Dynamic DNA Methylation in Plant Growth and Development

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
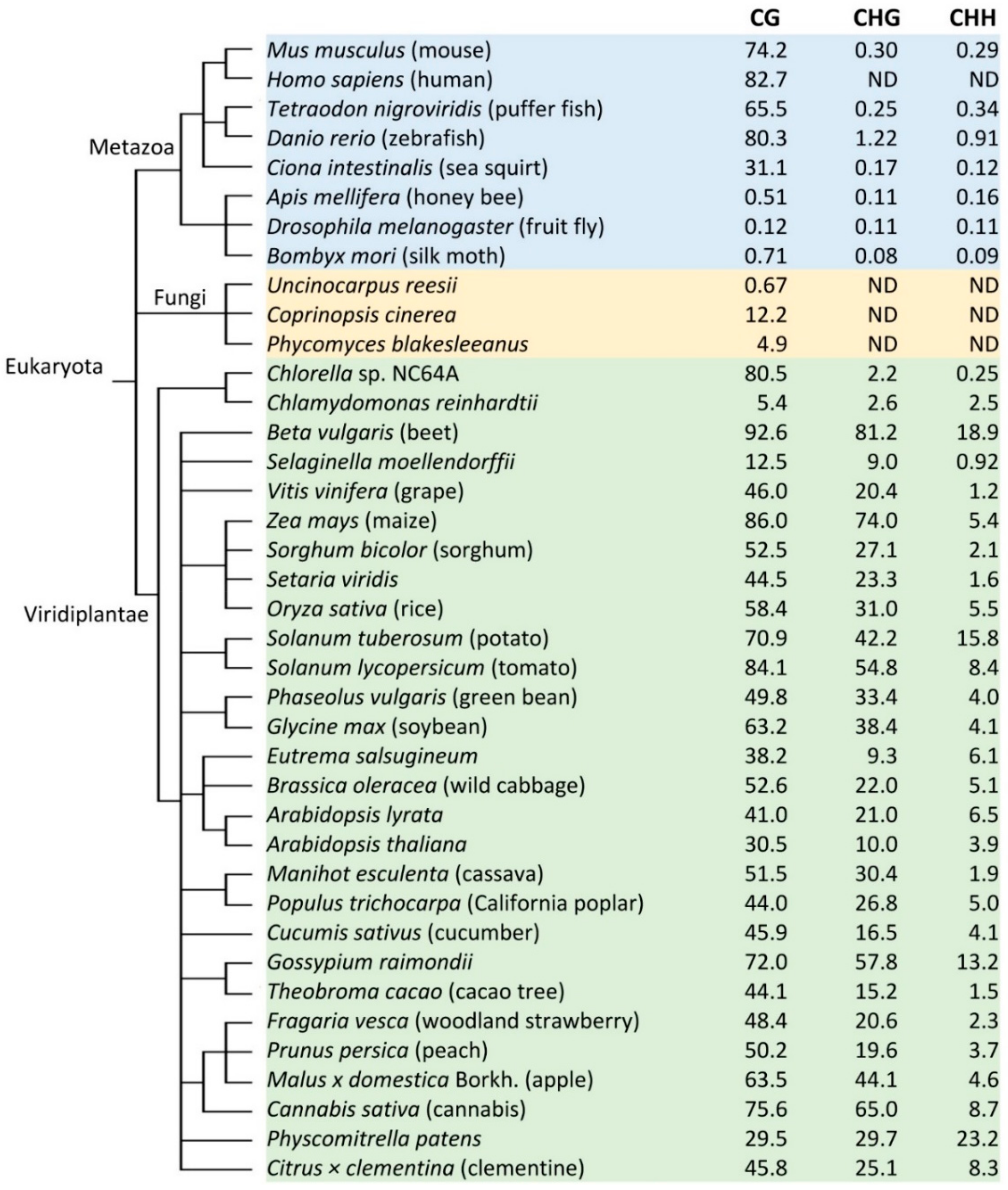
2. Variations of DNA Methylation in Different Plant Species
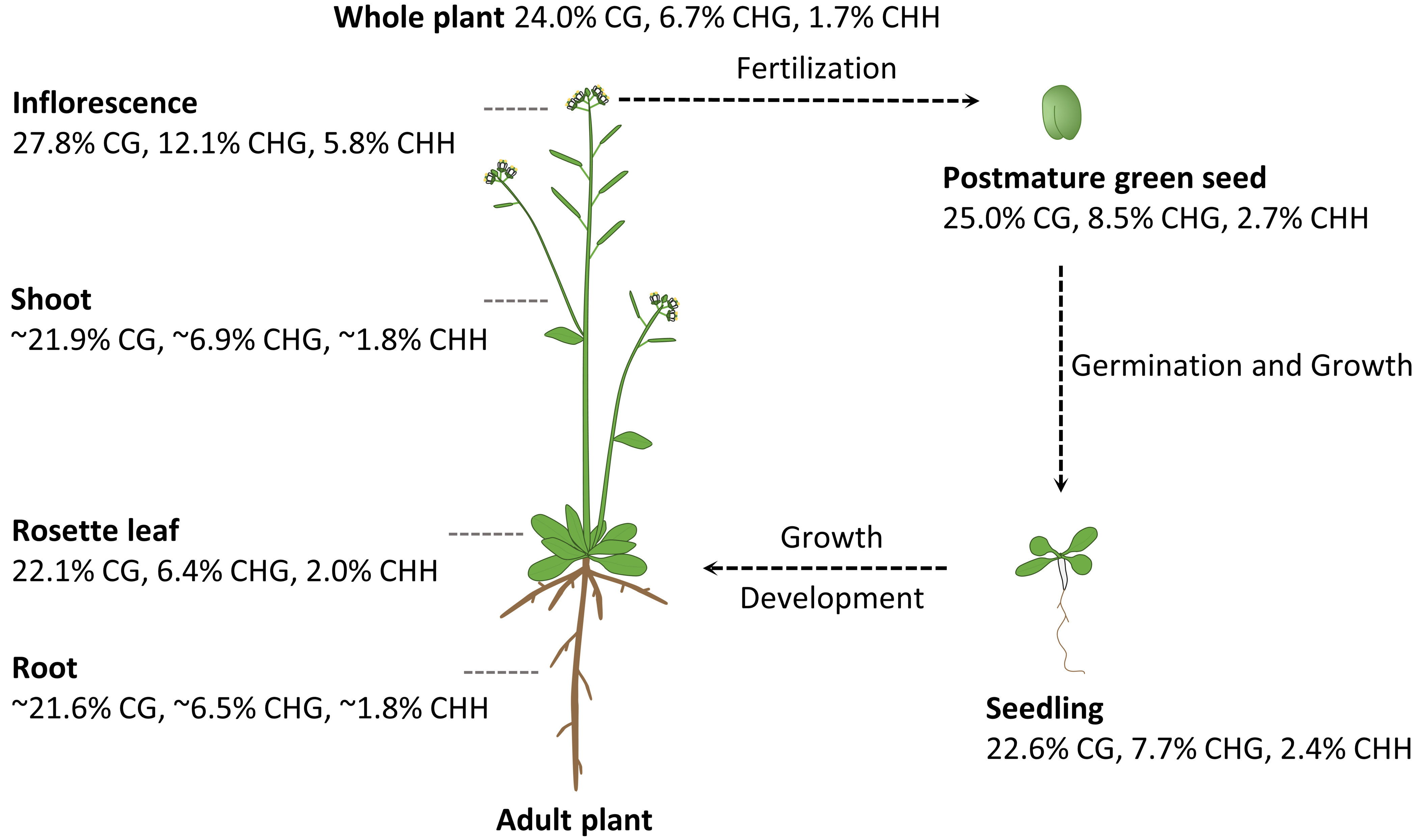
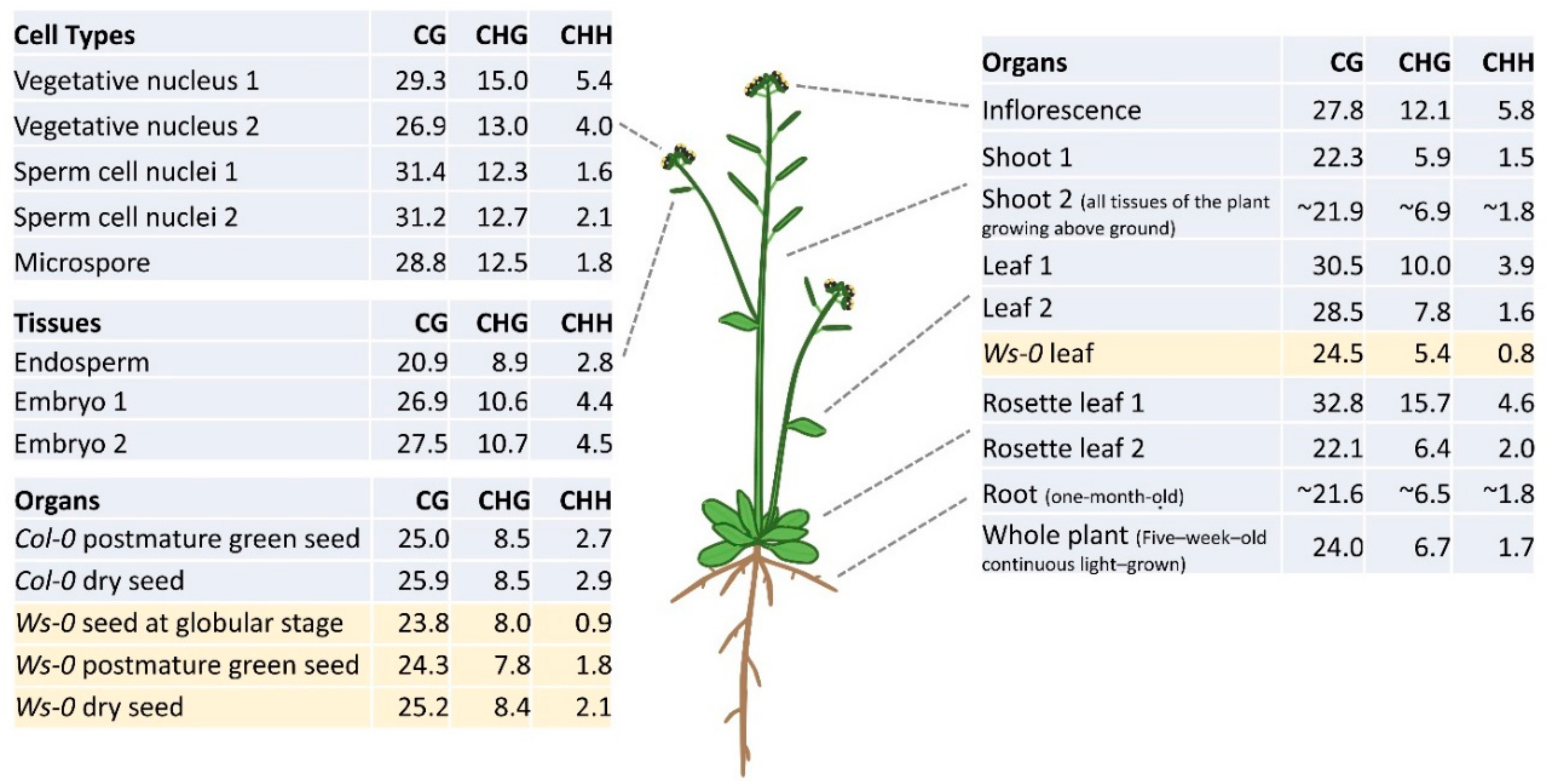
3. DNA Methylation Profiles in Different Plant Organs and Tissues
4. Reprogramming of DNA Methylation in Gametogenesis
5. Dynamic DNA Methylation during Seed Development and Germination
6. Alteration of DNA Methylation in Response to Environmental Stimuli
7. Mechanisms of Dynamic DNA Methylation
8. Conclusions and Discussion
Author Contributions
Acknowledgments
Conflicts of interests
Abbreviations
| 5mC | 5-methylcytosine |
| AGO | ARGONAUTE |
| CLSY1-4 | CLASSY1-4 |
| CMT2 | CHROMOMETHYLASE 2 |
| CMT3 | CHROMOMETHYLASE 3 |
| DCL | DICER-LIKE |
| ddc | the drm1 drm2 cmt3 mutant |
| ddcc | the drm1 drm2 cmt2 cmt3 mutant |
| DME | DEMETER |
| DML2 | DEMETER-LIKE 2 |
| DML3 | DEMETER-LIKE 3 |
| Dnmt1 | DNA methyltransferase 1 |
| DRM1 | DOMAINS REARRANGED METHYLTRANSFERASE 1 |
| DRM2 | DOMAINS REARRANGED METHYLTRANSFERASE 2 |
| KTF1 | KOW DOMAIN-CONTAINING TRANSCRIPTION FACTOR 1 |
| MEG | maternally expressed imprinted gene |
| MET1 | DNA METHYLTRANSFERASE 1 in plants |
| NRPE1 | NUCLEAR RNA POLYMERASE E1 |
| PcG | Polycomb group proteins |
| PEG | paternally expressed imprinted gene |
| RdDM | RNA-directed DNA methylation |
| RDR | RNA-dependent RNA polymerase |
| ROS1 | REPRESSOR OF SILENCING 1 |
| siRNA | small interference RNA |
| TET | Ten-eleven-translocation enzyme |
| TSS | transcriptional start site |
| TTS | transcriptional termination site |
References
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martienssen, R.A.; Colot, V. DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science 2001, 293, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. DNA methylation, a key regulator of plant development and other processes. Curr. Opin. Genet. Dev. 2000, 10, 217–223. [Google Scholar] [CrossRef]
- Richards, E.J. Inherited epigenetic variation—Revisiting soft inheritance. Nat. Rev. Genet. 2006, 7, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Li, E. Chromatin Modification and Epigenetic Reprogramming in Mammalian Development. Nat. Rev. Genet. 2002, 3, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Vongs, A.; Kakutani, T.; Martienssen, R.A.; Richards, E.J. Arabidopsis thaliana DNA methylation mutants. Science 1993, 260, 1926–1928. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, E.J.; Dennis, E.S. Isolation and identification by sequence homology of a putative cytosine methyltransferase from Arabidopsis thaliana. Nucleic Acids Res. 1993, 21, 2383–2388. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, E.J.; Kovac, K.A. Plant DNA Methyltransferases. Plant Mol. Biol. 2000, 43, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Kankel, M.W.; Ramsey, D.E.; Stokes, T.L.; Flowers, S.K.; Haag, J.R.; Jeddeloh, J.A.; Riddle, N.C.; Verbsky, M.L.; Richards, E.J. Arabidopsis MET1 Cytosine Methyltransferase Mutants. Genetics 2003, 163, 1109–1122. [Google Scholar] [PubMed]
- Lindroth, A.M.; Cao, X.; Jackson, J.P.; Zilberman, D.; McCallum, C.M.; Henikoff, S.; Jacobsen, S.E. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 2001, 292, 2077–2080. [Google Scholar] [CrossRef] [PubMed]
- Bartee, L.; Malagnac, F.; Bender, J. Arabidopsis CMT3 chromomethylase mutations block non-CG methylation and silencing of an endogenous gene. Genes Dev. 2001, 15, 1753–1758. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; Kim, M.Y.; Hsieh, P.H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Jacobsen, S.E. Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc. Natl. Acad. Sci. USA 2002, 99, 16491–16498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Jacobsen, S.E. Genetic analyses of DNA methyltransferases in Arabidopsis thaliana. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Teerawanichpan, P.; Krittanai, P.; Chauvatcharin, N.; Narangajavana, J. Purification and characterization of rice DNA methyltransferase. Plant Physiol. Biochem. 2009, 47, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.; Cummings, M.; Roberts, R.J.; Adams, R.L. Isolation, characterization and baculovirus-mediated expression of the cDNA encoding cytosine DNA methyltransferase from Pisum sativum. Nucleic Acids Res. 1998, 26, 1214–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steward, N.; Kusano, T.; Sano, H. Expression of ZmMET1, a gene encoding a DNA methyltransferase from maize, is associated not only with DNA replication in actively proliferating cells, but also with altered DNA methylation status in cold-stressed quiescent cells. Nucleic Acids Res. 2000, 28, 3250–3259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, R.; Sasaki, T.; Nishio, T. Characterization of DNA methyltransferase genes in Brassica rapa. Genes Genet. Syst. 2006, 81, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, C.M.; Springer, N.M.; Muszynski, M.G.; Meeley, R.; Kaeppler, S.M. Maize chromomethylase Zea methyltransferase2 is required for CpNpG methylation. Plant Cell 2001, 13, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Pavlopoulou, A.; Kossida, S. Plant cytosine-5 DNA methyltransferases: Structure, function, and molecular evolution. Genomics 2007, 90, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. Small RNAs in development—Insights from plants. Curr. Opin. Genet. Dev. 2012, 22, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.C.; Elmayan, T.; Vaucheret, H. MicroRNA maturation and action—The expanding roles of ARGONAUTEs. Curr. Opin. Plant Biol. 2008, 11, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Allen, E.; Wilken, A.; Carrington, J.C. DICER-LIKE 4 functions in trans-acting small interfering RNA biogenesis and vegetative phase change in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2005, 102, 12984–12989. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. Reduced DNA methylation in Arabidopsis results in abnormal plant development. Proc. Natl. Acad. Sci. USA 1996, 93, 8449–8454. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Gehring, M.; Choi, Y.; Margossian, L.; Pu, H.; Harada, J.J.; Goldberg, R.B.; Pennell, R.I.; Fischer, R.L. Imprinting of the MEA Polycomb gene is controlled by antagonism between MET1 methyltransferase and DME glycosylase. Dev. Cell 2003, 5, 891–901. [Google Scholar] [CrossRef]
- Xiao, W.; Custard, K.D.; Brown, R.C.; Lemmon, B.E.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. DNA methylation is critical for Arabidopsis embryogenesis and seed viability. Plant Cell 2006, 18, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Rea, M.; Zheng, W.; Chen, M.; Braud, C.; Bhangu, D.; Rognan, T.N.; Xiao, W. Histone H1 affects gene imprinting and DNA methylation in Arabidopsis. Plant J. 2012, 71, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Kakutani, T.; Jeddeloh, J.A.; Flowers, S.K.; Munakata, K.; Richards, E.J. Developmental abnormalities and epimutations associated with DNA hypomethylation mutations. Proc. Natl. Acad. Sci. USA 1996, 93, 12406–12411. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, S.E.; Sakai, H.; Finnegan, E.J.; Cao, X.; Meyerowitz, E.M. Ectopic hypermethylation of flower-specific genes in Arabidopsis. Curr. Biol. 2000, 10, 179–186. [Google Scholar] [CrossRef]
- Miura, A.; Yonebayashi, S.; Watanabe, K.; Toyama, T.; Shimada, H.; Kakutani, T. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 2001, 411, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Butenko, Y.; Ohad, N. Polycomb-group mediated epigenetic mechanisms through plant evolution. Biochim. Biophys. Acta 2011, 1809, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Ohr, H.; Lee, J.W.; Hyun, Y.; Fischer, R.L.; Choi, Y. Temporal and spatial downregulation of Arabidopsis MET1 activity results in global DNA hypomethylation and developmental defects. Mol. Cells 2008, 26, 611–615. [Google Scholar] [PubMed]
- Choi, Y.; Gehring, M.; Johnson, L.; Hannon, M.; Harada, J.J.; Goldberg, R.B.; Jacobsen, S.E.; Fischer, R.L. DEMETER, a DNA Glycosylase Domain Protein, Is Required for Endosperm Gene Imprinting and Seed Viability in Arabidopsis. Cell 2002, 110, 33–42. [Google Scholar] [CrossRef]
- Jullien, P.E.; Katz, A.; Oliva, M.; Ohad, N.; Berger, F. Polycomb group complexes self-regulate imprinting of the Polycomb group gene MEDEA in Arabidopsis. Curr. Biol. 2006, 16, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Brown, R.C.; Lemmon, B.E.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. Regulation of seed size by hypomethylation of maternal and paternal genomes. Plant Physiol. 2006, 142, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Vinkenoog, R.; Spielman, M.; Dickinson, H.G.; Scott, R.J. Parent-of-origin effects on seed development in Arabidopsis thaliana require DNA methylation. Development 2000, 127, 2493–2502. [Google Scholar] [PubMed]
- Luo, M.; Bilodeau, P.; Dennis, E.S.; Peacock, W.J.; Chaudhury, A. Expression and parent-of-origin effects for FIS2, MEA, and FIE in the endosperm and embryo of developing Arabidopsis seeds. Proc. Natl. Acad. Sci. USA 2000, 97, 10637–10642. [Google Scholar] [CrossRef] [PubMed]
- An, Y.C.; Goettel, W.; Han, Q.; Bartels, A.; Liu, Z.; Xiao, W. Dynamic Changes of Genome-Wide DNA Methylation during Soybean Seed Development. Sci. Rep. 2017, 7, 12263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Jacobsen, S.E.; Reik, W. Epigenetic reprogramming in plant and animal development. Science 2010, 330, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- Takuno, S.; Ran, J.H.; Gaut, B.S. Evolutionary patterns of genic DNA methylation vary across land plants. Nat. Plants 2016, 2, 15222. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.; Perez, R.; Bazaga, P.; Herrera, C.M. Global DNA cytosine methylation as an evolving trait: Phylogenetic signal and correlated evolution with genome size in angiosperms. Front. Genet. 2015, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gent, J.I.; Ellis, N.A.; Guo, L.; Harkess, A.E.; Yao, Y.; Zhang, X.; Dawe, R.K. CHH islands: De novo DNA methylation in near-gene chromatin regulation in maize. Genome Res. 2013, 23, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xie, J.; Hu, J.; Lan, B.; You, C.; Li, F.; Wang, Z.; Wang, H. Comparative epigenomics reveals evolution of duplicated genes in potato and tomato. Plant J. 2018, 93, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Messeguer, R.; Ganal, M.W.; Steffens, J.C.; Tanksley, S.D. Characterization of the level, target sites and inheritance of cytosine methylation in tomato nuclear DNA. Plant Mol. Biol. 1991, 16, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Garcia, L.; Cervera, M.; Martinez-Zapater, J. DNA methylation increases throughout Arabidopsis development. Planta 2005, 222, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Zluvova, J.; Janousek, B.; Vyskot, B. Immunohistochemical study of DNA methylation dynamics during plant development. J. Exp. Bot. 2001, 52, 2265–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitonti, M.B.; Cozza, R.; Chiappetta, A.; Giannino, D.; Castiglione, M.R.; Dewitte, W.; Mariotti, D.; Van Onckelen, H.; Innocenti, A.M. Distinct nuclear organization, DNA methylation pattern and cytokinin distribution mark juvenile, juvenile-like and adult vegetative apical meristems in peach (Prunus persica (L.) Batsch). J. Exp. Bot. 2002, 53, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Widman, N.; Feng, S.; Jacobsen, S.E.; Pellegrini, M. Epigenetic differences between shoots and roots in Arabidopsis reveals tissue-specific regulation. Epigenetics 2014, 9, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; Kim, M.Y.; Silva, P.; Rodrigues, J.A.; Dotson, B.; Brooks, M.D.; Zilberman, D. Local DNA hypomethylation activates genes in rice endosperm. Proc. Natl. Acad. Sci. USA 2010, 107, 18729–18734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, T.F.; Ibarra, C.A.; Silva, P.; Zemach, A.; Eshed-Williams, L.; Fischer, R.L.; Zilberman, D. Genome-wide demethylation of Arabidopsis endosperm. Science 2009, 324, 1451–1454. [Google Scholar] [CrossRef] [PubMed]
- Kawakatsu, T.; Stuart, T.; Valdes, M.; Breakfield, N.; Schmitz, R.J.; Nery, J.R.; Urich, M.A.; Han, X.; Lister, R.; Benfey, P.N. Unique cell-type-specific patterns of DNA methylation in the root meristem. Nat. Plants 2016, 2, 16058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Q.-X.; Lu, X.; Li, Q.-T.; Chen, H.; Hu, X.-Y.; Ma, B.; Zhang, W.-K.; Chen, S.-Y.; Zhang, J.-S. Genome-wide analysis of DNA methylation in soybean. Mol. Plant 2013, 6, 1961–1974. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.; Gao, H.; Zhang, J.; Aldridge, B.; Vickers, M.; Higgins, J.D.; Feng, X. Sexual-lineage-specific DNA methylation regulates meiosis in Arabidopsis. Nat. Genet. 2018, 50, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, T.; Berger, F. Epigenetic reprogramming in plant sexual reproduction. Nat. Rev. Genet. 2014, 15, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.-H.; He, S.; Buttress, T.; Gao, H.; Couchman, M.; Fischer, R.L.; Zilberman, D.; Feng, X. Arabidopsis male sexual lineage exhibits more robust maintenance of CG methylation than somatic tissues. Proc. Natl. Acad. Sci. USA 2016, 113, 15132–15137. [Google Scholar] [CrossRef] [PubMed]
- Calarco, J.P.; Borges, F.; Donoghue, M.T.; Van Ex, F.; Jullien, P.E.; Lopes, T.; Gardner, R.; Berger, F.; Feijo, J.A.; Becker, J.D.; et al. Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell 2012, 151, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, C.A.; Feng, X.; Schoft, V.K.; Hsieh, T.F.; Uzawa, R.; Rodrigues, J.A.; Zemach, A.; Chumak, N.; Machlicova, A.; Nishimura, T.; et al. Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science 2012, 337, 1360–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.Y.; Le, B.H.; Chen, M.; Henry, K.F.; Hur, J.; Hsieh, T.F.; Chen, P.Y.; Pelletier, J.M.; Pellegrini, M.; Fischer, R.L.; et al. Similarity between soybean and Arabidopsis seed methylomes and loss of non-CG methylation does not affect seed development. Proc. Natl. Acad. Sci. USA 2017, 114, E9730–E9739. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Hagmann, J.; Muller, J.; Koenig, D.; Stegle, O.; Borgwardt, K.; Weigel, D. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 2011, 480, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.R.; Crisp, P.A.; Eichten, S.R.; Pogson, B.J. The Arabidopsis DNA Methylome Is Stable under Transgenerational Drought Stress. Plant Physiol. 2017, 175, 1893–1912. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R. Do plants have a segregated germline? PLoS Biol. 2018, 16, e2005439. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Yadegari, R.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. Imprinting of the MEDEA polycomb gene in the Arabidopsis endosperm. Plant Cell 1999, 11, 1945–1952. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Miura, A.; Choi, Y.; Kinoshita, Y.; Cao, X.; Jacobsen, S.E.; Fischer, R.L.; Kakutani, T. One-way control of FWA imprinting in Arabidopsis endosperm by DNA methylation. Science 2004, 303, 521–523. [Google Scholar] [CrossRef] [PubMed]
- Jullien, P.E.; Kinoshita, T.; Ohad, N.; Berger, F. Maintenance of DNA methylation during the Arabidopsis life cycle is essential for parental imprinting. Plant Cell 2006, 18, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.F.; Shin, J.; Uzawa, R.; Silva, P.; Cohen, S.; Bauer, M.J.; Hashimoto, M.; Kirkbride, R.C.; Harada, J.J.; Zilberman, D.; et al. Regulation of imprinted gene expression in Arabidopsis endosperm. Proc. Natl. Acad. Sci. USA 2011, 108, 1755–1762. [Google Scholar] [CrossRef] [PubMed]
- Kohler, C.; Wolff, P.; Spillane, C. Epigenetic mechanisms underlying genomic imprinting in plants. Annu. Rev. Plant Biol. 2012, 63, 331–352. [Google Scholar] [CrossRef] [PubMed]
- Jahnke, S.; Scholten, S. Epigenetic Resetting of a Gene Imprinted in Plant Embryos. Curr. Biol. 2009, 19, 1677–1681. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Frost, J.M.; Park, K.; Ohr, H.; Park, G.T.; Kim, S.; Eom, H.; Lee, I.; Brooks, J.S.; Fischer, R.L.; et al. Control of DEMETER DNA demethylase gene transcription in male and female gamete companion cells in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2017, 114, 2078–2083. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, R.K.; Vaughn, M.; Borges, F.; Tanurdzic, M.; Becker, J.D.; Feijo, J.A.; Martienssen, R.A. Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 2009, 136, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Martinez, G.; Wolff, P.; Wang, Z.; Moreno-Romero, J.; Santos-Gonzalez, J.; Conze, L.L.; DeFraia, C.; Slotkin, R.K.; Kohler, C. Paternal easiRNAs regulate parental genome dosage in Arabidopsis. Nat. Genet. 2018, 50, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Klosinska, M.; Picard, C.L.; Gehring, M. Conserved imprinting associated with unique epigenetic signatures in the Arabidopsis genus. Nat. Plants 2016, 2, 16145. [Google Scholar] [CrossRef] [PubMed]
- Narsai, R.; Gouil, Q.; Secco, D.; Srivastava, A.; Karpievitch, Y.V.; Liew, L.C.; Lister, R.; Lewsey, M.G.; Whelan, J. Extensive transcriptomic and epigenomic remodelling occurs during Arabidopsis thaliana germination. Genome Biol. 2017, 18, 172. [Google Scholar] [CrossRef] [PubMed]
- Kawakatsu, T.; Nery, J.R.; Castanon, R.; Ecker, J.R. Dynamic DNA methylation reconfiguration during seed development and germination. Genome Biol. 2017, 18, 171. [Google Scholar] [CrossRef] [PubMed]
- Bouyer, D.; Kramdi, A.; Kassam, M.; Heese, M.; Schnittger, A.; Roudier, F.; Colot, V. DNA methylation dynamics during early plant life. Genome Biol. 2017, 18, 179. [Google Scholar] [CrossRef] [PubMed]
- Van Zanten, M.; Koini, M.A.; Geyer, R.; Liu, Y.; Brambilla, V.; Bartels, D.; Koornneef, M.; Fransz, P.; Soppe, W.J. Seed maturation in Arabidopsis thaliana is characterized by nuclear size reduction and increased chromatin condensation. Proc. Natl. Acad. Sci. USA 2011, 108, 20219–20224. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.Q.; Zhang, Y.J.; Zhou, S.R.; Hu, W.Y.; Wu, X.T.; Ye, Y.J.; Wu, X.X.; Xiao, Y.P.; Li, X.; Xue, H.W. Global Analysis Reveals the Crucial Roles of DNA Methylation during Rice Seed Development. Plant Physiol. 2015, 168, 1417–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grover, J.W.; Kendall, T.; Baten, A.; Burgess, D.; Freeling, M.; King, G.J.; Mosher, R.A. Maternal components of RNA-directed DNA methylation are required for seed development in Brassica rapa. Plant J. 2018, 94, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; Kawakatsu, T.; Kim, K.D.; Zhang, N.; Nguyen, C.T.; Khan, S.M.; Batek, J.M.; Joshi, T.; Schmutz, J.; Grimwood, J.; et al. Divergent cytosine DNA methylation patterns in single-cell, soybean root hairs. New Phytol. 2017, 214, 808–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Bilichak, A.; Golubov, A.; Kovalchuk, I. ddm1 plants are sensitive to methyl methane sulfonate and NaCl stresses and are deficient in DNA repair. Plant Cell Rep. 2012, 31, 1549–1561. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wang, Y.; Zheng, H.; Lu, W.; Wu, C.; Huang, J.; Yan, K.; Yang, G.; Zheng, C. Salt-induced transcription factor MYB74 is regulated by the RNA-directed DNA methylation pathway in Arabidopsis. J. Exp. Bot. 2015, 66, 5997–6008. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Conde, E.S.N.; Audonnet, L.; Servet, C.; Wei, W.; Zhou, D.X. Over-expression of histone H3K4 demethylase gene JMJ15 enhances salt tolerance in Arabidopsis. Front. Plant Sci. 2014, 5, 290. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.; Ramirez, V.; Garcia-Andrade, J.; Flors, V.; Vera, P. The RNA silencing enzyme RNA polymerase v is required for plant immunity. PLoS Genet. 2011, 7, e1002434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, A.; Lepere, G.; Jay, F.; Wang, J.; Bapaume, L.; Wang, Y.; Abraham, A.L.; Penterman, J.; Fischer, R.L.; Voinnet, O.; et al. Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. Proc. Natl. Acad. Sci. USA 2013, 110, 2389–2394. [Google Scholar] [CrossRef] [PubMed]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, A.; Daniel, X.; Flors, V.; Luna, E.; Hohn, B.; Mauch-Mani, B. Descendants of primed Arabidopsis plants exhibit resistance to biotic stress. Plant Physiol. 2012, 158, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Luna, E.; Ton, J. The epigenetic machinery controlling transgenerational systemic acquired resistance. Plant Signal. Behav. 2012, 7, 615–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez Sanchez, A.; Stassen, J.H.; Furci, L.; Smith, L.M.; Ton, J. The role of DNA (de)methylation in immune responsiveness of Arabidopsis. Plant J. 2016, 88, 361–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirouze, M.; Paszkowski, J. Epigenetic contribution to stress adaptation in plants. Curr. Opin. Plant Biol. 2011, 14, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Reinders, J.; Wulff, B.B.; Mirouze, M.; Mari-Ordonez, A.; Dapp, M.; Rozhon, W.; Bucher, E.; Theiler, G.; Paszkowski, J. Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes Dev. 2009, 23, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Lira-Medeiros, C.F.; Parisod, C.; Fernandes, R.A.; Mata, C.S.; Cardoso, M.A.; Ferreira, P.C. Epigenetic variation in mangrove plants occurring in contrasting natural environment. PLoS ONE 2010, 5, e10326. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Morales-Ruiz, T.; Ariza, R.R.; Roldan-Arjona, T.; David, L.; Zhu, J.-J. ROS1, a Repressor of Transcriptional Gene Silencing in Arabidopsis, Encodes a DNA Glycosylase/Lyase. Cell 2002, 111, 803–814. [Google Scholar] [CrossRef]
- Williams, B.P.; Pignatta, D.; Henikoff, S.; Gehring, M. Methylation-sensitive expression of a DNA demethylase gene serves as an epigenetic rheostat. PLoS Genet. 2015, 11, e1005142. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Zhang, H.; Julian, R.; Tang, K.; Xie, S.; Zhu, J.K. Regulatory link between DNA methylation and active demethylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2015, 112, 3553–3557. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Chen, X. Regulation of small RNA stability: Methylation and beyond. Cell Res. 2012, 22, 624–636. [Google Scholar] [CrossRef] [PubMed]
- He, X.J.; Hsu, Y.F.; Zhu, S.; Wierzbicki, A.T.; Pontes, O.; Pikaard, C.S.; Liu, H.L.; Wang, C.S.; Jin, H.; Zhu, J.K. An effector of RNA-directed DNA methylation in arabidopsis is an ARGONAUTE 4- and RNA-binding protein. Cell 2009, 137, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Bies-Etheve, N.; Pontier, D.; Lahmy, S.; Picart, C.; Vega, D.; Cooke, R.; Lagrange, T. RNA-directed DNA methylation requires an AGO4-interacting member of the SPT5 elongation factor family. EMBO Rep. 2009, 10, 649–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Ji, L.; Huang, Q.; Vassylyev, D.G.; Chen, X.; Ma, J.B. Structural insights into mechanisms of the small RNA methyltransferase HEN1. Nature 2009, 461, 823–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Palanca, A.M.S.; Law, J.A. Locus-specific control of the de novo DNA methylation pathway in Arabidopsis by the CLASSY family. Nat. Genet. 2018, 50, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Goel, S.; Meeley, R.B.; Dantec, C.; Parrinello, H.; Michaud, C.; Leblanc, O.; Grimanelli, D. Production of viable gametes without meiosis in maize deficient for an ARGONAUTE protein. Plant Cell 2011, 23, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.M.; Nakamura, M.; Calarco, J.P.; Susaki, D.; Lim, P.Q.; Kinoshita, T.; Higashiyama, T.; Martienssen, R.A.; Berger, F. RNA-directed DNA methylation regulates parental genomic imprinting at several loci in Arabidopsis. Development 2013, 140, 2953–2960. [Google Scholar] [CrossRef] [PubMed]
- Manning, K.; Tor, M.; Poole, M.; Hong, Y.; Thompson, A.J.; King, G.J.; Giovannoni, J.J.; Seymour, G.B. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 2006, 38, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, K.J.; Jansen, J.J.; van Dijk, P.J.; Biere, A. Stress-induced DNA methylation changes and their heritability in asexual dandelions. New Phytol. 2010, 185, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Suter, L.; Widmer, A. Environmental heat and salt stress induce transgenerational phenotypic changes in Arabidopsis thaliana. PLoS ONE 2013, 8, e60364. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Coleman-Derr, D.; Ballinger, T.; Henikoff, S. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature 2008, 456, 125–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seymour, D.K.; Koenig, D.; Hagmann, J.; Becker, C.; Weigel, D. Evolution of DNA methylation patterns in the Brassicaceae is driven by differences in genome organization. PLoS Genet. 2014, 10, e1004785. [Google Scholar] [CrossRef] [PubMed]
- Keller, T.E.; Yi, S.V. DNA methylation and evolution of duplicate genes. Proc. Natl. Acad. Sci. USA 2014, 111, 5932–5937. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.-F.; An, Y.-Q.C.; et al. Dynamic DNA Methylation in Plant Growth and Development. Int. J. Mol. Sci. 2018, 19, 2144. https://doi.org/10.3390/ijms19072144
Bartels A, Han Q, Nair P, Stacey L, Gaynier H, Mosley M, Huang QQ, Pearson JK, Hsieh T-F, An Y-QC, et al. Dynamic DNA Methylation in Plant Growth and Development. International Journal of Molecular Sciences. 2018; 19(7):2144. https://doi.org/10.3390/ijms19072144
Chicago/Turabian StyleBartels, Arthur, Qiang Han, Pooja Nair, Liam Stacey, Hannah Gaynier, Matthew Mosley, Qi Qing Huang, Jacob K. Pearson, Tzung-Fu Hsieh, Yong-Qiang Charles An, and et al. 2018. "Dynamic DNA Methylation in Plant Growth and Development" International Journal of Molecular Sciences 19, no. 7: 2144. https://doi.org/10.3390/ijms19072144
APA StyleBartels, A., Han, Q., Nair, P., Stacey, L., Gaynier, H., Mosley, M., Huang, Q. Q., Pearson, J. K., Hsieh, T. -F., An, Y. -Q. C., & Xiao, W. (2018). Dynamic DNA Methylation in Plant Growth and Development. International Journal of Molecular Sciences, 19(7), 2144. https://doi.org/10.3390/ijms19072144