Real-Time Imaging Reveals Augmentation of Glutamate-Induced Ca2+ Transients by the NO-cGMP Pathway in Cerebellar Granule Neurons

 ,
, 
Abstract
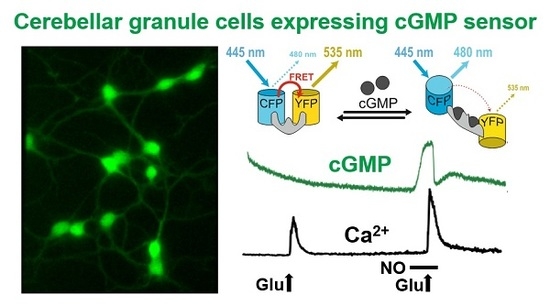
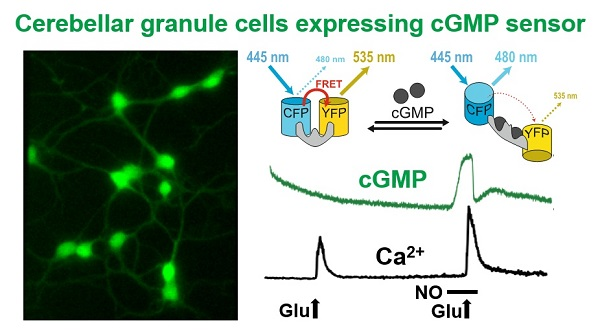
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
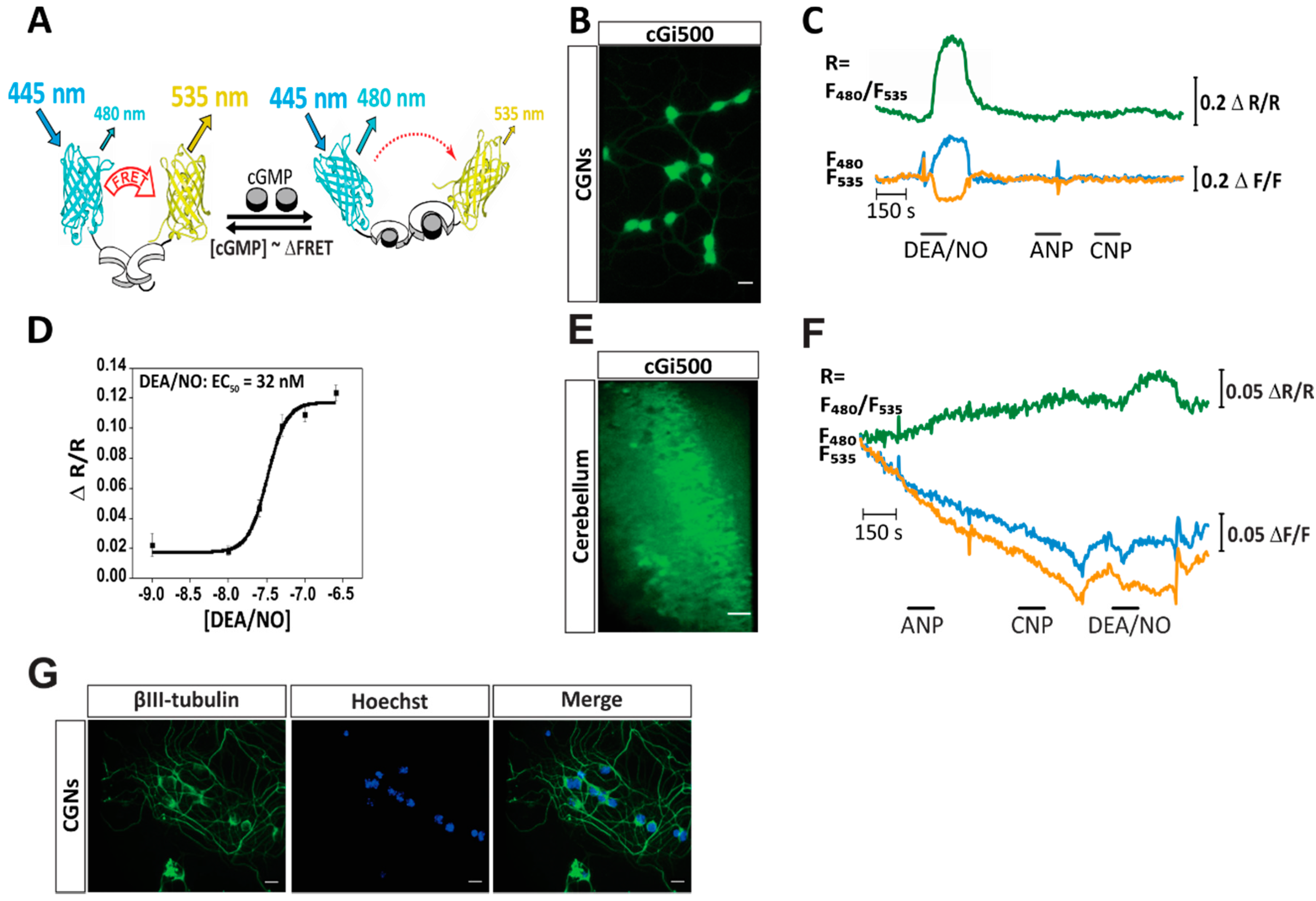
2.1. cGMP Imaging in CGNs and Acute Cerebellar Slices Reveals a NO-Induced cGMP Response
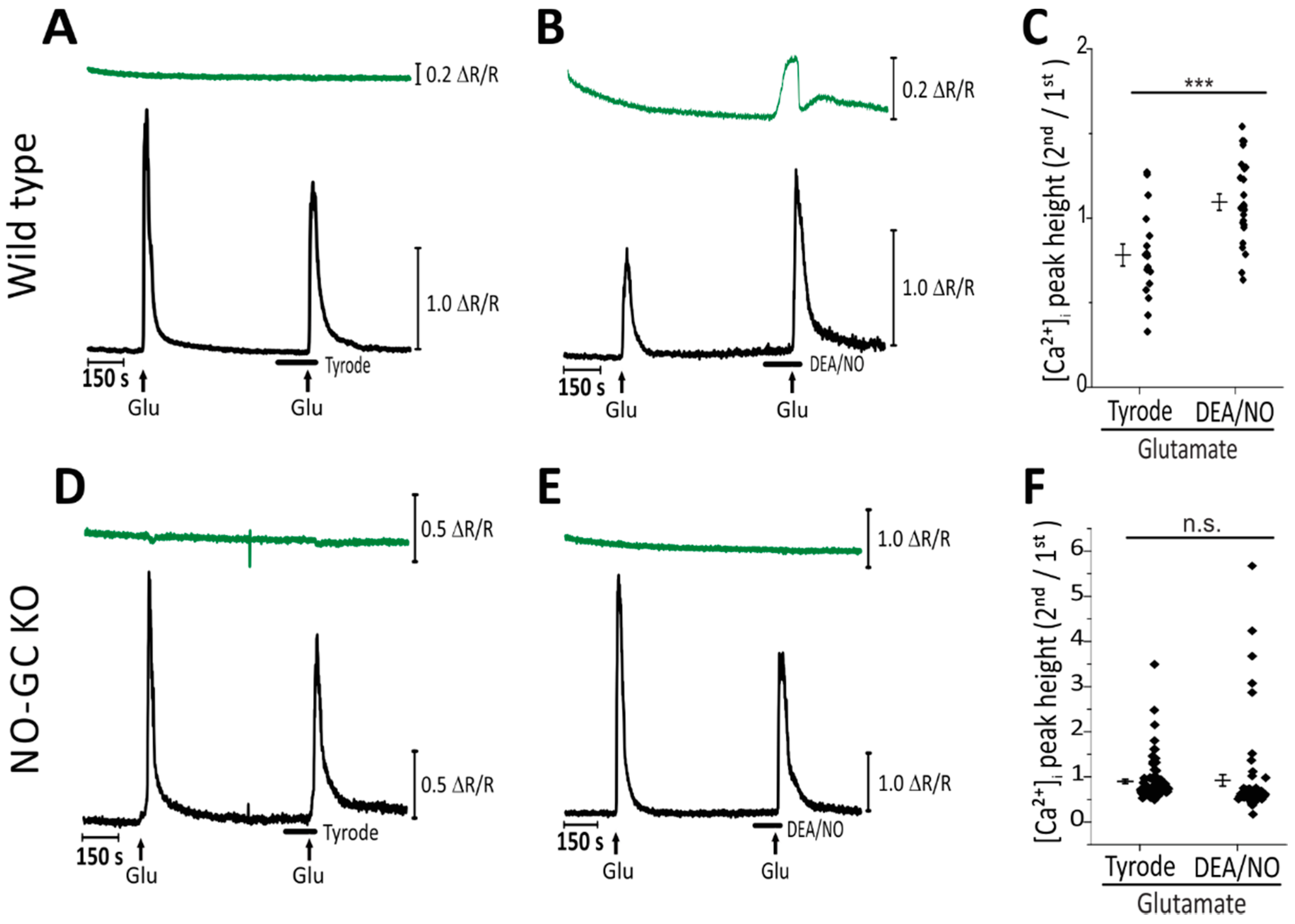
2.2. Simultaneous Imaging Implicates the NO-cGMP Pathway in Potentiation of Glutamate-Induced Ca2+ Transients in CGNs
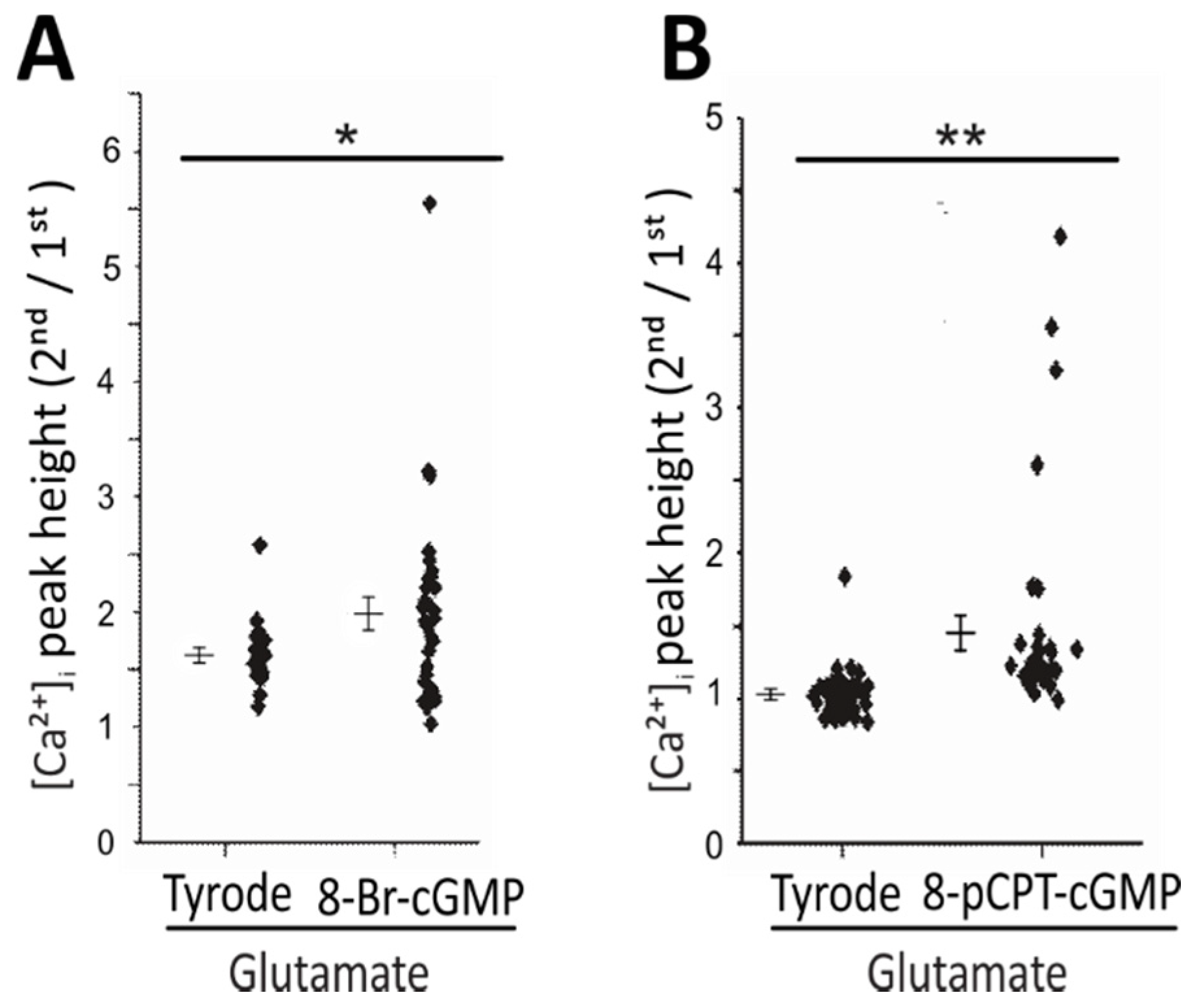
2.3. Use of cGMP Analogues Confirms cGMP-Dependent Increase of Glutamate-Induced Ca2+ Transients
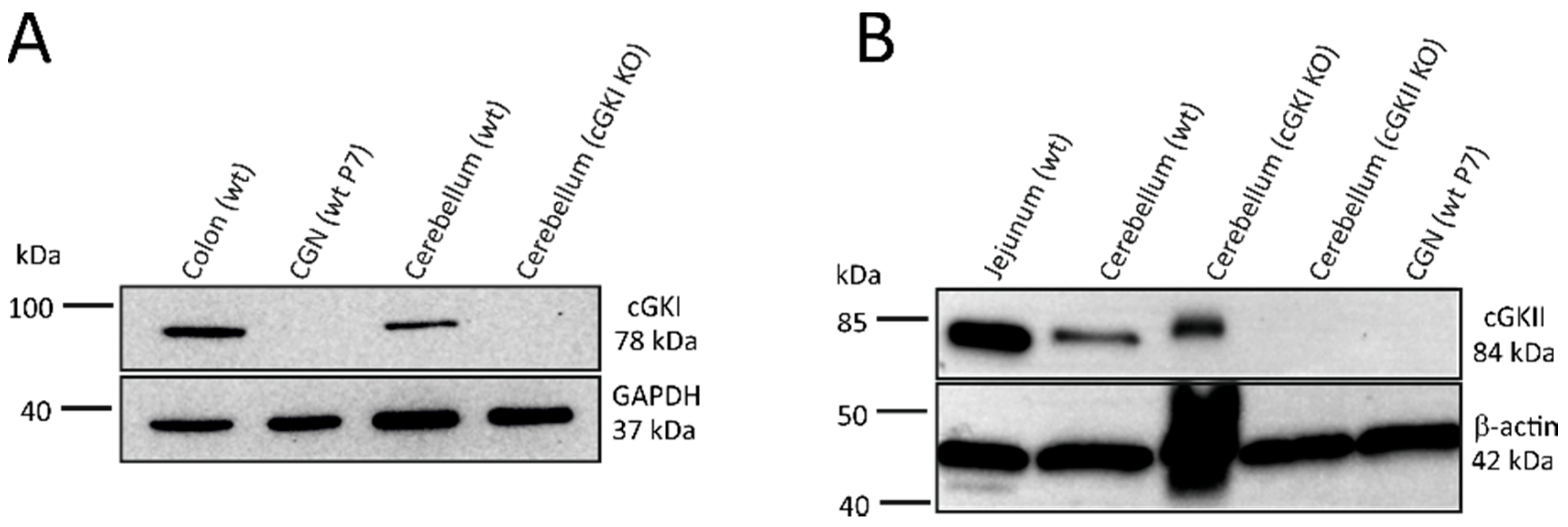
2.4. Western Blot Analysis Does Not Detect cGKI or cGKII Expression in CGNs
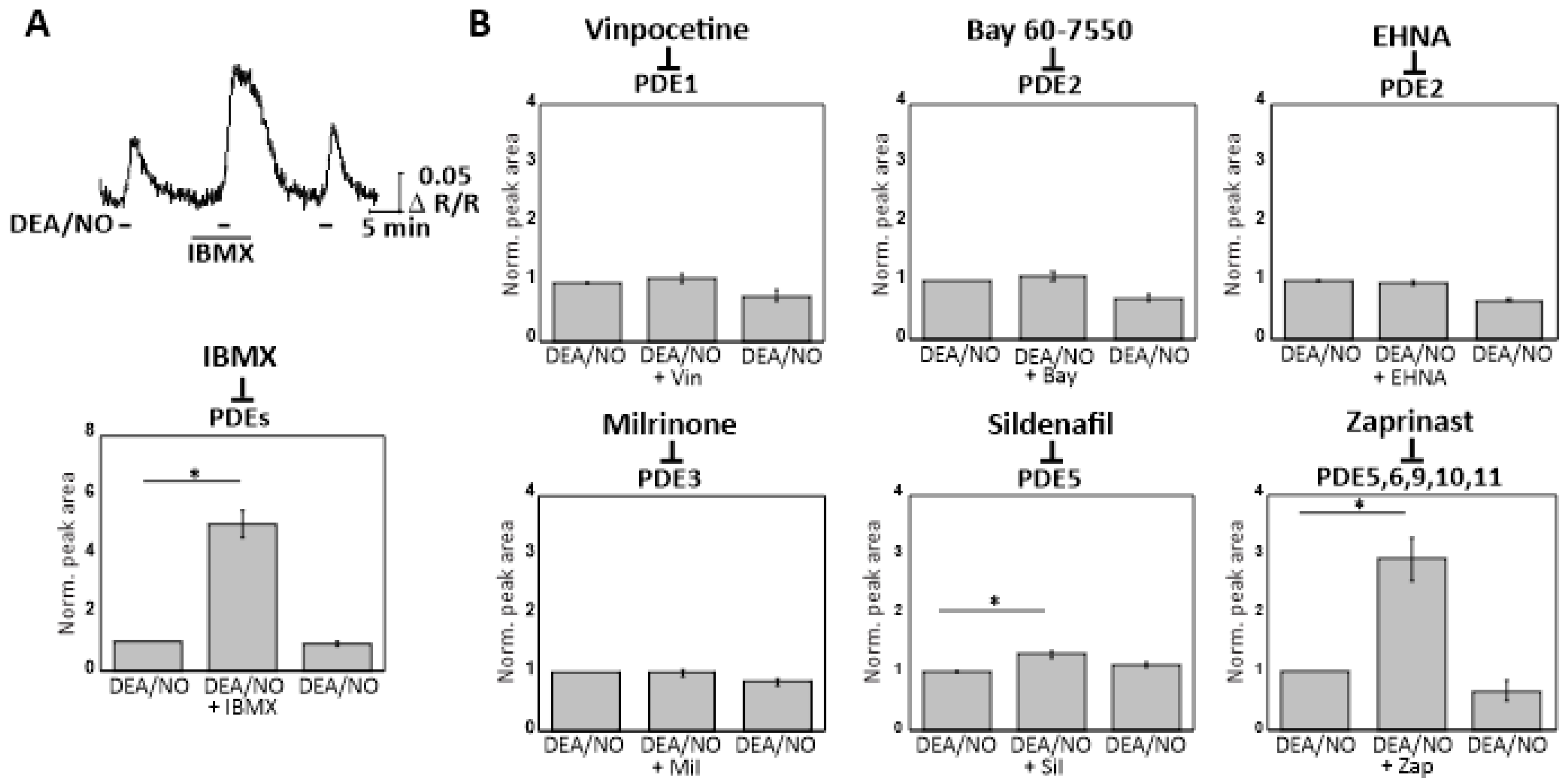
2.5. Real-Time cGMP Imaging in CGNs Reveals cGMP Degradation via Zaprinast-Sensitive PDEs
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Drugs
4.3. Cell Culture
4.4. Imaging
4.5. PDE Inhibitor Experiments
4.6. Protein Isolation and Western Blotting
4.7. Immunostaining of Cells
4.8. Data Analysis and Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANP | Atrial natriuretic peptide |
| BSA | Bovine serum albumin |
| [Ca2+]i | Intracellular Ca2+ concentration |
| CAG | CMV immediate early enhancer, chicken β-actin, and rabbit β-globin |
| CFP | Cyan fluorescent protein |
| cGi | cGMP indicator |
| cGK | cGMP-dependent protein kinase |
| cGMP | Cyclic 3′ 5′ guanosine monophosphate |
| CGN | Cerebellar granule neuron |
| CNG | Cyclic nucleotide gated |
| CNP | C-type natriuretic peptide |
| CNS | Central nervous system |
| DEA/NO | 2-(N,N-diethylamino)-diazenolate-2-oxide diethylammonium salt |
| FRET | Förster/Fluorescence resonance energy transfer |
| IBMX | 3-Isobutyl-1-methylxanthin |
| NMDA | N-methyl-d-aspartate |
| NO | Nitric oxide |
| NO-GC | NO-sensitive guanylyl cyclase |
| NO-GC KO | NO-sensitive guanylyl cyclase knockout |
| PBS | Phosphate buffered saline |
| PDE | Phosphodiesterase |
| YFP | Yellow fluorescent protein |
References
- Garthwaite, J.; Boulton, C. Nitric oxide signaling in the central nervous system. Annu. Rev. Physiol. 1995, 57, 683–706. [Google Scholar] [CrossRef] [PubMed]
- Kleppisch, T.; Feil, R. cGMP signalling in the mammalian brain: Role in synaptic plasticity and behaviour. Handb. Exp. Pharmacol. 2009, 191, 549–579. [Google Scholar] [CrossRef]
- Friebe, A.; Koesling, D. Regulation of nitric oxide-sensitive guanylyl cyclase. Circ. Res. 2003, 93, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Pyriochou, A.; Papapetropoulos, A. Soluble guanylyl cyclase: More secrets revealed. Cell Signal. 2005, 17, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Budworth, J.; Meillerais, S.; Charles, I.; Powell, K. Tissue distribution of human soluble guanylate cyclases. Biochem. Biophys. Res. Commun. 1999, 263, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. Molecular physiology of membrane guanylyl cyclase receptors. Physiol. Rev. 2016, 96, 751–804. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Lohmann, S.M.; de Jonge, H.; Walter, U.; Hofmann, F. Cyclic GMP-dependent protein kinases and the cardiovascular system: Insights from genetically modified mice. Circ. Res. 2003, 93, 907–916. [Google Scholar] [CrossRef] [PubMed]
- El-Husseini, A.; Bladen, C.; Vincent, S. Molecular characterization of a type II cyclic GMP-dependent protein kinase expressed in the rat brain. J. Neurochem. 1995, 64, 2814–2817. [Google Scholar] [CrossRef] [PubMed]
- Kleppisch, T.; Pfeifer, A.; Klatt, P.; Ruth, P.; Montkowski, A.; Fässler, R.; Hofmann, F. Long-term potentiation in the hippocampal CA1 region of mice lacking cGMP-dependent kinases is normal and susceptible to inhibition of nitric oxide synthase. J. Neurosci. 1999, 19, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Feil, S.; Zimmermann, P.; Knorn, A.; Brummer, S.; Schlossmann, J.; Hofmann, F.; Feil, R. Distribution of cGMP-dependent protein kinase type I and its isoforms in the mouse brain and retina. Neuroscience 2005, 135, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Zufall, F.; Shepherd, G.; Barnstable, C. Cyclic nucleotide gated channels as regulators of CNS development and plasticity. Curr. Opin. Neurobiol. 1997, 7, 404–412. [Google Scholar] [CrossRef]
- Biel, M.; Michalakis, S. Cyclic nucleotide-gated channels. Handb. Exp. Pharmacol. 2009, 191, 111–136. [Google Scholar] [CrossRef]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef] [PubMed]
- Brescia, M.; Zaccolo, M. Modulation of compartmentalised cyclic nucleotide signalling via local inhibition of phosphodiesterase activity. Int. J. Mol. Sci. 2016, 17, 1672. [Google Scholar] [CrossRef] [PubMed]
- Polito, M.; Klarenbeek, J.; Jalink, K.; Paupardin-Tritsch, D.; Vincent, P.; Castro, L.R. The NO/cGMP pathway inhibits transient cAMP signals through the activation of PDE2 in striatal neurons. Front. Cell. Neurosci. 2013, 7, 211. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, P.; Lacy, B.E.; Chey, W.D.; Lembo, A.J.; Kurtz, C.B.; Reasner, D.S.; Bochenek, W.; Tripp, K.; Currie, M.G.; Fox, S.M.; et al. Low-dose Linaclotide (72 mug) for chronic idiopathic constipation: A 12-week, tandomized, double-blind, placebo-controlled trial. Am. J. Gastroenterol. 2018, 113, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Kemp-Harper, B. cGMP signalling: From bench to bedside. Conference on cGMP generators, effectors and therapeutic implications. EMBO Rep. 2006, 7, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Oettrich, J.M.; Dao, V.T.; Frijhoff, J.; Kleikers, P.; Casas, A.I.; Hobbs, A.J.; Schmidt, H.H. Clinical relevance of cyclic GMP modulators: A translational success story of network pharmacology. Clin. Pharmacol. Ther. 2016, 99, 360–362. [Google Scholar] [CrossRef] [PubMed]
- Follmann, M.; Griebenow, N.; Hahn, M.G.; Hartung, I.; Mais, F.J.; Mittendorf, J.; Schafer, M.; Schirok, H.; Stasch, J.P.; Stoll, F.; et al. The chemistry and biology of soluble guanylate cyclase stimulators and activators. Angew. Chem. Int. Ed. Engl. 2013, 52, 9442–9462. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Hartmann, J.; Luo, C.; Wolfsgruber, W.; Schilling, K.; Feil, S.; Barski, J.J.; Meyer, M.; Konnerth, A.; De Zeeuw, C.I.; et al. Impairment of LTD and cerebellar learning by Purkinje cell-specific ablation of cGMP-dependent protein kinase I. J. Cell Biol. 2003, 163, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Jurado, S.; Sanchez-Prieto, J.; Torres, M. Elements of the nitric oxide/cGMP pathway expressed in cerebellar granule cells: Biochemical and functional characterisation. Neurochem. Int. 2004, 45, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Contestabile, A. Cerebellar granule cells as a model to study mechanisms of neuronal apoptosis or survival in vivo and in vitro. Cerebellum 2002, 1, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Garthwaite, J.; Garthwaite, G.; Hajós, F. Amino acid neurotoxicity: Relationship to neuronal depolarization in rat cerebellar slices. Neuroscience 1986, 18, 449–460. [Google Scholar] [CrossRef]
- Pearce, I.; Cambray-Deakin, M.; Burgoyne, R. Glutamate acting on NMDA receptors stimulates neurite outgrowth from cerebellar granule cells. FEBS Lett. 1987, 223, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Dickie, B.; Lewis, M.; Davies, J. NMDA-induced release of nitric oxide potentiates aspartate overflow from cerebellar slices. Neurosci. Lett. 1992, 138, 145–148. [Google Scholar] [CrossRef]
- Contestabile, A. Roles of NMDA receptor activity and nitric oxide production in brain development. Brain Res. Brain Res. Rev. 2000, 32, 476–509. [Google Scholar] [CrossRef]
- Thunemann, M.; Wen, L.; Hillenbrand, M.; Vachaviolos, A.; Feil, S.; Ott, T.; Han, X.; Fukumura, D.; Jain, R.K.; Russwurm, M.; et al. Transgenic mice for cGMP imaging. Circ. Res. 2013, 113, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Russwurm, M.; Mullershausen, F.; Friebe, A.; Jager, R.; Russwurm, C.; Koesling, D. Design of fluorescence resonance energy transfer (FRET)-based cGMP indicators: A systematic approach. Biochem. J. 2007, 407, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Peters, S.; Frank, K.; Wen, L.; Feil, R.; Rathjen, F.G. Dorsal root ganglion axon bifurcation tolerates increased cyclic GMP levels: The role of phosphodiesterase 2A and scavenger receptor Npr3. Eur. J. Neurosci. 2016, 44, 2991–3000. [Google Scholar] [CrossRef] [PubMed]
- Friebe, A.; Mergia, E.; Dangel, O.; Lange, A.; Koesling, D. Fatal gastrointestinal obstruction and hypertension in mice lacking nitric oxide-sensitive guanylyl cyclase. Proc. Natl. Acad. Sci. USA 2007, 104, 7699–7704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collado-Alsina, A.; Ramirez-Franco, J.; Sanchez-Prieto, J.; Torres, M. The regulation of synaptic vesicle recycling by cGMP-dependent protein kinase type II in cerebellar granule cells under strong and sustained stimulation. J. Neurosci. 2014, 34, 8788–8799. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Paolillo, M.; Mergia, E.; Koesling, D.; Kennel, L.; Schmidtko, A.; Russwurm, M.; Feil, R. cGMP imaging in brain slices reveals brain region-specific activity of NO-sensitive guanylyl cyclases (NO-GCs) and NO-GC stimulators. Int. J. Mol. Sci 2018, in press. [Google Scholar]
- Nonaka, S.; Hough, C.J.; Chuang, D.M. Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-d-aspartate receptor-mediated calcium influx. Proc. Natl. Acad. Sci. USA 1998, 95, 2642–2647. [Google Scholar] [CrossRef] [PubMed]
- Baltrons, M.A.; Saadoun, S.; Agullo, L.; García, A. Regulation by calcium of the nitric oxide/cyclic GMP system in cerebellar granule cells and astroglia in culture. J. Neurosci. Res. 1997, 49, 333–341. [Google Scholar] [CrossRef]
- Kouzoukas, D.E.; Bhalla, R.C.; Pantazis, N.J. Activation of cyclic GMP-dependent protein kinase blocks alcohol-mediated cell death and calcium disruption in cerebellar granule neurons. Neurosci. Lett. 2018, 676, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.; Strassmaier, T.; Brady, J.; Karpen, J. The pharmacology of cyclic nucleotide-gated channels: Emerging from the darkness. Curr. Pharm. Des. 2006, 12, 3597–3613. [Google Scholar] [CrossRef] [PubMed]
- Poppe, H.; Rybalkin, S.D.; Rehmann, H.; Hinds, T.R.; Tang, X.B.; Christensen, A.E.; Schwede, F.; Genieser, H.G.; Bos, J.L.; Doskeland, S.O.; et al. Cyclic nucleotide analogs as probes of signaling pathways. Nat. Methods 2008, 5, 277–278. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Jimenez, M.E.; Gonzalez, J.C.; Lizasoain, I.; Sanchez-Prieto, J.; Hernandez-Guijo, J.M.; Torres, M. Functional cGMP-gated channels in cerebellar granule cells. J. Cell. Physiol. 2012, 227, 2252–2263. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, R.; Gonzalez, D.; Valenzuela, C.; Brown, N.; Zuniga, L. Expression and cellular localization of HCN channels in rat cerebellar granule neurons. Biochem. Biophys. Res. Commun. 2016, 478, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Kleppisch, T.; Wolfsgruber, W.; Feil, S.; Allmann, R.; Wotjack, C.T.; Goebbels, S.; Nave, K.A.; Hofmann, F.; Feil, R. Hippocampal cGMP-dependent protein kinase I supports an age- and protein synthesis-dependent component of long-term potentiation but is not essential for spatial reference and contextual memory. J. Neurosci. 2003, 23, 6005–6012. [Google Scholar] [CrossRef] [PubMed]
- Langmesser, S.; Franken, P.; Feil, S.; Emmenegger, Y.; Albrecht, U.; Feil, R. cGMP-dependent protein kinase type I is implicated in the regulation of the timing and quality of sleep and wakefulness. PLoS ONE 2009, 4, e4238. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.M.; Gnugge, R.; Dhayade, S.; Thunemann, M.; Krippeit-Drews, P.; Drews, G.; Feil, R. H2O2 lowers the cytosolic Ca2+ concentration via activation of cGMP-dependent protein kinase Iα. Free Radic. Biol. Med. 2012, 53, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Feil, R. Functional reconstitution of vascular smooth muscle cells with cGMP-dependent protein kinase I isoforms. Circ. Res. 2002, 90, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, A.; Klatt, P.; Massberg, S.; Ny, L.; Sausbier, M.; Hirneiss, C.; Wang, G.X.; Korth, M.; Aszódi, A.; Andersson, K.E.; et al. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 1998, 17, 3045–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collado-Alsina, A.; Hofmann, F.; Sanchez-Prieto, J.; Torres, M. Altered synaptic membrane retrieval after strong stimulation of cerebellar granule neurons in cyclic GMP-dependent protein kinase II (cGKII) knockout mice. Int. J. Mol. Sci. 2017, 18, 2281. [Google Scholar] [CrossRef] [PubMed]
- Jurado, S.; Sanchez-Prieto, J.; Torres, M. Expression of cGMP-dependent protein kinases (I and II) and neuronal nitric oxide synthase in the developing rat cerebellum. Brain Res. Bull. 2005, 65, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.P. Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell Signal. 2018, 42, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Menniti, F.S.; Faraci, W.S.; Schmidt, C.J. Phosphodiesterases in the CNS: Targets for drug development. Nat. Rev. Drug Discov. 2006, 5, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Kleppisch, T. Phosphodiesterases in the central nervous system. Handb. Exp. Pharmacol. 2009, 191, 71–92. [Google Scholar] [CrossRef]
- van Staveren, W.C.; Glick, J.; Markerink-van Ittersum, M.; Shimizu, M.; Beavo, J.A.; Steinbusch, H.W.; de Vente, J. Cloning and localization of the cGMP-specific phosphodiesterase type 9 in the rat brain. J. Neurocytol. 2002, 31, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Irisarri, E.; Markerink-Van Ittersum, M.; Mengod, G.; de Vente, J. Expression of the cGMP-specific phosphodiesterases 2 and 9 in normal and Alzheimer’s disease human brains. Eur. J. Neurosci. 2007, 25, 3332–3338. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.; Lehner, T.; Luo, Y.; Loth, J.; Shao, W.; Sharpe, L.; Alexander, J.; Das, K.; Simon, R.; Fieve, R.; et al. A possible vulnerability locus for bipolar affective disorder on chromosome 21q22.3. Nat. Genet. 1994, 8, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Sano, H.; Nagai, Y.; Miyakawa, T.; Shigemoto, R.; Yokoi, M. Increased social interaction in mice deficient of the striatal medium spiny neuron-specific phosphodiesterase 10A2. J. Neurochem. 2008, 105, 546–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, C.J.; Chapin, D.S.; Cianfrogna, J.; Corman, M.L.; Hajos, M.; Harms, J.F.; Hoffman, W.E.; Lebel, L.A.; McCarthy, S.A.; Nelson, F.R.; et al. Preclinical characterization of selective phosphodiesterase 10A inhibitors: A new therapeutic approach to the treatment of schizophrenia. J. Pharmacol. Exp. Ther. 2008, 325, 681–690. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.L.; MacMullen, C.; Liu, D.J.; Leal, S.M.; Davis, R.L. Genetic association of cyclic AMP signaling genes with bipolar disorder. Transl. Psychiatry 2012, 2, e169. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Whelan, F.; Deloukas, P.; Whittaker, P.; Delgado, M.; Cantor, R.; McCann, S.; Licinio, J. Phosphodiesterase genes are associated with susceptibility to major depression and antidepressant treatment response. Proc. Natl. Acad. Sci. USA 2006, 103, 15124–15129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, M.P.; Adamowicz, W.; Bove, S.; Hartman, A.J.; Mariga, A.; Pathak, G.; Reinhart, V.; Romegialli, A.; Kleiman, R.J. Select 3′,5′-cyclic nucleotide phosphodiesterases exhibit altered expression in the aged rodent brain. Cell Signal. 2014, 26, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.S.; Klett, J.; Pilarzyk, K.; Lee, D.I.; Kass, D.; Menniti, F.S.; Kelly, M.P. Identification of new PDE9A isoforms and how their expression and subcellular compartmentalization in the brain change across the life span. Neurobiol. Aging 2018, 65, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.S.; Hamdani, N.; Holewinski, R.; Jo, S.H.; Danner, T.; Zhang, M.; Rainer, P.P.; et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelly, M.; Lim, B.K.; Cancedda, L.; Heilshorn, S.C.; Gao, H.; Poo, M.M. Local and long-range reciprocal regulation of cAMP and cGMP in axon/dendrite formation. Science 2010, 327, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Pietrobon, M.; Zamparo, I.; Maritan, M.; Franchi, S.A.; Pozzan, T.; Lodovichi, C. Interplay among cGMP, cAMP, and Ca2+ in living olfactory sensory neurons in vitro and in vivo. J. Neurosci. 2011, 31, 8395–8405. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Nagase, F.; Hotta, K.; Oka, K. Crosstalk between second messengers predicts the motility of the growth cone. Sci. Rep. 2013, 3, 3118. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Fukuda, T.; Tojima, T.; Nikolaev, V.O.; Kamiguchi, H. Cyclic nucleotide control of microtubule dynamics for axon guidance. J. Neurosci. 2016, 36, 5636–5649. [Google Scholar] [CrossRef] [PubMed]
- Thunemann, M.; Fomin, N.; Krawutschke, C.; Russwurm, M.; Feil, R. Visualization of cGMP with cGi biosensors. Methods Mol. Biol. 2013, 1020, 89–120. [Google Scholar] [CrossRef] [PubMed]
- Valtcheva, N.; Nestorov, P.; Beck, A.; Russwurm, M.; Hillenbrand, M.; Weinmeister, P.; Feil, R. The commonly used cGMP-dependent protein kinase type I (cGKI) inhibitor Rp-8-Br-PET-cGMPS can activate cGKI in vitro and in intact cells. J. Biol. Chem. 2009, 284, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Schramm, A.; Schinner, E.; Huettner, J.P.; Kees, F.; Tauber, P.; Hofmann, F.; Schlossmann, J. Function of cGMP-dependent protein kinase II in volume load-induced diuresis. Pflugers Arch. 2014, 466, 2009–2018. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paolillo, M.; Peters, S.; Schramm, A.; Schlossmann, J.; Feil, R. Real-Time Imaging Reveals Augmentation of Glutamate-Induced Ca2+ Transients by the NO-cGMP Pathway in Cerebellar Granule Neurons. Int. J. Mol. Sci. 2018, 19, 2185. https://doi.org/10.3390/ijms19082185
Paolillo M, Peters S, Schramm A, Schlossmann J, Feil R. Real-Time Imaging Reveals Augmentation of Glutamate-Induced Ca2+ Transients by the NO-cGMP Pathway in Cerebellar Granule Neurons. International Journal of Molecular Sciences. 2018; 19(8):2185. https://doi.org/10.3390/ijms19082185
Chicago/Turabian StylePaolillo, Michael, Stefanie Peters, Andrea Schramm, Jens Schlossmann, and Robert Feil. 2018. "Real-Time Imaging Reveals Augmentation of Glutamate-Induced Ca2+ Transients by the NO-cGMP Pathway in Cerebellar Granule Neurons" International Journal of Molecular Sciences 19, no. 8: 2185. https://doi.org/10.3390/ijms19082185
APA StylePaolillo, M., Peters, S., Schramm, A., Schlossmann, J., & Feil, R. (2018). Real-Time Imaging Reveals Augmentation of Glutamate-Induced Ca2+ Transients by the NO-cGMP Pathway in Cerebellar Granule Neurons. International Journal of Molecular Sciences, 19(8), 2185. https://doi.org/10.3390/ijms19082185