54] |
| Predicting protein-nucleic acid binding affinity changes upon mutations | |||
| mCSM-NA | ΔΔG relying on graph-based signatures and can predict the effects of single mutations on protein-nucleic acid binding | http://biosig.unimelb.edu.au/mcsm_na/ | [55] |
| SAMPDI | ΔΔG combining modified MM-PBSA based energy terms with knowledge based terms for predicting the protein-DNA binding affinity changes upon single mutations | http://compbio.clemson.edu/SAMPDI/ | [56] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, F.; Zheng, L.; Goncearenco, A.; Panchenko, A.R.; Li, M. Computational Approaches to Prioritize Cancer Driver Missense Mutations. Int. J. Mol. Sci. 2018, 19, 2113. https://doi.org/10.3390/ijms19072113
Zhao F, Zheng L, Goncearenco A, Panchenko AR, Li M. Computational Approaches to Prioritize Cancer Driver Missense Mutations. International Journal of Molecular Sciences. 2018; 19(7):2113. https://doi.org/10.3390/ijms19072113
Chicago/Turabian StyleZhao, Feiyang, Lei Zheng, Alexander Goncearenco, Anna R. Panchenko, and Minghui Li. 2018. "Computational Approaches to Prioritize Cancer Driver Missense Mutations" International Journal of Molecular Sciences 19, no. 7: 2113. https://doi.org/10.3390/ijms19072113
APA StyleZhao, F., Zheng, L., Goncearenco, A., Panchenko, A. R., & Li, M. (2018). Computational Approaches to Prioritize Cancer Driver Missense Mutations. International Journal of Molecular Sciences, 19(7), 2113. https://doi.org/10.3390/ijms19072113