Hedgehog Signaling Pathway and Autophagy in Cancer

Abstract
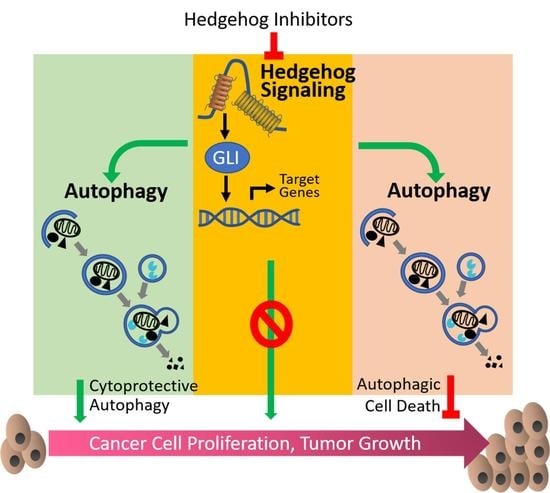
:
1. Introduction
2. Hedgehog Signaling Pathway at a Glance
2.1. Canonical and Non-Canonical Hedgehog (Hh) Signaling Transductions
2.2. Clinical Advances in the Combination of Hh Signaling Inhibition and Other Targeted Therapies
3. Autophagy in Cancer
3.1. Regulation of Autophagy in Cancer Cells
3.2. Context-Dependent Roles of Autophagy in Cancer
4. Crosstalk between Hedgehog Signaling Pathway and Autophagy
4.1. Hh Signaling Inhibits Autophagy
4.2. Hh Signaling Upregulates Autophagy
5. Combined Targeting Hh Pathway and Autophagy: A Therapeutic Opportunity for Cancer Therapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3-MA | 3-Methyladenine |
| AKT | Protein kinase B |
| AR | Androgen receptor |
| ATG | Autophagy-related protein |
| BCC | Basal cell carcinoma |
| BCL | B-cell lymphoma |
| BCR-ABL | BCR-ABL fusion gene |
| BDMC | Bisdemethoxycurcumin |
| cAMP | 3′,5′-cyclic adenosine monophosphate |
| CDK | Cyclin-dependent kinase |
| CML | Chronic myeloid leukemia |
| CQ | Chloroquine |
| DHH | Desert hedgehog |
| EGFR | Epidermal growth factor receptor |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| FOLFOX | A chemotherapy regimen that consists of folinic acid, fluorouracil, and oxaliplatin |
| GLI | Glioma-associated oncogene |
| GSK | Glycogen synthase kinase |
| HCC | Hepatocellular carcinoma |
| HCQ | Hydroxychloroquine |
| HDAC | Histone deacetylases |
| Hh | Hedgehog |
| IHH | Indian hedgehog |
| JAK | Janus kinase |
| KRAS | Kirsten rat sarcoma 2 viral oncogene homolog |
| LC3 | Microtubule-associated proteins 1A/1B light chain 3B |
| MAPK | Mitogen-activated protein kinase |
| MB | Medulloblastoma |
| MEF | Mouse embryonic fibroblasts |
| MEK | Meiosis-specific serine/threonine-protein kinase |
| mTOR | Mammalian target of rapamycin |
| OGD | Oxygen-glucose deprivation |
| PD-1 | Programmed death-ligand 1 |
| PDGFR | Platelet-derived growth factor receptor |
| PERK | PKR-like endoplasmic reticulum kinase |
| PI3K | Phosphatidylinositol 3-kinase |
| PTCH | Patched |
| RAF | Raf serine/threonine kinase |
| RBCC1 | RB1-inducible coiled-coil protein 1 |
| SHH | Sonic hedgehog |
| SMO | Smoothened |
| SUFU | Suppressor of fused |
| SMAD3 | Sma- and Mad-related Protein 3 |
| SRC | Src kinase |
| SUFU | Suppressor of fused |
| TGFβ | Transforming growth factor-β |
| ULK | Unc-51-like kinase |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| WNT | Wingless-related integration site |
| XELOX | A chemotherapy combination that combines capecitabine and oxaliplatin |
References
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Beachy, P.A.; Karhadkar, S.S.; Berman, D.M. Tissue repair and stem cell renewal in carcinogenesis. Nature 2004, 432, 324–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briscoe, J.; Therond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Ji, X.; De La Cruz, L.K.; Thareja, S.; Wang, B. Strategies to target the Hedgehog signaling pathway for cancer therapy. Med. Res. Rev. 2018, 38, 870–913. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Yauch, R.L.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 2009, 361, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; Chang, I.; Darbonne, W.C.; et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin. Cancer Res. 2011, 17, 2502–2511. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Mancias, J.D.; Kimmelman, A.C. The Role of Autophagy in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 19–39. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 1 July 2018).
- Wang, Y.; Ding, Q.; Yen, C.J.; Xia, W.; Izzo, J.G.; Lang, J.Y.; Li, C.W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, B.; Lu, Y.; Teng, K.Y.; Nuovo, G.; Li, X.; Shapiro, C.L.; Majumder, S. Hedgehog signaling is a novel therapeutic target in tamoxifen-resistant breast cancer aberrantly activated by PI3K/AKT pathway. Cancer Res. 2012, 72, 5048–5059. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Caiping, S.; Qing, Z.; Xiaojing, W. Sonic hedgehog-Gli1 signals promote epithelial-mesenchymal transition in ovarian cancer by mediating PI3K/AKT pathway. Med. Oncol. 2015, 32, 368. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.; Pedone, C.A.; del Valle, L.; Reiss, K.; Holland, E.C.; Fults, D.W. Sonic hedgehog and insulin-like growth factor signaling synergize to induce medulloblastoma formation from nestin-expressing neural progenitors in mice. Oncogene 2004, 23, 6156–6162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennler, S.; Andre, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.J.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef] [PubMed]
- Rajurkar, M.; de Jesus-Monge, W.E.; Driscoll, D.R.; Appleman, V.A.; Huang, H.; Cotton, J.L.; Klimstra, D.S.; Zhu, L.J.; Simin, K.; Xu, L.; et al. The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E1038–E1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Ingram, L.; Tolosa, E.J.; Vera, R.E.; Li, Q.; Kim, S.; Ma, Y.; Spyropoulos, D.D.; Beharry, Z.; Huang, J.; et al. Gli Transcription Factors Mediate the Oncogenic Transformation of Prostate Basal Cells Induced by a Kras-Androgen Receptor Axis. J. Biol. Chem. 2016, 291, 25749–25760. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.L.; Schumacher, D.; Staudte, S.; Steffen, A.; Haybaeck, J.; Keilholz, U.; Schweiger, C.; Golob-Schwarzl, N.; Mumberg, D.; Henderson, D.; et al. Non-Canonical Hedgehog Signaling Is a Positive Regulator of the WNT Pathway and Is Required for the Survival of Colon Cancer Stem Cells. Cell Rep. 2017, 21, 2813–2828. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Shah, A.S.; Moninger, T.O.; Ostedgaard, L.S.; Lu, L.; Tang, X.X.; Thornell, I.M.; Reznikov, L.R.; Ernst, S.E.; Karp, P.H.; et al. Motile cilia of human airway epithelia contain hedgehog signaling components that mediate noncanonical hedgehog signaling. Proc. Natl. Acad. Sci. USA 2018, 115, 1370–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blotta, S.; Jakubikova, J.; Calimeri, T.; Roccaro, A.M.; Amodio, N.; Azab, A.K.; Foresta, U.; Mitsiades, C.S.; Rossi, M.; Todoerti, K.; et al. Canonical and noncanonical Hedgehog pathway in the pathogenesis of multiple myeloma. Blood 2012, 120, 5002–5013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.C.; Peaceman, D.; Ozdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Investig. 2011, 121, 148–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Hou, Y.C.; Huang, J.; Fang, J.Y.; Xiong, H. Itraconazole induces apoptosis and cell cycle arrest via inhibiting Hedgehog signaling in gastric cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 50. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Aftab, B.T.; Tang, J.Y.; Kim, D.; Lee, A.H.; Rezaee, M.; Kim, J.; Chen, B.; King, E.M.; Borodovsky, A.; et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 2013, 23, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Bowles, D.W.; Keysar, S.B.; Eagles, J.R.; Wang, G.; Glogowska, M.J.; McDermott, J.D.; Le, P.N.; Gao, D.; Ray, C.E.; Rochon, P.J.; et al. A pilot study of cetuximab and the hedgehog inhibitor IPI-926 in recurrent/metastatic head and neck squamous cell carcinoma. Oral Oncol. 2016, 53, 74–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybstein, M.D.; Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. The autophagic network and cancer. Nat. Cell Biol. 2018, 20, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Li, Y.; Fan, J.; Zhao, H.; Xian, Z.; Sun, Y.; Wang, Z.; Wang, S.; Zhang, G.; Ju, D. Recombinant human arginase induced caspase-dependent apoptosis and autophagy in non-Hodgkin’s lymphoma cells. Cell Death Dis. 2013, 4, e840. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Ye, L.; Fan, J.; Li, Y.; Zeng, X.; Wang, Z.; Wang, S.; Zhang, G.; Yang, P.; Cao, Z.; et al. Asparaginase induces apoptosis and cytoprotective autophagy in chronic myeloid leukemia cells. Oncotarget 2015, 6, 3861–3873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, W.; Fan, J.; Wang, S.; Xian, Z.; Luan, J.; Li, Y.; Wang, Y.; Nan, Y.; Luo, M.; et al. Disrupting CD47-SIRPalpha axis alone or combined with autophagy depletion for the therapy of glioblastoma. Carcinogenesis 2018, 39, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, S.; Wang, Z.; Qian, X.; Fan, J.; Zeng, X.; Sun, Y.; Song, P.; Feng, M.; Ju, D. Cationic poly(amidoamine) dendrimers induced cyto-protective autophagy in hepatocellular carcinoma cells. Nanotechnology 2014, 25, 365101. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Munz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.Y.; Sanchez-Lopez, E.; Karin, M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Marinković, M.; Šprung, M.; Buljubašić, M.; Novak, I. Autophagy Modulation in Cancer: Current Knowledge on Action and Therapy. Oxid. Med. Cell. Longev. 2018, 2018, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, A.; Kanehisa, A.; Martins, I.; Senovilla, L.; Chargari, C.; Dugue, D.; Marino, G.; Kepp, O.; Michaud, M.; Perfettini, J.L.; et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. 2014, 21, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.T.; Pellegatti, P.; Shen, S.S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Brechbiel, J.; Miller-Moslin, K.; Adjei, A.A. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat. Rev. 2014, 40, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, S.; Stecca, B. Cooperative integration between HEDGEHOG-GLI signalling and other oncogenic pathways: Implications for cancer therapy. Expert Rev. Mol. Med. 2015, 17, e5. [Google Scholar] [CrossRef] [PubMed]
- Rovida, E.; Stecca, B. Mitogen-activated protein kinases and Hedgehog-GLI signaling in cancer: A crosstalk providing therapeutic opportunities? Semin. Cancer Biol. 2015, 35, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Sanchez, M.; Menzies, F.M.; Chang, Y.Y.; Simecek, N.; Neufeld, T.P.; Rubinsztein, D.C. The Hedgehog signalling pathway regulates autophagy. Nat. Commun. 2012, 3, 1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Rouschop, K.M.A.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 2007, 25, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, D.M.; Karhadkar, S.S.; Hallahan, A.R.; Pritchard, J.I.; Eberhart, C.G.; Watkins, D.N.; Chen, J.K.; Cooper, M.K.; Taipale, J.; Olson, J.M.; et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science 2002, 297, 1559–1561. [Google Scholar] [CrossRef] [PubMed]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernández-del, C.C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Rodova, M.; Roy, S.K.; Sharma, J.; Singh, K.P.; Srivastava, R.K.; Shankar, S. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 2013, 330, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benvenuto, M.; Masuelli, L.; De Smaele, E.; Fantini, M.; Mattera, R.; Cucchi, D.; Bonanno, E.; Di Stefano, E.; Frajese, G.V.; Orlandi, A.; et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the HedgehogGLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget 2016, 7, 9250–9270. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Toftgard, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A.; et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J. 2015, 34, 200–217. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Deng, L.; Chen, Q.; Wang, Y.; Xu, R.; Shi, C.; Shao, J.; Hu, G.; Gao, M.; Rao, H.; et al. Inhibition of Hedgehog signaling pathway impedes cancer cell proliferation by promotion of autophagy. Eur. J. Cell Biol. 2015, 94, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Han, C.; Lu, L.; Magliato, S.; Wu, T. Hedgehog signaling pathway regulates autophagy in human hepatocellular carcinoma cells. Hepatology 2013, 58, 995–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wei, S.; Zhao, Y.; Shi, C.; Liu, P.; Zhang, C.; Lei, Y.; Zhang, B.; Bai, B.; Huang, Y.; et al. Anti-proliferation of breast cancer cells with itraconazole: Hedgehog pathway inhibition induces apoptosis and autophagic cell death. Cancer Lett. 2017, 385, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Villegas, V.E.; Rondon-Lagos, M.; Annaratone, L.; Castellano, I.; Grismaldo, A.; Sapino, A.; Zaphiropoulos, P.G. Tamoxifen Treatment of Breast Cancer Cells: Impact on Hedgehog/GLI1 Signaling. Int. J. Mol. Sci. 2016, 17, 308. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, J.; Zhang, T.; Zou, L.; Chen, Y.; Wang, K.; Lei, Y.; Yuan, K.; Li, Y.; Lan, J.; et al. Itraconazole suppresses the growth of glioblastoma through induction of autophagy: Involvement of abnormal cholesterol trafficking. Autophagy 2014, 10, 1241–1255. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Walter, V.; Hayes, D.N.; Onaitis, M. Hedgehog-GLI signaling inhibition suppresses tumor growth in squamous lung cancer. Clin. Cancer Res. 2014, 20, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Pietanza, M.C.; Litvak, A.M.; Varghese, A.M.; Krug, L.M.; Fleisher, M.; Teitcher, J.B.; Holodny, A.I.; Sima, C.S.; Woo, K.M.; Ng, K.K.; et al. A phase I trial of the Hedgehog inhibitor, sonidegib (LDE225), in combination with etoposide and cisplatin for the initial treatment of extensive stage small cell lung cancer. Lung Cancer 2016, 99, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, Y.; Tanaka, N. The Hedgehog Signaling Networks in Lung Cancer: The Mechanisms and Roles in Tumor Progression and Implications for Cancer Therapy. BioMed Res. Int. 2016, 2016, 7969286. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.H.; Yang, H.P.; Zhou, X.D.; Wang, H.J.; Gong, L.; Tang, C.L. Autophagy Accompanied with Bisdemethoxycurcumin-induced Apoptosis in Non-small Cell Lung Cancer Cells. Biomed. Environ. Sci. 2015, 28, 105–115. [Google Scholar] [PubMed]
- Fan, J.; Ju, D.; Li, Y.; Wang, S.; Wang, Z. A novel approach to overcome non-small-cell lung cancer: Co-inhibition of autophagy and Hedgehog pathway. Ann. Oncol. 2015, 26 (Suppl. 7), vii106–vii151. [Google Scholar] [CrossRef]
- Fan, J.; Zhang, X.; Wang, S.; Chen, W.; Li, Y.; Zeng, X.; Wang, Y.; Luan, J.; Li, L.; Sun, X.; et al. Regulating autophagy facilitated therapeutic efficacy of Sonic hedgehog pathway inhibition on lung adenocarcinoma through Gli 2 suppression and ROS production. Cell Death Dis. 2018. under review. [Google Scholar]
- Amantini, C.; Morelli, M.B.; Nabissi, M.; Cardinali, C.; Santoni, M.; Gismondi, A.; Santoni, G. Capsaicin triggers autophagic cell survival which drives epithelial mesenchymal transition and chemoresistance in bladder cancer cells in an Hedgehog-dependent manner. Oncotarget 2016, 7, 50180–50194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; An, Y.; Wang, X.; Zha, W.; Li, X. Inhibition of the Hedgehog pathway induces autophagy in pancreatic ductal adenocarcinoma cells. Oncol. Rep. 2014, 31, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiet, T.D.; Hopyan, S.; Nadesan, P.; Gokgoz, N.; Poon, R.; Lin, A.C.; Yan, T.; Andrulis, I.L.; Alman, B.A.; Wunder, J.S. Constitutive hedgehog signaling in chondrosarcoma up-regulates tumor cell proliferation. Am. J. Pathol. 2006, 168, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Campbell, V.T.; Nadesan, P.; Ali, S.A.; Wang, C.Y.; Whetstone, H.; Poon, R.; Wei, Q.; Keilty, J.; Proctor, J.; Wang, L.W.; et al. Hedgehog pathway inhibition in chondrosarcoma using the smoothened inhibitor IPI-926 directly inhibits sarcoma cell growth. Mol. Cancer Ther. 2014, 13, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Guo, W.; Ren, T.; Liang, W.; Zhou, W.; Lu, Q.; Jiao, G.; Yan, T. Gli1 inhibition suppressed cell growth and cell cycle progression and induced apoptosis as well as autophagy depending on ERK1/2 activity in human chondrosarcoma cells. Cell Death Dis. 2014, 5, e979. [Google Scholar] [CrossRef] [PubMed]
- Jiao, G.; Ren, T.; Guo, W.; Ren, C.; Yang, K. Arsenic trioxide inhibits growth of human chondrosarcoma cells through G2/M arrest and apoptosis as well as autophagy. Tumour Biol. 2015, 36, 3969–3977. [Google Scholar] [CrossRef] [PubMed]
- Zibat, A.; Missiaglia, E.; Rosenberger, A.; Pritchard-Jones, K.; Shipley, J.; Hahn, H.; Fulda, S. Activation of the hedgehog pathway confers a poor prognosis in embryonal and fusion gene-negative alveolar rhabdomyosarcoma. Oncogene 2010, 29, 6323–6330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitzki, F.; Cuvelier, N.; Drager, J.; Schneider, A.; Braun, T.; Hahn, H. Hedgehog/Patched-associated rhabdomyosarcoma formation from delta1-expressing mesodermal cells. Oncogene 2016, 35, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Ridzewski, R.; Rettberg, D.; Dittmann, K.; Cuvelier, N.; Fulda, S.; Hahn, H. Hedgehog Inhibitors in Rhabdomyosarcoma: A Comparison of Four Compounds and Responsiveness of Four Cell Lines. Front. Oncol. 2015, 5, 130. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.U.; Shohet, J.M. Neuroblastoma: Molecular pathogenesis and therapy. Annu. Rev. Med. 2015, 66, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.X.; Huang, C.; Gao, C.; Xing, T.Y.; Liu, S.G.; Li, X.J.; Zhao, Q.; Wang, X.S.; Zhao, W.; Jin, M.; et al. MYCN amplification predicts poor prognosis based on interphase fluorescence in situ hybridization analysis of bone marrow cells in bone marrow metastases of neuroblastoma. Cancer Cell. Int. 2017, 17, 43. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.S.; Wang, X.W.; Wan, J.H.; Li, T.; Gong, X.Y.; Zhang, K.; Yi, L.; Xiang, Z.H.; Xu, M.H.; Cui, H.J. Sonic Hedgehog pathway is essential for neuroblastoma cell proliferation and tumor growth. Mol. Cell Biochem. 2012, 364, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gu, S.; Huang, J.; Chen, S.; Zhang, Z.; Xu, M. Inhibition of autophagy potentiates the efficacy of Gli inhibitor GANT-61 in MYCN-amplified neuroblastoma cells. BMC Cancer 2014, 14, 768. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, S.; Tian, R.; Chen, J.; Gao, H.; Xie, C.; Shan, Y.; Zhang, Z.; Gu, S.; Xu, M. The protective autophagy activated by GANT-61 in MYCN amplified neuroblastoma cells is mediated by PERK. Oncotarget 2018, 9, 14413–14427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016, 131, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Liu, K.W.; Wang, J.; Garancher, A.; Tao, R.; Esparza, L.A.; Maier, D.L.; Udaka, Y.T.; Murad, N.; Morrissy, S.; et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell 2016, 29, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, N.K.; Kling, M.J.; Coulter, D.W.; McGuire, T.R.; Ray, S.; Kesherwani, V.; Joshi, S.S.; Sharp, J.G. Improved therapy for medulloblastoma: Targeting hedgehog and PI3K-mTOR signaling pathways in combination with chemotherapy. Oncotarget 2018, 9, 16619–16633. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.; Rothe, K.; Woolfson, A.; Jiang, X. SMO and GLI2 are key regulators mediating resistance of CML stem/progenitor cells to tyrosine kinase inhibitors. Exp. Hematol. 2017, 53, S62. [Google Scholar] [CrossRef]
- Liu, X.; Rothe, K.; Yen, R.; Fruhstorfer, C.; Maetzig, T.; Chen, M.; Forrest, D.L.; Humphries, R.K.; Jiang, X. A novel AHI-1-BCR-ABL-DNM2 complex regulates leukemic properties of primitive CML cells through enhanced cellular endocytosis and ROS-mediated autophagy. Leukemia 2017, 31, 2376–2387. [Google Scholar] [CrossRef] [PubMed]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Irvine, D.A.; Copland, M. Targeting hedgehog in hematologic malignancy. Blood 2012, 119, 2196–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.; Zhao, H.; Li, Y.; Fan, J.; Sun, Y.; Wang, S.; Wang, Z.; Song, P.; Ju, D. Targeting Hedgehog signaling pathway and autophagy overcomes drug resistance of BCR-ABL-positive chronic myeloid leukemia. Autophagy 2015, 11, 355–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Zeng, X.; Li, Y.; Wang, S.; Yang, P.; Cao, Z.; Wang, Z.; Song, P.; Mei, X.; Ju, D. A novel therapeutic approach against B-cell non-Hodgkin’s lymphoma through co-inhibition of Hedgehog signaling pathway and autophagy. Tumour Biol. 2016, 37, 7305–7314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, Z.; Sattva, S.N.; Jorge, R.; Nami, M. Hedgehog inhibitors selectively target cell migration and adhesion of mantle cell lymphoma in bone marrow microenvironment. Oncotarget 2016, 7, 14350–14365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Wu, G.; Zeng, R.; Wang, J.; Cai, R.; Ho, J.C.-M.; Zhang, J.; Zheng, Y. Chromium contributes to human bronchial epithelial cell carcinogenesis by activating Gli2 and inhibiting autophagy. Toxicol. Res. 2017, 6, 324–332. [Google Scholar] [CrossRef]
- Granato, M.; Zompetta, C.; Vescarelli, E.; Rizzello, C.; Cardi, A.; Valia, S.; Antonelli, G.; Marchese, C.; Torrisi, M.R.; Faggioni, A.; et al. HCV derived from sera of HCV-infected patients induces pro-fibrotic effects in human primary fibroblasts by activating GLI2. Sci. Rep. 2016, 6, 30649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.Y.; He, Y.J.; Yang, Q.H.; Huang, W.; Liu, Z.H.; Ye, G.R.; Tang, S.H.; Shu, J.C. Induction of autophagy and apoptosis by miR-148a through the sonic hedgehog signaling pathway in hepatic stellate cells. Am. J. Cancer Res. 2015, 5, 2569–2589. [Google Scholar] [PubMed]
- Li, J.; Zhang, L.; Xia, Q.; Fu, J.; Zhou, Z.; Lin, F. Hedgehog signaling inhibitor GANT61 induces endoplasmic reticulum stress-mediated protective autophagy in hepatic stellate cells. Biochem. Biophys. Res. Commun. 2017, 493, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Duan, N.N.; Liu, X.J.; Wu, J. Palmitic acid elicits hepatic stellate cell activation through inflammasomes and hedgehog signaling. Life Sci. 2017, 176, 42–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.K.; Baena, A.; Ng, T.W.; Venkataswamy, M.M.; Kennedy, S.C.; Kunnath-Velayudhan, S.; Carreno, L.J.; Xu, J.; Chan, J.; Larsen, M.H.; et al. Suppression of autophagy and antigen presentation by Mycobacterium tuberculosis PE_PGRS47. Nat. Microbiol. 2016, 1, 16133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, P.A.; Furuta, K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015, 15, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Bah, A.; Vergne, I. Macrophage Autophagy and Bacterial Infections. Front. Immunol. 2017, 8, 1483. [Google Scholar] [CrossRef] [PubMed]
- Holla, S.; Kurowska-Stolarska, M.; Bayry, J.; Balaji, K.N. Selective inhibition of IFNG-induced autophagy by Mir155- and Mir31-responsive WNT5A and SHH signaling. Autophagy 2014, 10, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Milla, L.A.; Gonzalez-Ramirez, C.N.; Palma, V. Sonic Hedgehog in cancer stem cells: A novel link with autophagy. Biol. Res. 2012, 45, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Lo Re, A.E.; Fernandez-Barrena, M.G.; Almada, L.L.; Mills, L.D.; Elsawa, S.F.; Lund, G.; Ropolo, A.; Molejon, M.I.; Vaccaro, M.I.; Fernandez-Zapico, M.E. Novel AKT1-GLI3-VMP1 pathway mediates KRAS oncogene-induced autophagy in cancer cells. J. Biol. Chem. 2012, 287, 25325–25334. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—What challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Fu, J.; Srivastava, R.K.; Shankar, S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: Molecular mechanisms. PLoS ONE 2011, 6, e27306. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Venugopal, C.; Manoranjan, B.; McFarlane, N.; O’Farrell, E.; Nolte, S.; Gunnarsson, T.; Hollenberg, R.; Kwiecien, J.; Northcott, P.; et al. Sonic hedgehog regulates Bmi1 in human medulloblastoma brain tumor-initiating cells. Oncogene 2012, 31, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Po, A.; Ferretti, E.; Miele, E.; de Smaele, E.; Paganelli, A.; Canettieri, G.; Coni, S.; di Marcotullio, L.; Biffoni, M.; Massimi, L.; et al. Hedgehog controls neural stem cells through p53-independent regulation of Nanog. EMBO J. 2010, 29, 2646–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Delomenie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013, 32, 2261–2272, 2272e1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, D.; Liu, Y.; Su, Z.; Zhang, L.; Chen, F.; Zhou, Y.; Wu, Y.; Yu, M.; Zhang, Z.; et al. Role of the Hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells. Cancer Cell. Int. 2013, 13, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spivak-Kroizman, T.R.; Hostetter, G.; Posner, R.; Aziz, M.; Hu, C.; Demeure, M.J.; Von Hoff, D.; Hingorani, S.R.; Palculict, T.B.; Izzo, J.; et al. Hypoxia triggers hedgehog-mediated tumor-stromal interactions in pancreatic cancer. Cancer Res. 2013, 73, 3235–3247. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Li, J.J.; Li, Y.N.; Singh, P.; Cao, L.; Xu, L.J.; Li, D.; Wang, Y.B.; Xie, Z.P.; Gui, Y.; et al. Sonic hedgehog promotes autophagy of vascular smooth muscle cells. Am. J. Physiol.-Heart C 2012, 303, H1319–H1331. [Google Scholar] [CrossRef] [PubMed]
- Gagne-Sansfacon, J.; Allaire, J.M.; Jones, C.; Boudreau, F.; Perreault, N. Loss of Sonic hedgehog leads to alterations in intestinal secretory cell maturation and autophagy. PLoS ONE 2014, 9, e98751. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Yang, Y.; Qin, Y.; He, Y.H.; Chen, K.X.; Zhu, J.W.; Zhang, G.P.; Luo, J.D. AMP-activated protein kinase-dependent autophagy mediated the protective effect of sonic hedgehog pathway on oxygen glucose deprivation-induced injury of cardiomyocytes. Biochem. Biophys. Res. Commun. 2015, 457, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Petralia, R.S.; Schwartz, C.M.; Wang, Y.X.; Kawamoto, E.M.; Mattson, M.P.; Yao, P.J. Sonic hedgehog promotes autophagy in hippocampal neurons. Biol. Open 2013, 2, 499–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, K.Y.; Kim, G.Y.; Lim, S.J.; Sung, J.Y.; Kim, Y.W.; Park, Y.K.; Lee, J.; Choi, H.S. Autophagy is related to the hedgehog signaling pathway in human gastric adenocarcinoma: Prognostic significance of Beclin-1 and Gli2 expression in human gastric adenocarcinoma. Pathol. Res. Pract. 2015, 211, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, C.B.; DeVorkin, L.; Bosc, D.; Rothe, K.; Singh, J.; Bally, M.; Jiang, X.; Young, R.N.; Lum, J.J.; Gorski, S.M. Precision autophagy: Will the next wave of selective autophagy markers and specific autophagy inhibitors feed clinical pipelines? Autophagy 2015, 11, 1949–1952. [Google Scholar] [CrossRef] [PubMed]
- Bortnik, S.; Gorski, S.M. Clinical Applications of Autophagy Proteins in Cancer: From Potential Targets to Biomarkers. Int. J. Mol. Sci. 2017, 18, 1496. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Huang, S.; Wu, T.T.; Foster, N.R.; Sinicrope, F.A. Prognostic impact of Beclin 1, p62/sequestosome 1 and LC3 protein expression in colon carcinomas from patients receiving 5-fluorouracil as adjuvant chemotherapy. Cancer Biol. Ther. 2013, 14, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Jiang, Y.Z.; Huang, L.; Zhou, R.J.; Yu, K.D.; Liu, Y.; Shao, Z.M. The residual tumor autophagy marker LC3B serves as a prognostic marker in local advanced breast cancer after neoadjuvant chemotherapy. Clin. Cancer Res. 2013, 19, 6853–6862. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Hang, D.; Jiang, Y.; Chen, J.; Han, J.; Zhou, W.; Jin, G.; Ma, H.; Dai, J. Evaluation of genetic variants in autophagy pathway genes as prognostic biomarkers for breast cancer. Gene 2017, 627, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.Y.; Ellis, R.A.; Lovat, P.E. Prognostic Impact of Autophagy Biomarkers for Cutaneous Melanoma. Front. Oncol. 2016, 6, 236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lu, X.; Wang, N.; Wang, J.; Cao, Y.; Wang, T.; Zhou, X.; Jiao, Y.; Yang, L.; Wang, X.; et al. Autophagy-related gene expression is an independent prognostic indicator of glioma. Oncotarget 2017, 8, 60987–61000. [Google Scholar] [CrossRef] [PubMed] [Green Version]


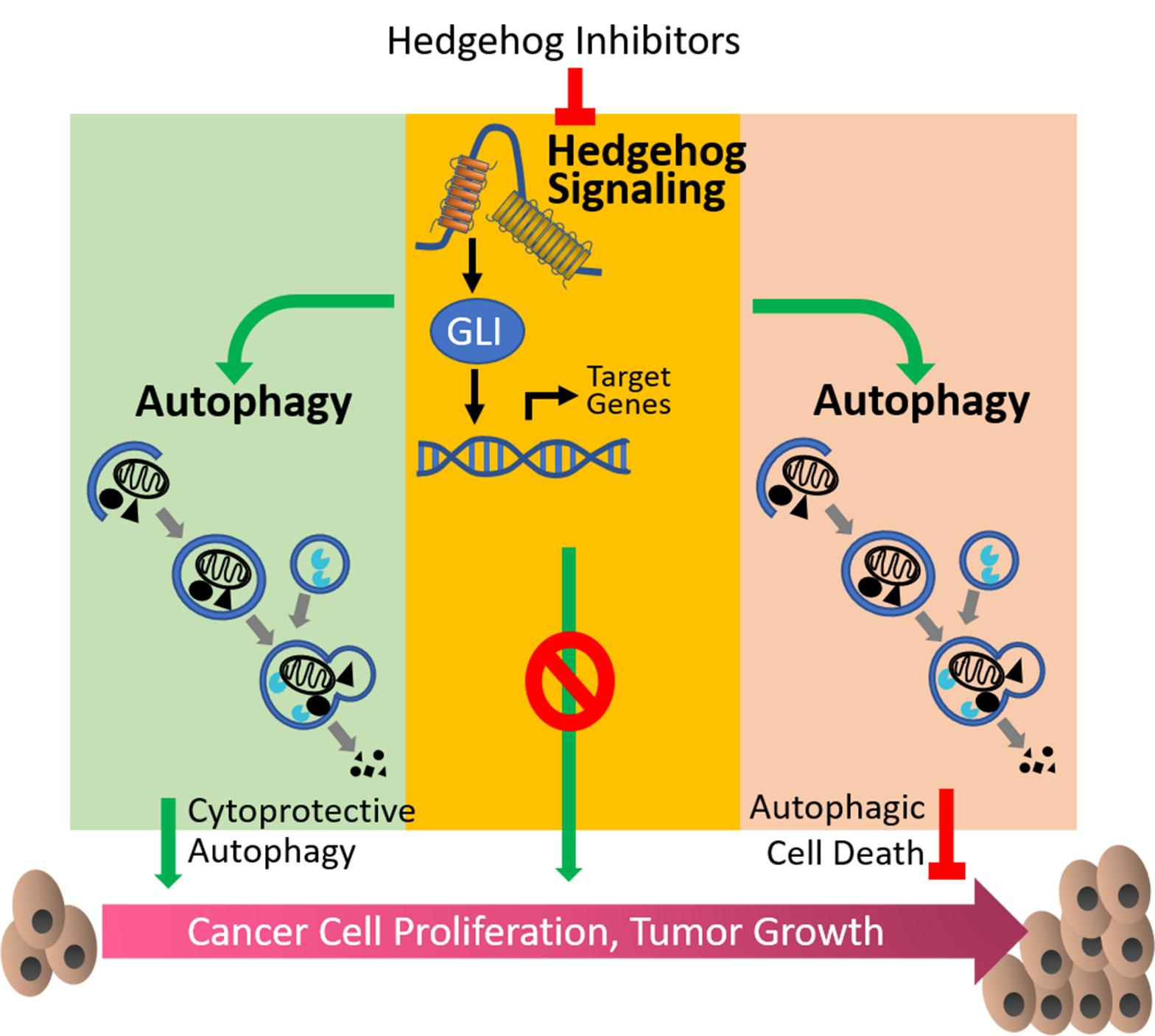{kind=link}
{kind=link}
| Combined Drugs | Combined Targets | Disease Indications | Clinical Trial Stage | Clinical Trial Status | ClinicalTrials.gov Accession |
|---|---|---|---|---|---|
| Sonidegib + Ruxolitinib | SMO + JAK1/2 | Myelofibrosis | Phase 1/2 | Active, not recruiting | NCT01787552 |
| Sonidegib + Buparlisib | SMO + PI3K | Advanced Solid Tumors | Phase 1 | Completed | NCT01576666 |
| Sonidegib + Ribociclib | SMO + CDK4/6 | Refractory or Recurrent Medulloblastoma | Phase 1 | Recruiting | NCT03434262 |
| Vismodegib + RO4929097 | SMO + Gamma-Secretase | Advanced or Metastatic Sarcoma | Phase 1/2 | Completed | NCT01154452 |
| Vismodegib + Bevacizumab | SMO + VEGF | Metastatic Colorectal Cancer | Phase 2 | Completed | NCT00636610 |
| Vismodegib + Sirolimus | SMO + mTOR | Metastatic Pancreatic Cancer | Phase 1 | Suspended | NCT01537107 |
| Vismodegib + Pembrolizumab | SMO + PD-1 | Metastatic or Unresectable Basal Cell Skin Cancer | Phase 1/2 | Active, not recruiting | NCT02690948 |
| Vismodegib + Erlotinib + Gemcitabine | SMO + EGFR | Metastatic Pancreatic Cancer | Phase 1 | Active, not recruiting | NCT00878163 |
| Saridegib + Cetuximab | SMO + EGFR | Recurrent Head and Neck Cancer | Phase 1 | Completed | NCT01255800 |
| Sonidegib + Nilotinib | SMO + BCR-ABL | Chronic or Accelerated Phase Myeloid Leukemia | Phase 1 | Completed | NCT01456676 |
| BMS-833923 + Dasatinib | SMO + BCR-ABL/SRC | Chronic Myeloid Leukemia | Phase 1/2 | Completed | NCT01218477 |
| BMS-833923 + Dasatinib | SMO + BCR-ABL/SRC | Chronic Myeloid Leukemia | Phase 2 | Terminated | NCT01357655 |
| ATO + Icotinib | GLI + EGFR | EGFR-TKI Resistant Non-Small Cell Lung Cancer | Phase 1 | Unknown | NCT02066870 |
| ATO + Gleevec | GLI + BCR-ABL | CML Who Fail Gleevec | Phase 2 | Completed | NCT00250042 |
| ATO + GO | GLI + CD33 | Advanced Myelodysplastic Syndromes | Phase 2 | Completed | NCT00274781 |
| ATO + GO + ATRA | GLI + CD33 | Acute Promyelocytic Leukemia | Phase 2 | Recruiting | NCT01409161 |
| Combined Drugs | Combined Targets | Disease Indications | Clinical Trial Stage | Clinical Trial Status | ClinicalTrials.gov Accession |
|---|---|---|---|---|---|
| Vorinostat + HCQ | HDAC + Autophagy | Advanced Solid Tumors | Phase 1 | Recruiting | NCT01023737 |
| Vorinostat + HCQ | HDAC + Autophagy | Advanced Cancer | Phase 1 | Active, not recruiting | NCT01266057 |
| Vorinostat + HCQ | HDAC + Autophagy | Colorectal Cancer | Phase 2 | Recruiting | NCT02316340 |
| Sorafenib + HCQ | VEGFR/PDGFR/RAF + Autophagy | Refractory or Relapsed Solid Tumors | Phase 1 | Completed | NCT01634893 |
| Sorafenib + HCQ | VEGFR/PDGFR/RAF + Autophagy | Hepatocellular Cancer | Phase 2 | Recruiting | NCT03037437 |
| RAD001 + HCQ | MTOR + Autophagy | Renal Cell Carcinoma | Phase 1/2 | Active, not recruiting | NCT01510119 |
| MK2206 + HCQ | AKT + Autophagy | Advanced Solid Tumors, Melanoma, Prostate or Kidney Cancer | Phase 1 | Active, not recruiting | NCT01480154 |
| Trametinib + HCQ | MEK1/2 + Autophagy | Advanced BRAF Mutant Melanoma | Phase 1/2 | Unknown | NCT02257424 |
| FOLFOX6/XELOX + Bevacizumab + HCQ | VEGF + Autophagy | Metastatic Colorectal Cancer | Phase 2 | Completed | NCT01006369 |
| Abiraterone + Navitoclax + HCQ | BCL-2/BCL-xL/BCL-w + Autophagy | Progressive Metastatic Castrate Refractory Prostate Cancer | Phase 2 | Terminated | NCT01828476 |
| Chemical Modulator Name | Target (Mode of Action) | Clinical Indication Examples | Maximum Developmental Stage |
|---|---|---|---|
| Vismodegib (GDC-0449) | SMO (Antagonist) | Approved: BCC Clinical Trials: Pancreatic ductal adenocarcinoma (NCT01096732); MB (NCT00939484, NCT01239316, NCT00822458); Advanced/metastatic sarcoma (NCT01154452); Ovarian cancer (NCT00739661, NCT00959647); AML (NCT01880437); | Approved |
| Sonidegib (Erismodegib, NVP-LDE225, LDE-225) | SMO (Antagonist) | Approved: BCC Clinical Trials: Prostate Cancer (NCT02111187); Pancreatic Adenocarcinoma (NCT01431794); Multiple Myeloma (NCT02254551, NCT02086552); Ovarian Cancer (NCT02195973); Breast Cancer (NCT01757327); Small Cell Lung Cancer (NCT01579929); | Approved |
| Saridegib (IPI-926, Patidegib) | SMO (Antagonist) | Clinical Trials: Solid Tumors (NCT00761696); Myelofibrosis (NCT01371617); Chondrosarcoma (NCT01310816); Metastatic Pancreatic Cancer (NCT01130142); Gorlin Syndrome (NCT02762084); | Phase 2 |
| Arsenic Trioxide (ATO) | GLI (Antagonist) | Approved: Acute promyelocytic leukemia (APL) Clinical Trials: Non-Small Cell Lung Cancer (NCT00075426); CML (NCT00250042); AML (NCT00005795); Myelodysplastic Syndrome (NCT00225992); | Approved 1 |
| Itraconazole | SMO (Antagonist) | Clinical Trials: BCC (NCT01108094); | Approved 2 |
| Cyclopamine | SMO (Antagonist) | Indication Evidence From In Vivo Studies: Small Cell Lung Cancer [52]; Glioblastoma [53]; CML [54]; Medulloblastoma [55]; Prostate [56]; Digestive tract tumors [57]; Pancreatic cancer [58]; | Experimental Stage |
| GANT61 | GLI (Antagonist) | Indication Evidence From In Vivo Studies: Pancreatic cancer [59]; Breast cancer [60]; Prostate Cancer [61] | Experimental Stage |
| Glabrescione B (GlaB) | GLI1 3 (Antagonist) | Indication Evidence From In Vivo Studies: MB [62] | Experimental Stage |
| SAG | SMO (Agonist) | Indication Evidence From In Vivo Studies: Pancreatic cancer [63] 4 | Experimental Stage |
| Purmorphamine | SMO (Agonist) | -- | -- |
| Cell Lines Used (Related Diseases) | Role of Hh-Related Autophagy and Supporting Evidence | Therapeutic Implications | References |
|---|---|---|---|
| H4 (Glioma), ES2 (Ovarian cancer), MKN45 (Gastric cancer), HT29 (Colon cancer) | Role: Autophagic cell death; Evidence: Inhibiting autophagy by 3-MA or knockout ATG5 partially rescued HH inhibition-induced cell proliferation. | Hh inhibitor + autophagy inducer | [64] |
| MCF-7, SKBR-3 (Breast cancer) | Role: Autophagic cell death Evidence: Inhibition of autophagy by 3-MA or ATG5 silencing impedes itraconazole-induced cell death. | Hh inhibitors + autophagy inducer | [66] |
| A549, NCI-H1975 (Lung cancer) | Role: Cytoprotective; Evidence: Inhibiting autophagy by inhibitors CQ or ATG5 or ATG7 siRNA strengthened vismodegib-induced cytotoxicity in cell lines. Co-administration of vismodegib-induced tumor-shrinkage in xenograft mouse model. | Hh inhibitor + autophagy inhibitor | [73,74] |
| HC-a, SW1353, JJ012 (Chondrosarcoma) | Role: Autophagic cell death; Evidence: Inhibiting autophagy by inhibitors (3-MA or CQ) or gene knockdown (ATG7 or Beclin1 siRNA) prevented GLI1 inhibition-induced cell death. | Hh inhibitor + autophagy inducer | [81] |
| Huh7, Hep3B, HepG2 (Liver cancer) | Role: Autophagic cell death; Evidence: inhibition autophagy by 3-MA or Beclin1 siRNA hampered GANT61-induced apoptosis and cytotoxicity; 3-MA co-administration weakened the tumor-shrinkage induced by GANT61 in Huh7 xenograft mouse model. | Hh inhibitor + autophagy inducer | [65] |
| MYCN-amplified NBL-W-S and SK-N-BE cell lines (neuroblastoma) | Role: Cytoprotective; Evidence: Inhibiting autophagy by 3-MA or gene knockdown (ATG5 or ATG7 siRNA) enhanced GANT61-induced apoptosis. | Hh inhibitor + autophagy inhibitor | [90] |
| K562, BaF3-BCR-ABLWT, BaF3-BCR-ABLY253F, BaF3-BCR-ABLT315I (drug resistant CML) | Role: Cytoprotective; Evidence: Inhibiting autophagy by CQ or gene knockdown (ATG5 or ATG7 siRNA) enhanced vismodegib-induced apoptosis. | Hh inhibitor + autophagy inhibitor | [99] |
| Raji (non-Hodgkin’s lymphoma) | Role: Cytoprotective; Evidence: Inhibiting autophagy by inhibitors (CQ or bafilomycin A) or ATG5 siRNA enhanced vismodegib-induced apoptosis. | Hh inhibitor + autophagy inhibitor | [100] |
| SP53 (Mantle cell lymphoma), Jeko, REC1, Pt1, Pt2 | Role: Cytoprotective; Evidence: Combination with 3-MA significantly increased LDE255-induced cytotoxicity. | Hh inhibitor + autophagy inhibitor | [101] |
| Hepatic stellate cell line LX-2 (Liver fibrosis) | Role: Cytoprotective Evidence: inhibiting autophagy with 3-MA or CQ can enhance GANT61-induced cytotoxicity. | Hh inhibitors + autophagy inhibitor | [106] |
| CFPAC-1 (pancreatic cancer) | Role: Autophagic cell death; Evidence: Inhibiting autophagy by 3-MA reversed GANT61-induced cytotoxicity in cell lines and anticancer effect in in vivo mouse model. | Hh inhibitor + autophagy inducer | [76] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, X.; Ju, D. Hedgehog Signaling Pathway and Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 2279. https://doi.org/10.3390/ijms19082279
Zeng X, Ju D. Hedgehog Signaling Pathway and Autophagy in Cancer. International Journal of Molecular Sciences. 2018; 19(8):2279. https://doi.org/10.3390/ijms19082279
Chicago/Turabian StyleZeng, Xian, and Dianwen Ju. 2018. "Hedgehog Signaling Pathway and Autophagy in Cancer" International Journal of Molecular Sciences 19, no. 8: 2279. https://doi.org/10.3390/ijms19082279
APA StyleZeng, X., & Ju, D. (2018). Hedgehog Signaling Pathway and Autophagy in Cancer. International Journal of Molecular Sciences, 19(8), 2279. https://doi.org/10.3390/ijms19082279