Myeloid-Derived Suppressor Cells and Pulmonary Hypertension


Abstract
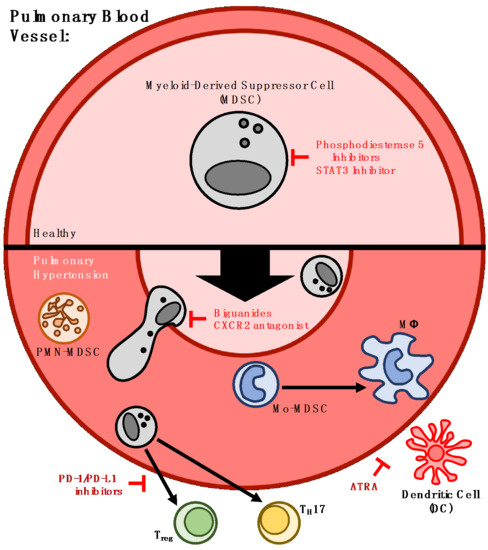
:
1. Introduction
2. Pulmonary Hypertension and Myeloid Cell Disorders
2.1. Stem Cell Transplantation and Pulmonary Hypertension
2.2. Myelodysplastic Syndromes and Pulmonary Hypertension
2.3. Abnormalities in Myeloid Cells in Pulmonary Hypertension
3. Myeloid-Derived Suppressor Cells and Pulmonary Hypertension
3.1. Molecular Mechanisms
3.1.1. C–X–C Motif Chemokine Receptor Type 2 (CXCR2)
3.1.2. Arginase–1 (Arg1)
3.1.3. Inducible Nitric Oxide Synthase (iNOS)
3.1.4. Indoleamine-Pyrrole 2,3-Dioxygenase (IDO)
3.1.5. Signal Transducer and Activator of Transcription 3 (STAT3)
3.1.6. Hypoxia-Inducible Factor (HIF)
3.2. Cellular Mechanisms
3.2.1. Dendritic Cells (DCs)
3.2.2. Macrophages
3.2.3. Regulatory T Cells (Treg) and T helper 17 Cells (TH17)
3.3. MDSCs and Metabolism
4. Strategies for Therapeutic Targeting of MDSCs in Pulmonary Hypertension
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CXCR2 | C-X-C motif chemokine receptor type 2 |
| IL–8 | Interleukin 8 |
| PD-1 | Programmed cell death protein-1 |
| PD-L1 | Programmed death-ligand 1 |
| Arg1 | Arginase-1 |
| iNOS | Inducible nitric oxide synthase |
| IDO | Indoleamine-pyrrole 2,3-dioxygenase |
| HIF | Hypoxia-inducible factor |
| STAT3 | Signal transducer and activator of transcription 3 |
| PMN-MDSC | Polymorphonuclear myeloid-derived suppressor cell |
| Mo-MDSC | Monocytic myeloid-derived suppressor cell |
| DC | Dendritic cell |
| Mϕ | Macrophage |
| Treg | Regulatory T cell |
| TH17 | T helper 17 cell |
| VEGF | Vascular endothelial growth factor |
| IL-10 | Interleukin 10 |
| IL-6 | Interleukin 6 |
| IL-23 | Interleukin 23 |
| TGF-β | Transforming growth factor beta |
References
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Rieber, N.; Brand, A.; Hector, A.; Graepler-Mainka, U.; Ost, M.; Schafer, I.; Wecker, I.; Neri, D.; Wirth, A.; Mays, L.; et al. Flagellin induces myeloid-derived suppressor cells: Implications for Pseudomonas aeruginosa infection in cystic fibrosis lung disease. J. Immunol. 2013, 190, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Rieber, N.; Singh, A.; Oz, H.; Carevic, M.; Bouzani, M.; Amich, J.; Ost, M.; Ye, Z.; Ballbach, M.; Schafer, I.; et al. Pathogenic fungi regulate immunity by inducing neutrophilic myeloid-derived suppressor cells. Cell Host Microbe 2015, 17, 507–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du Plessis, N.; Loebenberg, L.; Kriel, M.; von Groote-Bidlingmaier, F.; Ribechini, E.; Loxton, A.G.; van Helden, P.D.; Lutz, M.B.; Walzl, G. Increased frequency of myeloid-derived suppressor cells during active tuberculosis and after recent mycobacterium tuberculosis infection suppresses T.–cell function. Am. J. Respir. Crit. Care Med. 2013, 188, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Knaul, J.K.; Jorg, S.; Oberbeck-Mueller, D.; Heinemann, E.; Scheuermann, L.; Brinkmann, V.; Mollenkopf, H.J.; Yeremeev, V.; Kaufmann, S.H.; Dorhoi, A. Lung-residing myeloid-derived suppressors display dual functionality in murine pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lei, G.S.; Shao, S.; Jung, H.W.; Durant, P.J.; Lee, C.H. Accumulation of myeloid-derived suppressor cells in the lungs during Pneumocystis pneumonia. Infect. Immun. 2012, 80, 3634–3641. [Google Scholar] [CrossRef] [PubMed]
- De Santo, C.; Salio, M.; Masri, S.H.; Lee, L.Y.; Dong, T.; Speak, A.O.; Porubsky, S.; Booth, S.; Veerapen, N.; Besra, G.S.; et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J. Clin. Investing. 2008, 118, 4036–4048. [Google Scholar] [CrossRef] [PubMed]
- Kolahian, S.; Oz, H.H.; Zhou, B.; Griessinger, C.M.; Rieber, N.; Hartl, D. The emerging role of myeloid-derived suppressor cells in lung diseases. Eur. Respir. J. 2016, 47, 967–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeager, M.E.; Nguyen, C.M.; Belchenko, D.D.; Colvin, K.L.; Takatsuki, S.; Ivy, D.D.; Stenmark, K.R. Circulating myeloid-derived suppressor cells are increased and activated in pulmonary hypertension. Chest 2012, 141, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Bryant, A.J.; Shenoy, V.; Fu, C.; Marek, G.; Lorentsen, K.J.; Herzog, E.L.; Brantly, M.L.; Avram, D.; Scott, E.W. Myeloid-derived Suppressor Cells are Necessary for Development of Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2017, 58, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 121–150. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Shi, L.; Venkataramani, M.; Abdelaal, A.M.; Culpepper, C.; Kidder, K.; Liang, H.; Zen, K.; Liu, Y. Tumor conditions induce bone marrow expansion of granulocytic, but not monocytic, immunosuppressive leukocytes with increased CXCR2 expression in mice. Eur. J. Immunol. 2018, 48, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Mishalian, I.; Singhal, S.; Cheng, G.; Kapoor, V.; Horng, W.; Fridlender, G.; Bayuh, R.; Worthen, G.S. Transcriptomic analysis comparing tumor-associated neutrophils with granulocytic myeloid-derived suppressor cells and normal neutrophils. PLoS ONE 2012, 7, e31524. [Google Scholar] [CrossRef] [PubMed]
- Donkor, M.K.; Lahue, E.; Hoke, T.A.; Shafer, L.R.; Coskun, U.; Solheim, J.C.; Gulen, D.; Bishay, J.; Talmadge, J.E. Mammary tumor heterogeneity in the expansion of myeloid-derived suppressor cells. Int. Immunopharmacol. 2009, 9, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Seguchi, M.; Hirabayashi, N.; Fujii, Y.; Azuno, Y.; Fujita, N.; Takeda, K.; Sato, Y.; Nishimura, M.; Yamada, K.; Oka, Y. Pulmonary hypertension associated with pulmonary occlusive vasculopathy after allogeneic bone marrow transplantation. Transplantation 2000, 69, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Pate, A.; Rotz, S.; Warren, M.; Hirsch, R.; Cash, M.; Myers, K.C.; El-Bietar, J.; Nelson, A.; Wallace, G.; Filipovich, A.H.; et al. Pulmonary hypertension associated with bronchiolitis obliterans after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2016, 51, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Mattei, D.; Feola, M.; Orzan, F.; Mordini, N.; Rapezzi, D.; Gallamini, A. Reversible dasatinib-induced pulmonary arterial hypertension and right ventricle failure in a previously allografted CML patient. Bone Marrow Transplant. 2009, 43, 967–968. [Google Scholar] [CrossRef] [PubMed]
- Binks, M.; Passweg, J.R.; Furst, D.; McSweeney, P.; Sullivan, K.; Besenthal, C.; Finke, J.; Peter, H.H.; van Laar, J.; Breedveld, F.C.; et al. Phase I/II trial of autologous stem cell transplantation in systemic sclerosis: Procedure related mortality and impact on skin disease. Ann. Rheum. Dis. 2001, 60, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Vonk, M.C.; Marjanovic, Z.; van den Hoogen, F.H.; Zohar, S.; Schattenberg, A.V.; Fibbe, W.E.; Larghero, J.; Gluckman, E.; Preijers, F.W.; van Dijk, A.P.; et al. Long-term follow-up results after autologous haematopoietic stem cell transplantation for severe systemic sclerosis. Ann. Rheum. Dis. 2008, 67, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.M.; Goldmuntz, E.A.; Keyes-Elstein, L.; McSweeney, P.A.; Pinckney, A.; Welch, B.; Mayes, M.D.; Nash, R.A.; Crofford, L.J.; Eggleston, B.; et al. Myeloablative Autologous Stem-Cell Transplantation for Severe Scleroderma. N. Engl. J. Med. 2018, 378, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.; Oyama, Y.; Statkute, L.; Traynor, A.; Satkus, J.; Quigley, K.; Yaung, K.; Barr, W.; Bucha, J.; Gheorghiade, M.; et al. Autologous hematopoietic stem cell transplantation in systemic lupus erythematosus patients with cardiac dysfunction: Feasibility and reversibility of ventricular and valvular dysfunction with transplant-induced remission. Bone Marrow Transplant 2007, 40, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Traynor, A.E.; Corbridge, T.C.; Eagan, A.E.; Barr, W.G.; Liu, Q.; Oyama, Y.; Burt, R.K. Prevalence and reversibility of pulmonary dysfunction in refractory systemic lupus: Improvement correlates with disease remission following hematopoietic stem cell transplantation. Chest 2005, 127, 1680–1689. [Google Scholar] [CrossRef] [PubMed]
- Pittman, C.; Hsieh, M.M.; Coles, W.; Tisdale, J.F.; Weir, N.A.; Fitzhugh, C.D. Reversal of pre-capillary pulmonary hypertension in a patient with sickle cell anemia who underwent haploidentical peripheral blood stem cell transplantation. Bone Marrow Transplant 2017, 52, 641–642. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.D.; Courtman, D.W.; Deng, Y.; Kugathasan, L.; Zhang, Q.; Stewart, D.J. Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: Efficacy of combined cell and eNOS gene therapy in established disease. Circ. Res. 2005, 96, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Kanki-Horimoto, S.; Horimoto, H.; Mieno, S.; Kishida, K.; Watanabe, F.; Furuya, E.; Katsumata, T. Implantation of mesenchymal stem cells overexpressing endothelial nitric oxide synthase improves right ventricular impairments caused by pulmonary hypertension. Circulation 2006, 114, I181–1185. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Kagaya, Y.; Nakano, M.; Ito, Y.; Ohta, J.; Tada, H.; Karibe, A.; Minegishi, N.; Suzuki, N.; Yamamoto, M.; et al. Important role of endogenous erythropoietin system in recruitment of endothelial progenitor cells in hypoxia-induced pulmonary hypertension in mice. Circulation 2006, 113, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Raoul, W.; Wagner-Ballon, O.; Saber, G.; Hulin, A.; Marcos, E.; Giraudier, S.; Vainchenker, W.; Adnot, S.; Eddahibi, S.; Maitre, B. Effects of bone marrow-derived cells on monocrotaline- and hypoxia-induced pulmonary hypertension in mice. Respir. Res. 2007, 8, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Sahara, M.; Sata, M.; Morita, T.; Nakamura, K.; Hirata, Y.; Nagai, R. Diverse contribution of bone marrow-derived cells to vascular remodeling associated with pulmonary arterial hypertension and arterial neointimal formation. Circulation 2007, 115, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Aliotta, J.M.; Keaney, P.J.; Warburton, R.R.; DelTatto, M.; Dooner, M.S.; Passero, M.A.; Quesenberry, P.J.; Klinger, J.R. Marrow cell infusion attenuates vascular remodeling in a murine model of monocrotaline-induced pulmonary hypertension. Stem Cells Dev. 2009, 18, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Umar, S.; de Visser, Y.P.; Steendijk, P.; Schutte, C.I.; Laghmani el, H.; Wagenaar, G.T.; Bax, W.H.; Mantikou, E.; Pijnappels, D.A.; Atsma, D.E.; et al. Allogenic stem cell therapy improves right ventricular function by improving lung pathology in rats with pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1606–H1616. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Zhang, Z.H.; Wei, D.E.; Lu, Y.; Wang, Y.B. Effects of autologous bone marrow mononuclear cells implantation in canine model of pulmonary hypertension. Circ. J. 2012, 76, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Chen, X.; Talati, M.; Nunley, B.W.; Gladson, S.; Blackwell, T.; Cogan, J.; Austin, E.; Wheeler, F.; Loyd, J.; et al. Bone Marrow-derived Cells Contribute to the Pathogenesis of Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2016, 193, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Asosingh, K.; Wanner, N.; Weiss, K.; Queisser, K.; Gebreab, L.; Kassa, B.; Stuehr, E.; Graham, B.; Erzurum, S. Bone marrow transplantation prevents right ventricle disease in the caveolin-1-deficient mouse model of pulmonary hypertension. Blood. Adv. 2017, 1, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Dingli, D.; Utz, J.P.; Krowka, M.J.; Oberg, A.L.; Tefferi, A. Unexplained pulmonary hypertension in chronic myeloproliferative disorders. Chest 2001, 120, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Garypidou, V.; Vakalopoulou, S.; Dimitriadis, D.; Tziomalos, K.; Sfikas, G.; Perifanis, V. Incidence of pulmonary hypertension in patients with chronic myeloproliferative disorders. Haematologica 2004, 89, 245–246. [Google Scholar] [PubMed]
- Garcia-Manero, G.; Schuster, S.J.; Patrick, H.; Martinez, J. Pulmonary hypertension in patients with myelofibrosis secondary to myeloproliferative diseases. Am. J. Hematol. 1999, 60, 130–135. [Google Scholar] [CrossRef] [Green Version]
- Roach, E.C.; Park, M.M.; Tang, W.H.; Thomas, J.D.; Asosingh, K.; Kalaycio, M.; Erzurum, S.C.; Farha, S. Impaired right ventricular-pulmonary vascular function in myeloproliferative neoplasms. J. Heart Lung Transplant 2015, 34, 390–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattar, M.M.; Morad, M.A.; El Husseiny, N.M.; Ali, N.H.; El Demerdash, D.M. Correlation between JAK2 allele burden and pulmonary arterial hypertension and hematological parameters in Philadelphia negative JAK2 positive myeloproliferative neoplasms. An Egyptian experience. Ann. Hematol. 2016, 95, 1611–1616. [Google Scholar] [CrossRef] [PubMed]
- Popat, U.; Frost, A.; Liu, E.; Guan, Y.; Durette, A.; Reddy, V.; Prchal, J.T. High levels of circulating CD34 cells, dacrocytes, clonal hematopoiesis, and JAK2 mutation differentiate myelofibrosis with myeloid metaplasia from secondary myelofibrosis associated with pulmonary hypertension. Blood 2006, 107, 3486–3488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faiz, S.A.; Iliescu, C.; Lopez-Mattei, J.; Patel, B.; Bashoura, L.; Popat, U. Resolution of myelofibrosis-associated pulmonary arterial hypertension following allogeneic hematopoietic stem cell transplantation. Pulm. Circ. 2016, 6, 611–613. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Huang, J.; Wu, J.M.; Fallon, J.T.; Gewitz, M.H. Hematological disorders and pulmonary hypertension. World J. Cardiol. 2016, 8, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Adir, Y.; Elia, D.; Harari, S. Pulmonary hypertension in patients with chronic myeloproliferative disorders. Eur. Respir. Rev. 2015, 24, 400–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adir, Y.; Humbert, M. Pulmonary hypertension in patients with chronic myeloproliferative disorders. Eur. Respir. J. 2010, 35, 1396–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raychaudhuri, B.; Bonfield, T.L.; Malur, A.; Hague, K.; Kavuru, M.S.; Arroliga, A.C.; Thomassen, M.J. Circulating monocytes from patients with primary pulmonary hypertension are hyporesponsive. Clin. Immunol. 2002, 104, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Rose, F.; Hattar, K.; Gakisch, S.; Grimminger, F.; Olschewski, H.; Seeger, W.; Tschuschner, A.; Schermuly, R.T.; Weissmann, N.; Hanze, J.; et al. Increased neutrophil mediator release in patients with pulmonary hypertension--suppression by inhaled iloprost. Thromb. Haemost. 2003, 90, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Asosingh, K.; Aldred, M.A.; Vasanji, A.; Drazba, J.; Sharp, J.; Farver, C.; Comhair, S.A.; Xu, W.; Licina, L.; Huang, L.; et al. Circulating angiogenic precursors in idiopathic pulmonary arterial hypertension. Am. J. Pathol. 2008, 172, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Farha, S.; Asosingh, K.; Xu, W.; Sharp, J.; George, D.; Comhair, S.; Park, M.; Tang, W.H.; Loyd, J.E.; Theil, K.; et al. Hypoxia-inducible factors in human pulmonary arterial hypertension: A link to the intrinsic myeloid abnormalities. Blood 2011, 117, 3485–3493. [Google Scholar] [CrossRef] [PubMed]
- Asosingh, K.; Farha, S.; Lichtin, A.; Graham, B.; George, D.; Aldred, M.; Hazen, S.L.; Loyd, J.; Tuder, R.; Erzurum, S.C. Pulmonary vascular disease in mice xenografted with human BM progenitors from patients with pulmonary arterial hypertension. Blood 2012, 120, 1218–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frid, M.G.; Brunetti, J.A.; Burke, D.L.; Carpenter, T.C.; Davie, N.J.; Reeves, J.T.; Roedersheimer, M.T.; van Rooijen, N.; Stenmark, K.R. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am. J. Pathol. 2006, 168, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Jiang, X.; Tamosiuniene, R.; Sung, Y.K.; Qian, J.; Dhillon, G.; Gera, L.; Farkas, L.; Rabinovitch, M.; Zamanian, R.T.; et al. Blocking macrophage leukotriene b4 prevents endothelial injury and reverses pulmonary hypertension. Sci. Transl. Med. 2013, 5, 117–200. [Google Scholar] [CrossRef] [PubMed]
- Sawada, H.; Saito, T.; Nickel, N.P.; Alastalo, T.P.; Glotzbach, J.P.; Chan, R.; Haghighat, L.; Fuchs, G.; Januszyk, M.; Cao, A.; et al. Reduced BMPR2 expression induces GM-CSF translation and macrophage recruitment in humans and mice to exacerbate pulmonary hypertension. J. Exp. Med. 2014, 211, 263–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto-Kataoka, T.; Hosen, N.; Sonobe, T.; Arita, Y.; Yasui, T.; Masaki, T.; Minami, M.; Inagaki, T.; Miyagawa, S.; Sawa, Y.; et al. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc. Natl. Acad. Sci. USA 2015, 112, E2677–E2686. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Mickael, C.; Chabon, J.; Gebreab, L.; Rutebemberwa, A.; Garcia, A.R.; Koyanagi, D.E.; Sanders, L.; Gandjeva, A.; Kearns, M.T.; et al. The Causal Role of IL-4 and IL-13 in Schistosoma mansoni Pulmonary Hypertension. Am. J. Respir. Crit. Care. Med. 2015, 192, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Kitaichi, T.; Urata, M.; Kurobe, H.; Kanbara, T.; Motoki, T.; Kitagawa, T. Syngeneic bone marrow mononuclear cells improve pulmonary arterial hypertension through vascular endothelial growth factor upregulation. Ann. Thorac. Surg. 2009, 88, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Ke, M.W.; Cheng, C.C.; Chiou, S.H.; Wann, S.R.; Shu, C.W.; Chiou, K.R.; Tseng, C.J.; Pan, H.W.; Mar, G.Y.; et al. Therapeutic Benefits of Induced Pluripotent Stem Cells in Monocrotaline-Induced Pulmonary Arterial Hypertension. PLoS ONE 2016, 11, e0142476. [Google Scholar] [CrossRef] [PubMed]
- Talati, M.; West, J.; Zaynagetdinov, R.; Hong, C.C.; Han, W.; Blackwell, T.; Robinson, L.; Blackwell, T.S.; Lane, K. BMP pathway regulation of and by macrophages. PLoS ONE 2014, 9, e94119. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.R.; Mao, L.; Piantadosi, C.A.; Gunn, M.D. CCR2 deficiency, dysregulation of Notch signaling, and spontaneous pulmonary arterial hypertension. Am. J Respir. Cell Mol. Biol. 2013, 48, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Harbaum, L.; Baaske, K.M.; Simon, M.; Oqueka, T.; Sinning, C.; Glatzel, A.; Luneburg, N.; Sydow, K.; Bokemeyer, C.; Klose, H. Exploratory analysis of the neutrophil to lymphocyte ratio in patients with pulmonary arterial hypertension. BMC. Pulm. Med. 2017, 17, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Aldabbous, L.; Abdul-Salam, V.; McKinnon, T.; Duluc, L.; Pepke-Zaba, J.; Southwood, M.; Ainscough, A.J.; Hadinnapola, C.; Wilkins, M.R.; Toshner, M.; et al. Neutrophil Extracellular Traps Promote Angiogenesis: Evidence from Vascular Pathology in Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.; Bryan, A.C. Pulmonary hypertension after postlavage lung injury in rabbits: Possible role of polymorphonuclear leukocytes. J. Appl. Physiol. 1991, 71, 1990–1995. [Google Scholar] [CrossRef] [PubMed]
- Perkett, E.A.; Brigham, K.L.; Meyrick, B. Granulocyte depletion attenuates sustained pulmonary hypertension and increased pulmonary vasoreactivity caused by continuous air embolization in sheep. Am. Rev. Respir. Dis. 1990, 141, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Burton, V.J.; Ciuclan, L.I.; Holmes, A.M.; Rodman, D.M.; Walker, C.; Budd, D.C. Bone morphogenetic protein receptor II regulates pulmonary artery endothelial cell barrier function. Blood 2011, 117, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Burton, V.J.; Holmes, A.M.; Ciuclan, L.I.; Robinson, A.; Roger, J.S.; Jarai, G.; Pearce, A.C.; Budd, D.C. Attenuation of leukocyte recruitment via CXCR1/2 inhibition stops the progression of PAH in mice with genetic ablation of endothelial BMPR-II. Blood 2011, 118, 4750–4758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, L.M.; Jandl, K.; Grunig, G.; Foris, V.; Bashir, M.; Ghanim, B.; Klepetko, W.; Olschewski, H.; Olschewski, A.; Kwapiszewska, G. The inflammatory cell landscape in the lungs of patients with idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Launay, J.M.; Herve, P.; Callebert, J.; Mallat, Z.; Collet, C.; Doly, S.; Belmer, A.; Diaz, S.L.; Hatia, S.; Cote, F.; et al. Serotonin 5-HT2B receptors are required for bone-marrow contribution to pulmonary arterial hypertension. Blood 2012, 119, 1772–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerasimovskaya, E.; Kratzer, A.; Sidiakova, A.; Salys, J.; Zamora, M.; Taraseviciene-Stewart, L. Interplay of macrophages and T cells in the lung vasculature. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L1014–L1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maston, L.D.; Jones, D.T.; Giermakowska, W.; Howard, T.A.; Cannon, J.L.; Wang, W.; Wei, Y.; Xuan, W.; Resta, T.C.; Gonzalez Bosc, L.V. Central role of T helper 17 cells in chronic hypoxia-induced pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L609–L624. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.; Baron, D.M.; Bloch, K.D.; Luster, A.D.; Lee, J.J.; Medoff, B.D. Eosinophils are necessary for pulmonary arterial remodeling in a mouse model of eosinophilic inflammation-induced pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 301, L927–L936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenmark, K.R.; Frid, M.G.; Yeager, M.E. Fibrocytes: Potential new therapeutic targets for pulmonary hypertension? Eur. Respir. J. 2010, 36, 1232–1235. [Google Scholar] [CrossRef] [PubMed]
- Ormiston, M.L.; Deng, Y.; Stewart, D.J.; Courtman, D.W. Innate immunity in the therapeutic actions of endothelial progenitor cells in pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2010, 43, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Avogaro, A.; Ferraccioli, G.; Agostini, C. Endothelial progenitors in pulmonary hypertension: New pathophysiology and therapeutic implications. Eur. Respir. J. 2010, 35, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Sata, M. Role of circulating vascular progenitors in angiogenesis, vascular healing, and pulmonary hypertension: Lessons from animal models. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Maxova, H.; Herget, J.; Vizek, M. Lung mast cells and hypoxic pulmonary hypertension. Physiol. Res. 2012, 61, 1–11. [Google Scholar] [PubMed]
- Fu, J.; Chen, Y.F.; Zhao, X.; Creighton, J.R.; Guo, Y.; Hage, F.G.; Oparil, S.; Xing, D.D. Targeted delivery of pulmonary arterial endothelial cells overexpressing interleukin-8 receptors attenuates monocrotaline-induced pulmonary vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Wang, D.; Daikoku, T.; Sun, H.; Dey, S.K.; Dubois, R.N. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 2013, 24, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2016, 29, 832–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Ye, Y.L.; Li, M.X.; Ye, S.B.; Huang, W.R.; Cai, T.T.; He, J.; Peng, J.Y.; Duan, T.H.; Cui, J.; et al. CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene 2017, 36, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Amsellem, V.; Abid, S.; Poupel, L.; Parpaleix, A.; Rodero, M.; Gary-Bobo, G.; Latiri, M.; Dubois-Rande, J.L.; Lipskaia, L.; Combadiere, C.; et al. Roles for the CX3CL1/CX3CR1 and CCL2/CCR2 Chemokine Systems in Hypoxic Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2017, 56, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Florentin, J.; Coppin, E.; Vasamsetti, S.B.; Zhao, J.; Tai, Y.Y.; Tang, Y.; Zhang, Y.; Watson, A.; Sembrat, J.; Rojas, M.; et al. Inflammatory Macrophage Expansion in Pulmonary Hypertension Depends upon Mobilization of Blood-Borne Monocytes. J. Immunol. 2018, 200, 3612–3625. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.C.; Wedes, S.H.; Hsu, J.W.; Bohren, K.M.; Comhair, S.A.; Jahoor, F.; Erzurum, S.C. Arginine metabolic endotypes in pulmonary arterial hypertension. Pulm. Circ. 2015, 5, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Cowburn, A.S.; Crosby, A.; Macias, D.; Branco, C.; Colaco, R.D.; Southwood, M.; Toshner, M.; Crotty Alexander, L.E.; Morrell, N.W.; Chilvers, E.R.; et al. HIF2alpha-arginase axis is essential for the development of pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2016, 113, 8801–8806. [Google Scholar] [CrossRef] [PubMed]
- Trittmann, J.K.; Jin, Y.; Chicoine, L.G.; Liu, Y.; Chen, B.; Nelin, L.D. An arginase-1 SNP that protects against the development of pulmonary hypertension in bronchopulmonary dysplasia enhances NO-mediated apoptosis in lymphocytes. Physiol. Rep. 2016, 4, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Grun, K.; Betge, S.; Pernow, J.; Kelm, M.; Muessig, J.; Masyuk, M.; Kuethe, F.; Ndongson-Dongmo, B.; Bauer, R.; et al. Arginase Inhibition Reverses Monocrotaline-Induced Pulmonary Hypertension. Int. J. Mol. Sci. 2017, 18, 1609. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Abdelaal, A.M.; Shi, L.; Liang, H.; Xiong, L.; Kidder, K.; Venkataramani, M.; Culpepper, C.; Zen, K.; Liu, Y. Arginase-1 is neither constitutively expressed in nor required for myeloid-derived suppressor cell-mediated inhibition of T.-cell proliferation. Eur. J. Immunol. 2018, 48, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Azzaoui, I.; Uhel, F.; Rossille, D.; Pangault, C.; Dulong, J.; Le Priol, J.; Lamy, T.; Houot, R.; Le Gouill, S.; Cartron, G.; et al. T-cell defect in diffuse large B-cell lymphomas involves expansion of myeloid-derived suppressor cells. Blood 2016, 128, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, X.; Shi, M.; Chen, Y.; Yu, D.; Zhao, C.; Gu, Y.; Yang, B.; Guo, S.; Ding, G.; et al. CD13(hi) Neutrophil-like myeloid-derived suppressor cells exert immune suppression through Arginase 1 expression in pancreatic ductal adenocarcinoma. Oncoimmunology 2017, 6, e1258504. [Google Scholar] [CrossRef] [PubMed]
- Kostlin, N.; Vogelmann, M.; Spring, B.; Schwarz, J.; Feucht, J.; Hartel, C.; Orlikowsky, T.W.; Poets, C.F.; Gille, C. Granulocytic myeloid-derived suppressor cells from human cord blood modulate T-helper cell response towards an anti-inflammatory phenotype. Immunology 2017, 152, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Zuckerbraun, B.S.; George, P.; Gladwin, M.T. Nitrite in pulmonary arterial hypertension: Therapeutic avenues in the setting of dysregulated arginine/nitric oxide synthase signalling. Cardiovasc. Res. 2011, 89, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, T.; Tsutsui, M.; Kido, T.; Sakanashi, M.; Naito, K.; Oda, K.; Ishimoto, H.; Yamada, S.; Wang, K.Y.; Toyohira, Y.; et al. Protective Role of Myelocytic Nitric Oxide Synthases Against Hypoxic Pulmonary Hypertension in Mice. Am. J. Respir. Crit. Care Med. 2018, 3, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Seimetz, M.; Parajuli, N.; Pichl, A.; Veit, F.; Kwapiszewska, G.; Weisel, F.C.; Milger, K.; Egemnazarov, B.; Turowska, A.; Fuchs, B.; et al. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell 2011, 147, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Haverkamp, J.M.; Crist, S.A.; Elzey, B.D.; Cimen, C.; Ratliff, T.L. In vivo suppressive function of myeloid-derived suppressor cells is limited to the inflammatory site. Eur. J. Immunol. 2011, 41, 749–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaraman, P.; Parikh, F.; Lopez-Rivera, E.; Hailemichael, Y.; Clark, A.; Ma, G.; Cannan, D.; Ramacher, M.; Kato, M.; Overwijk, W.W.; et al. Tumor-expressed inducible nitric oxide synthase controls induction of functional myeloid-derived suppressor cells through modulation of vascular endothelial growth factor release. J. Immunol. 2012, 188, 5365–5376. [Google Scholar] [CrossRef] [PubMed]
- Hampl, V.; Bibova, J.; Banasova, A.; Uhlik, J.; Mikova, D.; Hnilickova, O.; Lachmanova, V.; Herget, J. Pulmonary vascular iNOS induction participates in the onset of chronic hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L11–L20. [Google Scholar] [CrossRef] [PubMed]
- Mondanelli, G.; Bianchi, R.; Pallotta, M.T.; Orabona, C.; Albini, E.; Iacono, A.; Belladonna, M.L.; Vacca, C.; Fallarino, F.; Macchiarulo, A.; et al. A Relay Pathway between Arginine and Tryptophan Metabolism Confers Immunosuppressive Properties on Dendritic Cells. Immunity 2017, 46, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Christou, H.; Liu, L.; Visner, G.; Mitsialis, S.A.; Kourembanas, S.; Liu, H. Endothelial indoleamine 2,3-dioxygenase protects against development of pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2013, 188, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.D.; Ngo, D.; Hemnes, A.R.; Farrell, L.; Domos, C.; Pappagianopoulos, P.P.; Dhakal, B.P.; Souza, A.; Shi, X.; Pugh, M.E.; et al. Metabolic Profiling of Right Ventricular-Pulmonary Vascular Function Reveals Circulating Biomarkers of Pulmonary Hypertension. J. Am. Coll. Cardiol. 2016, 67, 174–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, B.M.; Nagaraj, C.; Meinitzer, A.; Sharma, N.; Papp, R.; Foris, V.; Ghanim, B.; Kwapiszewska, G.; Kovacs, G.; Klepetko, W.; et al. Importance of kynurenine in pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L741–L751. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wang, Y.; Yan, F.; Zhang, P.; Li, H.; Zhao, H.; Yan, C.; Yan, F.; Ren, X. Noncanonical NF-kappaB activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J. Immunol. 2014, 193, 2574–2586. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef] [PubMed]
- Jitschin, R.; Braun, M.; Buttner, M.; Dettmer-Wilde, K.; Bricks, J.; Berger, J.; Eckart, M.J.; Krause, S.W.; Oefner, P.J.; Le Blanc, K.; et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood 2014, 124, 750–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoso, A.; Mazza, E.M.; Bicciato, S.; Mandruzzato, S.; Bronte, V.; Serafini, P.; Inverardi, L. Human fibrocytic myeloid-derived suppressor cells express IDO and promote tolerance via Treg-cell expansion. Eur. J. Immunol. 2014, 44, 3307–3319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgaard, R.B.; Zamarin, D.; Li, Y.; Gasmi, B.; Munn, D.H.; Allison, J.P.; Merghoub, T.; Wolchok, J.D. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell. Rep. 2015, 13, 412–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulin, R.; Courboulin, A.; Meloche, J.; Mainguy, V.; Dumas de la Roque, E.; Saksouk, N.; Cote, J.; Provencher, S.; Sussman, M.A.; Bonnet, S. Signal transducers and activators of transcription-3/pim1 axis plays a critical role in the pathogenesis of human pulmonary arterial hypertension. Circulation 2011, 123, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Paulin, R.; Meloche, J.; Jacob, M.H.; Bisserier, M.; Courboulin, A.; Bonnet, S. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1798–H1809. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Si, C.; Zhang, Q.; Yan, F.; Li, C.; Zhang, H.; Ma, Q.; Dai, J.; Li, Z.; Shi, H.; et al. Autophagy regulates accumulation and functional activity of granulocytic myeloid-derived suppressor cells via STAT3 signaling in endotoxin shock. Biochim. Biophys. Acta 2017, 1863, 2796–2807. [Google Scholar] [CrossRef] [PubMed]
- Paulin, R.; Meloche, J.; Bonnet, S. STAT3 signaling in pulmonary arterial hypertension. Jakstat 2012, 1, 223–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Du, H.; Li, Y.; Qu, P.; Yan, C. Signal transducer and activator of transcription 3 (Stat3C) promotes myeloid-derived suppressor cell expansion and immune suppression during lung tumorigenesis. Am. J. Pathol. 2011, 179, 2131–2141. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.Y.; Shimoda, L.A.; Iyer, N.V.; Huso, D.L.; Sun, X.; McWilliams, R.; Beaty, T.; Sham, J.S.; Wiener, C.M.; Sylvester, J.T.; et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J. Clin. Investig. 1999, 103, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Brusselmans, K.; Compernolle, V.; Tjwa, M.; Wiesener, M.S.; Maxwell, P.H.; Collen, D.; Carmeliet, P. Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J. Clin. Investig. 2003, 111, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.Q.; Saddouk, F.Z.; Ntokou, A.; Mazurek, R.; Greif, D.M. Cell Autonomous and Non-cell Autonomous Regulation of SMC Progenitors in Pulmonary Hypertension. Cell Rep. 2018, 23, 1152–1165. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, P.; Gilkes, D.M.; Takano, N.; Semenza, G.L. Hypoxia-inducible factor-dependent signaling between triple-negative breast cancer cells and mesenchymal stem cells promotes macrophage recruitment. Proc. Natl. Acad. Sci. USA 2014, 111, E2120-9. [Google Scholar] [CrossRef] [PubMed]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Gabrilovich, D.I. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology 2014, 143, 512–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubillos-Zapata, C.; Avendano-Ortiz, J.; Hernandez-Jimenez, E.; Toledano, V.; Casas-Martin, J.; Varela-Serrano, A.; Torres, M.; Almendros, I.; Casitas, R.; Fernandez-Navarro, I.; et al. Hypoxia-induced PD-L1/PD-1 crosstalk impairs T-cell function in sleep apnoea. Eur. Respir. J. 2017, 50, 34–36. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T. cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Savai, R.; Pullamsetti, S.S.; Kolbe, J.; Bieniek, E.; Voswinckel, R.; Fink, L.; Scheed, A.; Ritter, C.; Dahal, B.K.; Vater, A.; et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Perros, F.; Dorfmuller, P.; Souza, R.; Durand-Gasselin, I.; Mussot, S.; Mazmanian, M.; Herve, P.; Emilie, D.; Simonneau, G.; Humbert, M. Dendritic cell recruitment in lesions of human and experimental pulmonary hypertension. Eur. Respir. J. 2007, 29, 462–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, D.L.; Frid, M.G.; Kunrath, C.L.; Karoor, V.; Anwar, A.; Wagner, B.D.; Strassheim, D.; Stenmark, K.R. Sustained hypoxia promotes the development of a pulmonary artery-specific chronic inflammatory microenvironment. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L238–L250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yan, H.; Zhu, W.; Cui, Y.; Chen, J.; Wang, X.; Li, S.; Zhu, J. Impairment of monocyte-derived dendritic cells in idiopathic pulmonary arterial hypertension. J. Clin. Immunol. 2009, 29, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Sendo, S.; Saegusa, J.; Okano, T.; Takahashi, S.; Akashi, K.; Morinobu, A. CD11b+Gr-1(dim) Tolerogenic Dendritic Cell-Like Cells Are Expanded in Interstitial Lung Disease in SKG Mice. Arthritis Rheumatol. 2017, 69, 2314–2327. [Google Scholar] [CrossRef] [PubMed]
- Albeituni, S.H.; Ding, C.; Liu, M.; Hu, X.; Luo, F.; Kloecker, G.; Bousamra, M., 2nd; Zhang, H.G.; Yan, J. Yeast-Derived Particulate beta-Glucan Treatment Subverts the Suppression of Myeloid-Derived Suppressor Cells (MDSC) by Inducing Polymorphonuclear MDSC Apoptosis and Monocytic MDSC Differentiation to APC in Cancer. J. Immunol. 2016, 196, 2167–2180. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, P.; Clements, V.K.; Fulton, A.M.; Ostrand-Rosenberg, S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007, 67, 4507–4513. [Google Scholar] [CrossRef] [PubMed]
- Poschke, I.; Mao, Y.; Adamson, L.; Salazar-Onfray, F.; Masucci, G.; Kiessling, R. Myeloid-derived suppressor cells impair the quality of dendritic cell vaccines. Cancer Immunol. Immunother. 2012, 61, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.E.; Gan, J.; Zhang, R.D.; Cheng, Y.R.; Huang, G.J. Up-regulated myeloid-derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scand. J. Gastroenterol. 2011, 46, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Pullamsetti, S.S.; Savai, R. Macrophage Regulation during Vascular Remodeling: Implications for Pulmonary Hypertension Therapy. Am. J. Respir. Cell Mol. Biol. 2017, 56, 556–558. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Groves, B.; Badesch, D.B.; Voelkel, N.F. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am. J. Pathol. 1994, 144, 275–285. [Google Scholar] [PubMed]
- Vergadi, E.; Chang, M.S.; Lee, C.; Liang, O.D.; Liu, X.; Fernandez-Gonzalez, A.; Mitsialis, S.A.; Kourembanas, S. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation 2011, 123, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- El Kasmi, K.C.; Pugliese, S.C.; Riddle, S.R.; Poth, J.M.; Anderson, A.L.; Frid, M.G.; Li, M.; Pullamsetti, S.S.; Savai, R.; Nagel, M.A.; et al. Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J. Immunol. 2014, 193, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Ugel, S.; De Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [PubMed]
- Savage, N.D.; de Boer, T.; Walburg, K.V.; Joosten, S.A.; van Meijgaarden, K.; Geluk, A.; Ottenhoff, T.H. Human anti-inflammatory macrophages induce Foxp3+ GITR+ CD25+ regulatory T. cells, which suppress via membrane-bound TGFbeta-1. J. Immunol. 2008, 181, 2220–2226. [Google Scholar] [CrossRef] [PubMed]
- Thibodeau, J.; Bourgeois-Daigneault, M.C.; Huppe, G.; Tremblay, J.; Aumont, A.; Houde, M.; Bartee, E.; Brunet, A.; Gauvreau, M.E.; de Gassart, A.; et al. Interleukin-10-induced MARCH1 mediates intracellular sequestration of MHC class II in monocytes. Eur. J. Immunol. 2008, 38, 1225–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, E.D.; Rock, M.T.; Mosse, C.A.; Vnencak-Jones, C.L.; Yoder, S.M.; Robbins, I.M.; Loyd, J.E.; Meyrick, B.O. T lymphocyte subset abnormalities in the blood and lung in pulmonary arterial hypertension. Respir. Med. 2010, 104, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Rabieyousefi, M.; Soroosh, P.; Satoh, K.; Date, F.; Ishii, N.; Yamashita, M.; Oka, M.; McMurtry, I.F.; Shimokawa, H.; Nose, M.; et al. Indispensable roles of OX40L-derived signal and epistatic genetic effect in immune-mediated pathogenesis of spontaneous pulmonary hypertension. BMC. Immunol. 2011, 12, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Huertas, A.; Tu, L.; Gambaryan, N.; Girerd, B.; Perros, F.; Montani, D.; Fabre, D.; Fadel, E.; Eddahibi, S.; Cohen-Kaminsky, S.; Guignabert, C. Leptin and regulatory T-lymphocytes in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2012, 40, 895–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrich, S.; Nicolls, M.R.; Taraseviciene, L.; Speich, R.; Voelkel, N. Increased regulatory and decreased CD8+ cytotoxic T cells in the blood of patients with idiopathic pulmonary arterial hypertension. Respiration 2008, 75, 272–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taraseviciene-Stewart, L.; Nicolls, M.R.; Kraskauskas, D.; Scerbavicius, R.; Burns, N.; Cool, C.; Wood, K.; Parr, J.E.; Boackle, S.A.; Voelkel, N.F. Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. Am. J. Respir. Crit. Care Med. 2007, 175, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Daley, E.; Emson, C.; Guignabert, C.; de Waal Malefyt, R.; Louten, J.; Kurup, V.P.; Hogaboam, C.; Taraseviciene-Stewart, L.; Voelkel, N.F.; Rabinovitch, M.; et al. Pulmonary arterial remodeling induced by a Th2 immune response. J. Exp. Med. 2008, 205, 361–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuttica, M.J.; Langenickel, T.; Noguchi, A.; Machado, R.F.; Gladwin, M.T.; Boehm, M. Perivascular T-cell infiltration leads to sustained pulmonary artery remodeling after endothelial cell damage. Am. J. Respir. Cell Mol. Biol. 2011, 45, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Tamosiuniene, R.; Tian, W.; Dhillon, G.; Wang, L.; Sung, Y.K.; Gera, L.; Patterson, A.J.; Agrawal, R.; Rabinovitch, M.; Ambler, K.; et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ. Res. 2011, 109, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Hautefort, A.; Girerd, B.; Montani, D.; Cohen-Kaminsky, S.; Price, L.; Lambrecht, B.N.; Humbert, M.; Perros, F. T-helper 17 cell polarization in pulmonary arterial hypertension. Chest 2015, 147, 1610–1620. [Google Scholar] [CrossRef] [PubMed]
- Jasiewicz, M.; Moniuszko, M.; Pawlak, D.; Knapp, M.; Rusak, M.; Kazimierczyk, R.; Musial, W.J.; Dabrowska, M.; Kaminski, K.A. Activity of the kynurenine pathway and its interplay with immunity in patients with pulmonary arterial hypertension. Heart 2016, 102, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Harbaum, L.; Oqueka, T.; Glatzel, A.; Hennigs, J.K.; Luneburg, N.; Klose, H. Does circulating IL-17 identify a subset of patients with idiopathic pulmonary arterial hypertension? Chest 2015, 148, 131–132. [Google Scholar] [CrossRef] [PubMed]
- Huertas, A.; Phan, C.; Bordenave, J.; Tu, L.; Thuillet, R.; Le Hiress, M.; Avouac, J.; Tamura, Y.; Allanore, Y.; Jovan, R.; et al. Regulatory T Cell Dysfunction in Idiopathic, Heritable and Connective Tissue-Associated Pulmonary Arterial Hypertension. Chest 2016, 149, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Hoechst, B.; Gamrekelashvili, J.; Manns, M.P.; Greten, T.F.; Korangy, F. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood 2011, 117, 6532–6541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, J.; Xu, J.; Zhao, S.; Liu, F.; Qi, J.; Song, Y.; Ren, J.; Wang, T.; Dou, H.; Hou, Y. Myeloid-derived suppressor cells contribute to systemic lupus erythaematosus by regulating differentiation of Th17 cells and Tregs. Clin. Sci. 2016, 130, 1453–1467. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhen, Y.; Ma, Z.; Li, H.; Yu, J.; Xu, Z.G.; Wang, X.Y.; Yi, H.; Yang, Y.G. Arginase-1-dependent promotion of TH17 differentiation and disease progression by MDSCs in systemic lupus erythematosus. Sci. Transl. Med. 2016, 8, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T. cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Sangaletti, S.; Tripodo, C.; Santangelo, A.; Castioni, N.; Portararo, P.; Gulino, A.; Botti, L.; Parenza, M.; Cappetti, B.; Orlandi, R.; et al. Mesenchymal Transition of High-Grade Breast Carcinomas Depends on Extracellular Matrix Control of Myeloid Suppressor Cell Activity. Cell. Rep. 2016, 17, 233–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammami, I.; Chen, J.; Murschel, F.; Bronte, V.; De Crescenzo, G.; Jolicoeur, M. Immunosuppressive activity enhances central carbon metabolism and bioenergetics in myeloid-derived suppressor cells in vitro models. BMC Cell Biol. 2012, 13, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Valle, L.D.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Bi, Y.; Shen, B.; Yang, H.; Zhang, Y.; Wang, X.; Liu, H.; Lu, Y.; Liao, J.; Chen, X.; et al. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1alpha-dependent glycolysis. Cancer Res. 2014, 74, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lu, Y.; Zhang, Z.; Wang, J.; Yang, H.; Liu, G. Intercellular interplay between Sirt1 signalling and cell metabolism in immune cell biology. Immunology 2015, 145, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.; Li, J.; Qu, P.; Lin, P.C. C/EBP-delta positively regulates MDSC expansion and endothelial VEGFR2 expression in tumor development. Oncotarget 2017, 8, 50582–50593. [Google Scholar] [CrossRef] [PubMed]
- Bryant, A.J.; Carrick, R.P.; McConaha, M.E.; Jones, B.R.; Shay, S.D.; Moore, C.S.; Blackwell, T.R.; Gladson, S.; Penner, N.L.; Burman, A.; et al. Endothelial HIF signaling regulates pulmonary fibrosis-associated pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L249–L262. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Oh, S.H.; Kim, E.J.; Park, S.J.; Hong, S.P.; Cheon, J.H.; Kim, T.I.; Kim, W.H. The role of myofibroblasts in upregulation of S100A8 and S100A9 and the differentiation of myeloid cells in the colorectal cancer microenvironment. Biochem. Biophys. Res. Commun. 2012, 423, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ou, L.; Han, S.; Li, M.; Pena, M.M.; Pena, E.A.; Liu, C.; Nagarkatti, M.; Fan, D.; Ai, W. Deficiency of Kruppel-like factor KLF4 in myeloid-derived suppressor cells inhibits tumor pulmonary metastasis in mice accompanied by decreased fibrocytes. Oncogenesis 2014, 3, 129–166. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.Q.; Misra, A.; Rosas, I.O.; Adams, R.H.; Greif, D.M. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci. Transl. Med. 2015, 7, 308–359. [Google Scholar] [CrossRef] [PubMed]
- Shatat, M.A.; Tian, H.; Zhang, R.; Tandon, G.; Hale, A.; Fritz, J.S.; Zhou, G.; Martinez-Gonzalez, J.; Rodriguez, C.; Champion, H.C.; et al. Endothelial Kruppel-like factor 4 modulates pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2014, 50, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Cottrill, K.A.; Chan, S.Y. Metabolic dysfunction in pulmonary hypertension: The expanding relevance of the Warburg effect. Eur. J. Clin. Investig. 2013, 43, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Davis, L.A.; Graham, B.B. Targeting energetic metabolism: A new frontier in the pathogenesis and treatment of pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Egnatchik, R.A.; Brittain, E.L.; Shah, A.T.; Fares, W.H.; Ford, H.J.; Monahan, K.; Kang, C.J.; Kocurek, E.G.; Zhu, S.; Luong, T.; et al. Dysfunctional BMPR2 signaling drives an abnormal endothelial requirement for glutamine in pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 186–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fessel, J.P.; Hamid, R.; Wittmann, B.M.; Robinson, L.J.; Blackwell, T.; Tada, Y.; Tanabe, N.; Tatsumi, K.; Hemnes, A.R.; West, J.D. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm. Circ 2012, 2, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Rafikov, R.; Sun, X.; Rafikova, O.; Meadows, M.L.; Desai, A.A.; Khalpey, Z.; Yuan, J.X.; Fineman, J.R.; Black, S.M. Complex I dysfunction underlies the glycolytic switch in pulmonary hypertensive smooth muscle cells. Redox. Biol. 2015, 6, 278–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paffett, M.L.; Lucas, S.N.; Campen, M.J. Resveratrol reverses monocrotaline-induced pulmonary vascular and cardiac dysfunction: A potential role for atrogin-1 in smooth muscle. Vascul. Pharmacol. 2012, 56, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Tu, Y.; Jia, X.; Fang, K.; Liu, L.; Wan, L.; Xiang, C.; Wang, Y.; Sun, X.; Liu, T.; et al. Resveratrol Protects Against Pulmonary Arterial Hypertension in Rats via Activation of Silent Information Regulator 1. Cell Physiol. Biochem. 2017, 42, 55–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, C.; Sevko, A.; Ramacher, M.; Bazhin, A.V.; Falk, C.S.; Osen, W.; Borrello, I.; Kato, M.; Schadendorf, D.; Baniyash, M.; et al. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proc. Natl. Acad. Sci. USA 2011, 108, 17111–17116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafini, P.; Meckel, K.; Kelso, M.; Noonan, K.; Califano, J.; Koch, W.; Dolcetti, L.; Bronte, V.; Borrello, I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. Exp. Med. 2006, 203, 2691–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, L.H.; Alkayyal, A.A.; Leslie, A.L.; Sahi, S.; Bennett, S.; Tanese de Souza, C.; Baxter, K.; Angka, L.; Xu, R.; Kennedy, M.A.; et al. Phosphodiesterase-5 inhibition reduces postoperative metastatic disease by targeting surgery-induced myeloid derived suppressor cell-dependent inhibition of Natural Killer cell cytotoxicity. Oncoimmunology 2018, 7, e1431082. [Google Scholar] [CrossRef] [PubMed]
- Califano, J.A.; Khan, Z.; Noonan, K.A.; Rudraraju, L.; Zhang, Z.; Wang, H.; Goodman, S.; Gourin, C.G.; Ha, P.K.; Fakhry, C.; et al. Tadalafil augments tumor specific immunity in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Noonan, K.A.; Ghosh, N.; Rudraraju, L.; Bui, M.; Borrello, I. Targeting immune suppression with PDE5 inhibition in end-stage multiple myeloma. Cancer Immunol. Res. 2014, 2, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Iclozan, C.; Antonia, S.; Chiappori, A.; Chen, D.T.; Gabrilovich, D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol. Immunother. 2013, 62, 909–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, N.; Fishman, M.; Fricke, I.; Dunn, M.; Neuger, A.M.; Frost, T.J.; Lush, R.M.; Antonia, S.; Gabrilovich, D.I. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006, 66, 9299–9307. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhou, A.; Ben, X.; Shen, J.; Liang, Y.; Li, F. All-trans retinoic acid in pulmonary vascular structural remodeling in rats with pulmonary hypertension induced by monocrotaline. Chin. Med. J. 2001, 114, 462–465. [Google Scholar] [PubMed]
- Xin, Y.; Lv, J.Q.; Wang, Y.Z.; Zhang, J.; Zhang, X. Effect of all-trans retinoic acids (ATRA) on the expression of alpha-smooth muscle actin (alpha-SMA) in the lung tissues of rats with pulmonary arterial hypertension (PAH). Genet. Mol. Res. 2015, 14, 14308–14313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Jiang, B.; Yokochi, A.; Maruyama, J.; Mitani, Y.; Ma, N.; Maruyama, K. Effect of all-trans-retinoic acid on the development of chronic hypoxia-induced pulmonary hypertension. Circ. J. 2010, 74, 1696–1703. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hossain, D.M.; Duttagupta, P.; Moreira, D.; Zhao, X.; Won, H.; Buettner, R.; Nechaev, S.; Majka, M.; Zhang, B.; et al. Serum-resistant CpG-STAT3 decoy for targeting survival and immune checkpoint signaling in acute myeloid leukemia. Blood 2016, 127, 1687–1700. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314–385. [Google Scholar] [CrossRef] [PubMed]
- Tamosiuniene, R.; Manouvakhova, O.; Mesange, P.; Saito, T.; Qian, J.; Sanyal, M.; Lin, Y.C.; Nguyen, L.P.; Luria, A.; Tu, A.B.; et al. Dominant Role for Regulatory T Cells in Protecting Females Against Pulmonary Hypertension. Circ. Res. 2018, 122, 1689–1702. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Li, M.; Trousil, S.; Zhang, Y.; Pasca di Magliano, M.; Swanson, K.D.; Zheng, B. Phenformin Inhibits Myeloid-Derived Suppressor Cells and Enhances the Anti-Tumor Activity of PD-1 Blockade in Melanoma. J. Invest. Dermatol. 2017, 137, 1740–1748. [Google Scholar] [CrossRef] [PubMed]
- Fahlen, M.; Bergman, H.; Helder, G.; Ryden, L.; Wallentin, I.; Zettergren, L. Phenformin and pulmonary hypertension. Br. Heart J. 1973, 35, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Agard, C.; Rolli-Derkinderen, M.; Dumas-de-La-Roque, E.; Rio, M.; Sagan, C.; Savineau, J.P.; Loirand, G.; Pacaud, P. Protective role of the antidiabetic drug metformin against chronic experimental pulmonary hypertension. Br. J. Pharmacol. 2009, 158, 1285–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.C.; Tabima, D.M.; Dube, J.J.; Hughan, K.S.; Vanderpool, R.R.; Goncharov, D.A.; St Croix, C.M.; Garcia-Ocana, A.; Goncharova, E.A.; Tofovic, S.P.; et al. SIRT3-AMP-Activated Protein Kinase Activation by Nitrite and Metformin Improves Hyperglycemia and Normalizes Pulmonary Hypertension Associated with Heart Failure With Preserved Ejection Fraction. Circulation 2016, 133, 717–731. [Google Scholar] [PubMed]
- Qin, G.; Lian, J.; Huang, L.; Zhao, Q.; Liu, S.; Zhang, Z.; Chen, X.; Yue, D.; Li, L.; Li, F.; et al. Metformin blocks myeloid-derived suppressor cell accumulation through AMPK-DACH1-CXCL1 axis. Oncoimmunology 2018, 7, e1442167. [Google Scholar] [CrossRef] [PubMed]
- Di Mitri, D.; Toso, A.; Chen, J.J.; Sarti, M.; Pinton, S.; Jost, T.R.; D’Antuono, R.; Montani, E.; Garcia-Escudero, R.; Guccini, I.; et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature 2014, 515, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Blattner, C.; Fleming, V.; Weber, R.; Himmelhan, B.; Altevogt, P.; Gebhardt, C.; Schulze, T.J.; Razon, H.; Hawila, E.; Wildbaum, G.; et al. CCR5(+) Myeloid-Derived Suppressor Cells Are Enriched and Activated in Melanoma Lesions. Cancer Res. 2018, 78, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Nicolls, M.R.; Voelkel, N.F. The Roles of Immunity in the Prevention and Evolution of Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care. Med. 2017, 195, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Fleming, V.; Hu, X.; Weber, R.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front. Immunol. 2018, 9, 398–417. [Google Scholar] [CrossRef] [PubMed]


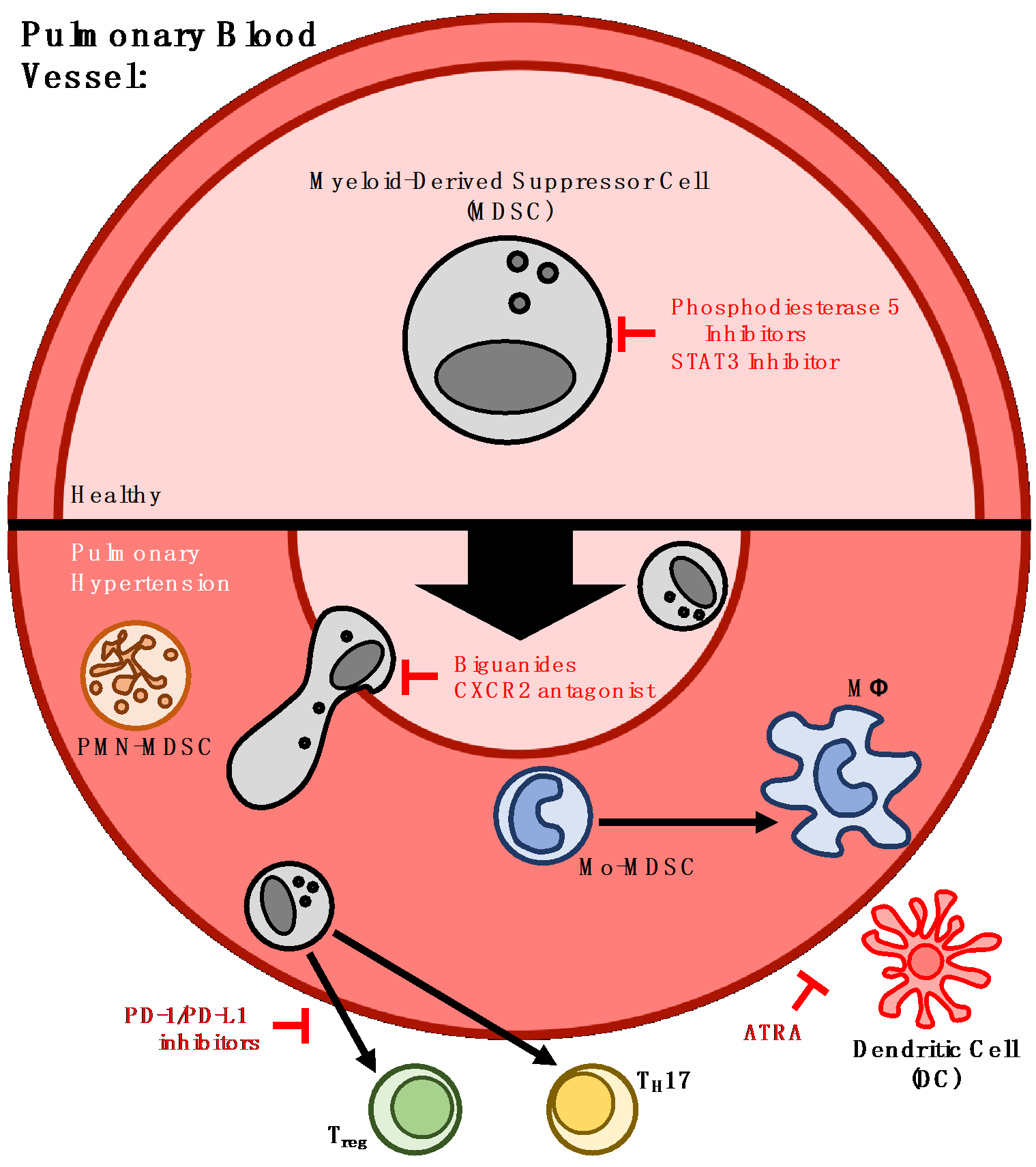{kind=link}
{kind=link}
| Molecular or Cellular Effector | Pulmonary Hypertension [ref.] | Myeloid-Derived Suppressor Cells (MDSCs) [ref.] |
|---|---|---|
| CXCR2/IL–8 | ⇧ [63,64] | ⇧ [10,76,77,78,79] |
| Arg1 | ⇧ [9,82,83,84,85] | ⇩ [86] and ⇧ [87,88,89] |
| iNOS | ⇧ [91,92,95] | ⇧ [99] |
| IDO | ⇩ [97,98,99] | ⇧ [100,101,102,103] |
| STAT3 | ⇧ [105,106] | ⇧ [107] |
| HIF | ⇧ [110,111,112] | ⇧ [116,117] (via PD–1/PD–L1 axis) |
| DC | ⇧ [118,119,120,121,122] | ⇧ [122,123,124,125,126,127] (immature DC) |
| MΦ | ⇧ [52,57,81,131] (M1 phenotype) | ⇧ [135,136] (M2/TAM phenotype) |
| Treg | ⇩ [139,141,144,148] ⇧ [137,138,140] | ⇩ [150,151] ⇧ [104,149] |
| TH17 | ⇧ [68,145,146] | ⇩ [149] and ⇧ [150,151] |
| Drug(s) | Mechanism or Pathway of Action | Expected Outcome |
|---|---|---|
| Sildenafil Tadalafil | Phosphodiesterase-5 inhibitor; downregulate Arg1 and iNOS expression in MDSC | In addition to vasodilatory effects, inhibits MDSC–mediated immunosuppression |
| All–Trans Retinoic Acid (ATRA) | Retinoic acid signal transduction | Differentiation of MDSC into macrophages and DC, and decrease collagen deposition |
| AZD9150 | STAT3 antisense oligonucleotide inhibitor | Inhibition of MDSC immunosuppressive activity and restoration of T cell function |
| Metformin Phenformin | Antidiabetic drug of biguanide class | Blocks accumulation of MDSC and enhances effect of PD-1 blockade |
| Nivolumab Pembrolizumab Atezolizumab | Monoclonal antibodies directed against immune checkpoint inhibitors PD–1 or PD–L1 | Decreased T cell exhaustion, arrest, and anergy |
| AZD5059 | CXCR2 antagonist | Decreased MDSC trafficking to site of inflammation and injury |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bryant, A.J.; Mehrad, B.; Brusko, T.M.; West, J.D.; Moldawer, L.L. Myeloid-Derived Suppressor Cells and Pulmonary Hypertension. Int. J. Mol. Sci. 2018, 19, 2277. https://doi.org/10.3390/ijms19082277
Bryant AJ, Mehrad B, Brusko TM, West JD, Moldawer LL. Myeloid-Derived Suppressor Cells and Pulmonary Hypertension. International Journal of Molecular Sciences. 2018; 19(8):2277. https://doi.org/10.3390/ijms19082277
Chicago/Turabian StyleBryant, Andrew J., Borna Mehrad, Todd M. Brusko, James D. West, and Lyle L. Moldawer. 2018. "Myeloid-Derived Suppressor Cells and Pulmonary Hypertension" International Journal of Molecular Sciences 19, no. 8: 2277. https://doi.org/10.3390/ijms19082277
APA StyleBryant, A. J., Mehrad, B., Brusko, T. M., West, J. D., & Moldawer, L. L. (2018). Myeloid-Derived Suppressor Cells and Pulmonary Hypertension. International Journal of Molecular Sciences, 19(8), 2277. https://doi.org/10.3390/ijms19082277