Na+, K+-ATPase Signaling and Bipolar Disorder

{kind=link}
{kind=link}
Abstract
:1. Depressive and Bipolar Disorder (BD)
2. Na+, K+-ATPase
3. Na+, K+-ATPase and Behavior
4. Cardiac Steroids (CS) and Endogenous CS (ECS)
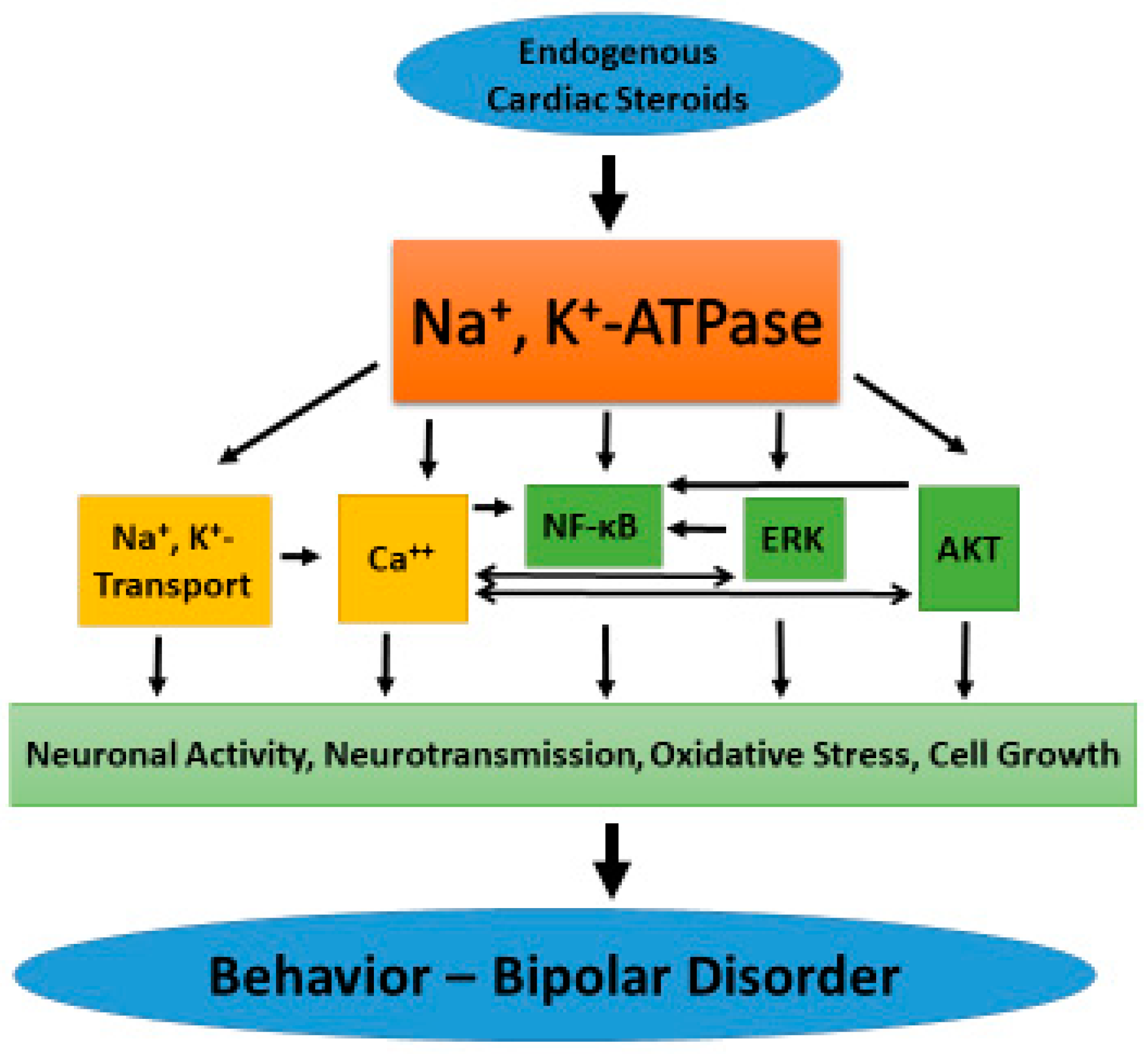
5. Na+, K+-ATPase-Induced Intracellular Signaling
6. Na+, K+-ATPase and ECS in BD
- An allelic association between BD and a Na+, K+-ATPase α subunit gene (ATP1A3) has been reported [77]. The significant association with BD of six single SNPs in the three genes of the Na+, K+-ATPase α isoforms, suggests that this enzyme plays a role in the etiology of the disease [78]. It was also shown that a genetic dysfunction of the neuron-specific Na+, K+-ATPase α3 isoform (Myshkin mice) induces manic-like behavior [79].
- BD has been consistently associated with abnormalities in Na+, K+-ATPase activity in erythrocytes [80,81]. Meta-analysis of erythrocyte Na+, K+-ATPase activity in bipolar illness showed a significant mood-state-related decrease in the enzyme’s activity in both manic and BD patients [82]. Furthermore, Na+, K+-ATPase density was significantly lower in BD patients than in major depressed and schizophrenic patients [83]. In addition, a reduction in brain Na+, K+-ATPase α1 isoform expression was found in mice treated with the mood stabilizer lithium [83].
- The plasma levels of endogenous CS were significantly reduced in manic individuals, compared with those in normal controls [84,85]. The levels of these compounds were increased in the parietal cortex of post mortem samples from BD patients, vs schizophrenic, major depressed, and normal individuals [86].
- Numerous studies have demonstrated that intracerebroventricular (i.c.v.) injection of ouabain induces hyperactive behavior in rats [87,88,89]. Actually, some studies refer to an ouabain-induced increase in activity as an animal model for mania [89,90,91]. Indeed, CS-induced hyperlocomotion is reduced following the administration of lithium or valporic acid, common mood stabilizers used in the treatment of bipolar disorder [92].
- The i.c.v. administration of highly specific and sensitive anti-ouabain antibodies, which lower brain ECS, resulted in anti-depressive effects, as measured in the forced swimming test in normal rats [86] as well as in the Flinder Sensitive Line (FSL) of genetically depressed rats [93]. In addition, administration of anti-ouabain antibodies also elicited anti-depressive effects in lipopolysaccharide-treated rats, another animal model of depression [86]. Furthermore, this treatment caused significant changes in catecholamine metabolism in the hippocampus and ventral tegmentum, two areas know to be associated with mood disorders [93].
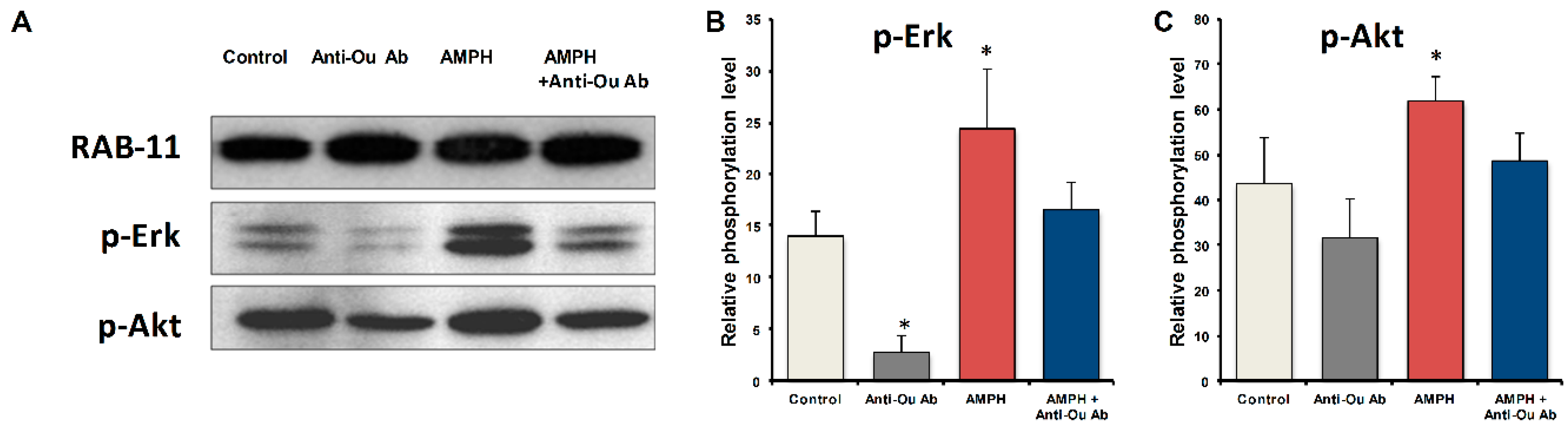
- Administration of amphetamine (AMPH), a potent central nervous system stimulant, to BALB/c and black Swiss mice, resulted in a marked increase in locomotor activity, accompanied by a threefold increase in brain ECS [94]. The reduction in brain ECS by i.c.v. administration of anti-ouabain antibodies prevented the AMPH-induced hyperactivity and the increase in brain ECS levels [94].
- AMPH caused oxidative stress in the hippocampus and frontal cortex, manifested by an increase in SOD and a decrease in CAT and GPx activity, and a reduction in NPSH and an increase in TBARS levels. The reduced brain ECS activity following i.c.v. administration of anti-ouabain antibodies protected against these AMPH-induced effects [95].
7. Na+, K+-ATPase Signaling and BD
8. Prospect and Future Directions
9. Search Strategy
Funding
Conflicts of Interest
References
- Lewis-Fernandez, R.; Aggarwal, N.K. Culture and psychiatric diagnosis. Adv. Psychosom. Med. 2013, 33, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Hirschfeld, R.M. Differential diagnosis of bipolar disorder and major depressive disorder. J. Affect. Disord. 2014, 169 (Suppl. 1), S12–S16. [Google Scholar] [CrossRef]
- Tondo, L.; Vazquez, G.H.; Baldessarini, R.J. Depression and Mania in Bipolar Disorder. Curr. Neuropharmacol. 2017, 15, 353–358. [Google Scholar] [CrossRef] [PubMed]
- De la Vega, D.; Pina, A.; Peralta, F.J.; Kelly, S.A.; Giner, L. A Review on the General Stability of Mood Disorder Diagnoses along the Lifetime. Curr. Psychiatry Rep. 2018, 20, 29. [Google Scholar] [CrossRef] [PubMed]
- Neale, B.M.; Sklar, P. Genetic analysis of schizophrenia and bipolar disorder reveals polygenicity but also suggests new directions for molecular interrogation. Curr. Opin. Neurobiol. 2015, 30, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Nierenberg, A.A.; Kansky, C.; Brennan, B.P.; Shelton, R.C.; Perlis, R.; Iosifescu, D.V. Mitochondrial modulators for bipolar disorder: A pathophysiologically informed paradigm for new drug development. Aust. N. Z. J. Psychiatry 2013, 47, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Morsel, A.M.; Morrens, M.; Sabbe, B. An overview of pharmacotherapy for bipolar I disorder. Expert Opin. Pharmacother. 2018, 19, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.C.; Pryor, W.W.; Gibbons, J.E.; Orgain, E.S. Depression and anxiety occurring during Rauwolfia therapy. J. Am. Med. Assoc. 1955, 159, 836–839. [Google Scholar] [CrossRef] [PubMed]
- Shore, P.A.; Silver, S.L.; Brodie, B.B. Interaction of reserpine, serotonin, and lysergic acid diethylamide in brain. Science 1955, 122, 284–285. [Google Scholar] [CrossRef] [PubMed]
- Hirschfeld, R.M. History and evolution of the monoamine hypothesis of depression. J. Clin. Psychiatry 2000, 61, 4–6. [Google Scholar] [PubMed]
- Dale, E.; Bang-Andersen, B.; Sanchez, C. Emerging mechanisms and treatments for depression beyond SSRIs and SNRIs. Biochem. Pharmacol. 2015, 95, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Iniesta, R.; Hodgson, K.; Stahl, D.; Malki, K.; Maier, W.; Rietschel, M.; Mors, O.; Hauser, J.; Henigsberg, N.; Dernovsek, M.Z.; et al. Antidepressant drug-specific prediction of depression treatment outcomes from genetic and clinical variables. Sci. Rep. 2018, 8, 5530. [Google Scholar] [CrossRef] [PubMed]
- Salomon, R.M.; Miller, H.L.; Krystal, J.H.; Heninger, G.R.; Charney, D.S. Lack of behavioral effects of monoamine depletion in healthy subjects. Biol. Psychiatry 1997, 41, 58–64. [Google Scholar] [CrossRef]
- Jeon, W.J.; Dean, B.; Scarr, E.; Gibbons, A. The Role of Muscarinic Receptors in the Pathophysiology of Mood Disorders: A Potential Novel Treatment? Curr. Neuropharmacol. 2015, 13, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Blacker, C.J.; Lewis, C.P.; Frye, M.A.; Veldic, M. Metabotropic glutamate receptors as emerging research targets in bipolar disorder. Psychiatry Res. 2017, 257, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Lener, M.S.; Niciu, M.J.; Ballard, E.D.; Park, M.; Park, L.T.; Nugent, A.C.; Zarate, C.A., Jr. Glutamate and Gamma-Aminobutyric Acid Systems in the Pathophysiology of Major Depression and Antidepressant Response to Ketamine. Biol. Psychiatry 2017, 81, 886–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrich, E.; Turncliff, R.; Du, Y.; Leigh-Pemberton, R.; Fernandez, E.; Jones, R.; Fava, M. Evaluation of opioid modulation in major depressive disorder. Neuropsychopharmacology 2015, 40, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Iasevoli, F.; Buonaguro, E.F.; De Berardis, D.; Fornaro, M.; Fiengo, A.L.; Martinotti, G.; Orsolini, L.; Valchera, A.; Di Giannantonio, M.; et al. Treating the Synapse in Major Psychiatric Disorders: The Role of Postsynaptic Density Network in Dopamine-Glutamate Interplay and Psychopharmacologic Drugs Molecular Actions. Int. J. Mol. Sci. 2017, 18, 135. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Walder, K.; McGee, S.L.; Dean, O.M.; Tye, S.J.; Maes, M.; Berk, M. A model of the mitochondrial basis of bipolar disorder. Neurosci. Biobehav. Rev. 2017, 74, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Cikankova, T.; Sigitova, E.; Zverova, M.; Fisar, Z.; Raboch, J.; Hroudova, J. Mitochondrial Dysfunctions in Bipolar Disorder: Effect of the Disease and Pharmacotherapy. CNS Neurol. Disord. Drug Targets 2017, 16, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Erdem, M.A.S.; Pan, E.; Kurt, Y.G. Bipolar Disorder and Oxidative Stress. J. Mood Disord. 2014, 4, 70–79. [Google Scholar] [CrossRef]
- De Berardis, D.; Campanella, D.; Gambi, F.; la Rovere, R.; Carano, A.; Conti, C.M.; Sivestrini, C.; Serroni, N.; Piersanti, D.; di Giuseppe, B.; et al. The role of C-reactive protein in mood disorders. Int. J. Immunopathol. Pharmacol. 2006, 19, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, N.; Doukhan, R.; Kurtlucan, O.; Tamouza, R.; Leboyer, M. Immunity, inflammation, and bipolar disorder: Diagnostic and therapeutic implications. Curr. Psychiatry Rep. 2013, 15, 387. [Google Scholar] [CrossRef] [PubMed]
- Gawryluk, J.W.; Wang, J.F.; Andreazza, A.C.; Shao, L.; Young, L.T. Decreased levels of glutathione, the major brain antioxidant, in post-mortem prefrontal cortex from patients with psychiatric disorders. Int. J. Neuropsychopharmacol. 2011, 14, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Benes, F.M.; Matzilevich, D.; Burke, R.E.; Walsh, J. The expression of proapoptosis genes is increased in bipolar disorder, but not in schizophrenia. Mol. Psychiatry 2006, 11, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Siwek, M.; Sowa-Kucma, M.; Styczen, K.; Misztak, P.; Szewczyk, B.; Topor-Madry, R.; Nowak, G.; Dudek, D.; Rybakowski, J.K. Thiobarbituric Acid-Reactive Substances: Markers of an Acute Episode and a Late Stage of Bipolar Disorder. Neuropsychobiology 2016, 73, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Diniz, B.S. Decreased Brain-Derived Neurotrophic Factor (BDNF) in Older Adults with Bipolar Disorder: Meaning and Utility? Am. J. Geriatr. Psychiatry 2016, 24, 602–603. [Google Scholar] [CrossRef] [PubMed]
- Munkholm, K.; Vinberg, M.; Kessing, L.V. Peripheral blood brain-derived neurotrophic factor in bipolar disorder: A comprehensive systematic review and meta-analysis. Mol. Psychiatry 2016, 21, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Muneer, A. Wnt and GSK3 Signaling Pathways in Bipolar Disorder: Clinical and Therapeutic Implications. Clin. Psychopharmacol. Neurosci. 2017, 15, 100–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, M.V.; Hilbers, F.; Poulsen, H. The Structure and Function of the Na,K-ATPase Isoforms in Health and Disease. Front. Physiol. 2017, 8, 371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, G.; Mercer, R.W. Isozymes of the Na-K-ATPase: Heterogeneity in structure, diversity in function. Am. J. Physiol. 1998, 275, F633–F650. [Google Scholar] [CrossRef] [PubMed]
- Hilbers, F.; Kopec, W.; Isaksen, T.J.; Holm, T.H.; Lykke-Hartmann, K.; Nissen, P.; Khandelia, H.; Poulsen, H. Tuning of the Na,K-ATPase by the beta subunit. Sci. Rep. 2016, 6, 20442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Langhans, S.A. Transcriptional regulators of Na,K-ATPase subunits. Front. Cell Dev. Biol. 2015, 3, 66. [Google Scholar] [CrossRef] [PubMed]
- McGrail, K.M.; Phillips, J.M.; Sweadner, K.J. Immunofluorescent localization of three Na,K-ATPase isozymes in the rat central nervous system: Both neurons and glia can express more than one Na,K-ATPase. J. Neurosci. 1991, 11, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Romanovsky, D.; Moseley, A.E.; Mrak, R.E.; Taylor, M.D.; Dobretsov, M. Phylogenetic preservation of α3 Na+,K+-ATPase distribution in vertebrate peripheral nervous systems. J. Comp. Neurol. 2007, 500, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Bøttger, P.; Tracz, Z.; Heuck, A.; Nissen, P.; Romero-Ramos, M.; Lykke-Hartmann, K. Distribution of Na/K-ATPase alpha 3 isoform, a sodium-potassium P-type pump associated with rapid-onset of dystonia parkinsonism (RDP) in the adult mouse brain. J. Comp. Neurol. 2010, 519, 376–404. [Google Scholar] [CrossRef] [PubMed]
- Blom, H.; Ronnlund, D.; Scott, L.; Spicarova, Z.; Widengren, J.; Bondar, A.; Aperia, A.; Brismar, H. Spatial distribution of Na+-K+-ATPase in dendritic spines dissected by nanoscale superresolution STED microscopy. BMC Neurosci. 2011, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Geering, K. FXYD proteins: New regulators of Na-K-ATPase. Am. J. Physiol. Renal Physiol. 2006, 290, F241–F250. [Google Scholar] [CrossRef] [PubMed]
- Moseley, A.E.; Williams, M.T.; Schaefer, T.L.; Bohanan, C.S.; Neumann, J.C.; Behbehani, M.M.; Vorhees, C.V.; Lingrel, J.B. Deficiency in Na,K-ATPase alpha isoform genes alters spatial learning, motor activity, and anxiety in mice. J. Neurosci. 2007, 27, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Lingrel, J.B.; Williams, M.T.; Vorhees, C.V.; Moseley, A.E. Na,K-ATPase and the role of alpha isoforms in behavior. J. Bioenerg. Biomembr. 2007, 39, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, T.L.; Lingrel, J.B.; Moseley, A.E.; Vorhees, C.V.; Williams, M.T. Targeted mutations in the Na,K-ATPase alpha 2 isoform confer ouabain resistance and result in abnormal behavior in mice. Synapse 2010, 65, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Bottger, P.; Glerup, S.; Gesslein, B.; Illarionova, N.B.; Isaksen, T.J.; Heuck, A.; Clausen, B.H.; Fuchtbauer, E.M.; Gramsbergen, J.B.; Gunnarson, E.; et al. Glutamate-system defects behind psychiatric manifestations in a familial hemiplegic migraine type 2 disease-mutation mouse model. Sci. Rep. 2016, 6, 22047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, T.H.; Lykke-Hartmann, K. Insights into the Pathology of the alpha3 Na+/K+-ATPase Ion Pump in Neurological Disorders; Lessons from Animal Models. Front. Physiol. 2016, 7, 209. [Google Scholar] [CrossRef] [PubMed]
- Kirshenbaum, G.S.; Idris, N.F.; Dachtler, J.; Roder, J.C.; Clapcote, S.J. Deficits in social behavioral tests in a mouse model of alternating hemiplegia of childhood. J. Neurogenet. 2016, 30, 42–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timothy, J.W.S.; Klas, N.; Sanghani, H.R.; Al-Mansouri, T.; Hughes, A.T.L.; Kirshenbaum, G.S.; Brienza, V.; Belle, M.D.C.; Ralph, M.R.; Clapcote, S.J.; et al. Circadian Disruptions in the Myshkin Mouse Model of Mania Are Independent of Deficits in Suprachiasmatic Molecular Clock Function. Biol. Psychiatry 2017. [Google Scholar] [CrossRef] [PubMed]
- Kirshenbaum, G.S.; Clapcote, S.J.; Duffy, S.; Burgess, C.R.; Petersen, J.; Jarowek, K.J.; Yucel, Y.H.; Cortez, M.A.; Snead, O.C., 3rd; Vilsen, B.; et al. Mania-like behavior induced by genetic dysfunction of the neuron-specific Na+,K+-ATPase α3 sodium pump. Proc. Natl. Acad. Sci. USA 2011. [Google Scholar] [CrossRef] [PubMed]
- Page, E. The Actions of Cardiac Glycosides on Heart Muscle Cells. Circulation 1964, 30, 237–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, D.M.; Gallapatthy, G.; Dunuwille, A.; Chan, B.S. Pharmacological treatment of cardiac glycoside poisoning. Br. J. Clin. Pharmacol. 2016, 81, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Chan, K.; Yeung, H.W. Simultaneous determination of bufadienolides in the traditional Chinese medicine preparation, liu-shen-wan, by liquid chromatography. J. Pharm. Pharmacol. 1992, 44, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Krenn, L.; Kopp, B. Bufadienolides from animal and plant sources. Phytochemistry 1998, 48, 1–29. [Google Scholar] [CrossRef]
- Hamlyn, J.M.; Blaustein, M.P. Endogenous Ouabain: Recent Advances and Controversies. Hypertension 2016, 68, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Goto, A.; Ishiguro, T.; Yamada, K.; Ishii, M.; Yoshioka, M.; Eguchi, C.; Shimora, M.; Sugimoto, T. Isolation of a urinary digitalis-like factor indistinguishable from digoxin. Biochem. Biophys. Res. Commun. 1990, 173, 1093–1101. [Google Scholar] [CrossRef]
- Lichtstein, D.; Gati, I.; Samuelov, S.; Berson, D.; Rozenman, Y.; Landau, L.; Deutsch, J. Identification of digitalis-like compounds in human cataractous lenses. Eur. J. Biochem. 1993, 216, 261–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilton, P.J.; White, R.W.; Lord, G.A.; Garner, G.V.; Gordon, D.B.; Hilton, M.J.; Forni, L.G.; McKinnon, W.; Ismail, F.M.; Keenan, M.; et al. An inhibitor of the sodium pump obtained from human placenta. Lancet 1996, 348, 303–305. [Google Scholar] [CrossRef]
- Schneider, R.; Antolovic, R.; Kost, H.; Sich, B.; Kirch, U.; Tepel, M.; Zidek, W.; Schoner, W. Proscillaridin A immunoreactivity: Its purification, transport in blood by a specific binding protein and its correlation with blood pressure. Clin. Exp. Hypertens. 1998, 20, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.V.; Zernetkina, V.I.; Shilova, V.Y.; Grigorova, Y.N.; Juhasz, O.; Wei, W.; Marshall, C.A.; Lakatta, E.G.; Bagrov, A.Y. Synthesis of an Endogenous Steroidal Na Pump Inhibitor Marinobufagenin, Implicated in Human Cardiovascular Diseases, Is Initiated by CYP27A1 via Bile Acid Pathway. Circ. Cardiovasc. Genet. 2015, 8, 736–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komiyama, Y.; Dong, X.H.; Nishimura, N.; Masaki, H.; Yoshika, M.; Masuda, M.; Takahashi, H. A novel endogenous digitalis, telocinobufagin, exhibits elevated plasma levels in patients with terminal renal failure. Clin. Biochem. 2005, 38, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Hamlyn, J.M.; Blaustein, M.P.; Bova, S.; DuCharme, D.W.; Harris, D.W.; Mandel, F.; Mathews, W.R.; Ludens, J.H. Identification and characterization of a ouabain-like compound from human plasma. Proc. Natl. Acad. Sci. USA 1991, 88, 6259–6263. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, M.P. The pump, the exchanger, and the holy spirit: Origins and 40-year evolution of ideas about the ouabain-Na+ pump endocrine system. Am. J. Physiol. Cell Physiol. 2018, 314, C3–C26. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.K.; Yandle, T.G.; Hilton, P.J.; Jensen, B.P.; Begg, E.J.; Nicholls, M.G. Endogenous ouabain is not ouabain. Hypertension 2014, 64, 680–683. [Google Scholar] [CrossRef] [PubMed]
- Baecher, S.; Kroiss, M.; Fassnacht, M.; Vogeser, M. No endogenous ouabain is detectable in human plasma by ultra-sensitive UPLC-MS/MS. Clin. Chim. Acta 2014, 431, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Vogeser, M. Letter to the editor: Comments on Blaustein (2018): “The pump, the exchanger, and the holy spirit: Origins and 40-year evolution of ideas about the ouabain-Na+ pump endocrine system”. Am. J. Physiol. Cell Physiol. 2018, 314, C640. [Google Scholar] [CrossRef] [PubMed]
- Hamlyn, J.M. Biosynthesis of endogenous cardiac glycosides by mammalian adrenocortical cells: Three steps forward. Clin. Chem. 2004, 50, 469–470. [Google Scholar] [CrossRef] [PubMed]
- Laredo, J.; Hamilton, B.P.; Hamlyn, J.M. Ouabain is secreted by bovine adrenocortical cells. Endocrinology 1994, 135, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Perrin, A.; Brasmes, B.; Chambaz, E.M.; Defaye, G. Bovine adrenocortical cells in culture synthesize an ouabain-like compound. Mol. Cell. Endocrinol. 1997, 126, 7–15. [Google Scholar] [CrossRef]
- Lichtstein, D.; Steinitz, M.; Gati, I.; Samuelov, S.; Deutsch, J.; Orly, J. Biosynthesis of digitalis-like compounds in rat adrenal cells: Hydroxycholesterol as possible precursor. Life Sci. 1998, 62, 2109–2126. [Google Scholar] [CrossRef]
- Nesher, M.; Shpolansky, U.; Rosen, H.; Lichtstein, D. The digitalis-like steroid hormones: New mechanisms of action and biological significance. Life Sci. 2007, 80, 2093–2107. [Google Scholar] [CrossRef] [PubMed]
- Buckalew, V.M. Endogenous digitalis-like factors: An overview of the history. Front. Endocrinol. (Lausanne) 2015, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Hamlyn, J.M.; Manunta, P. Endogenous cardiotonic steroids in kidney failure: A review and an hypothesis. Adv. Chronic Kidney Dis. 2015, 22, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Shapiro, J.I.; Fedorova, O.V. Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 2009, 61, 9–38. [Google Scholar] [CrossRef] [PubMed]
- Hodes, A.; Lichtstein, D. Natriuretic hormones in brain function. Front. Endocrinol. (Lausanne) 2014, 5, 201. [Google Scholar] [CrossRef] [PubMed]
- Buckalew, V.M. Role of endogenous digitalis-like factors in the clinical manifestations of severe preeclampsia: A sytematic review. Clin. Sci. (Lond.) 2018, 132, 1215–1242. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Askari, A.; Xie, Z. Involvement of Src and epidermal growth factor receptor in the signal-=transducing function of Na,K-ATPase. J. Biol. Chem. 2000, 275, 27832–27837. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xie, Z.J. The sodium pump and cardiotonic steroids-induced signal transduction protein kinases and calcium-signaling microdomain in regulation of transporter trafficking. Biochim. Biophys. Acta 2010, 1802, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Xie, Z. Protein Interaction and Na/K-ATPase-Mediated Signal Transduction. Molecules 2017, 22, 990. [Google Scholar]
- Buckalew, V. Is endogenous ouabain a physiological regulator of cardiovascular and renal function? Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1972–H1973. [Google Scholar] [CrossRef] [PubMed]
- Mynett-Johnson, L.; Murphy, V.; McCormack, J.; Shields, D.C.; Claffey, E.; Manley, P.; McKeon, P. Evidence for an allelic association between bipolar disorder and a Na+, K+ adenosine triphosphatase alpha subunit gene (ATP1A3). Biol. Psychiatry 1998, 44, 47–51. [Google Scholar] [CrossRef]
- Goldstein, I.; Lerer, E.; Laiba, E.; Mallet, J.; Mujaheed, M.; Laurent, C.; Rosen, H.; Ebstein, R.P.; Lichtstein, D. Association between sodium- and potassium-activated adenosine triphosphatase alpha isoforms and bipolar disorders. Biol. Psychiatry 2009, 65, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Kirshenbaum, G.S.; Burgess, C.R.; Dery, N.; Fahnestock, M.; Peever, J.H.; Roder, J.C. Attenuation of mania-like behavior in Na+,K+-ATPase α3 mutant mice by prospective therapies for bipolar disorder: Melatonin and exercise. Neuroscience 2014, 260, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Nurnberger, J., Jr.; Jimerson, D.C.; Allen, J.R.; Simmons, S.; Gershon, E. Red cell ouabain-sensitive Na+-K+-adenosine triphosphatase: A state marker in affective disorder inversely related to plasma cortisol. Biol. Psychiatry 1982, 17, 981–992. [Google Scholar] [PubMed]
- Naylor, G.J.; McNamee, H.B.; Moody, J.P. Changes in erythrocyte sodium and potassium on recovery from a depressive illness. Br. J. Psychiatry 1971, 118, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Looney, S.W.; El-Mallakh, R.S. Meta-analysis of erythrocyte Na,K-ATPase activity in bipolar illness. Depress. Anxiety 1997, 5, 53–65. [Google Scholar] [CrossRef]
- Chetcuti, A.; Adams, L.J.; Mitchell, P.B.; Schofield, P.R. Microarray gene expression profiling of mouse brain mRNA in a model of lithium treatment. Psychiatr. Genet. 2008, 18, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Grider, G.; El-Mallakh, R.S.; Huff, M.O.; Buss, T.J.; Miller, J.; Valdes, R.J. Endogenous digoxin-like immunoreactive factor (DLIF) serum concentrations are decreased in manic bipolar patients compared to normal controls. J. Affect. Disord. 1999, 54, 261–270. [Google Scholar] [CrossRef]
- El-Mallakh, R.S.; Stoddard, M.; Jortani, S.A.; El-Masri, M.A.; Sephton, S.; Valdes, R.J. regulation of endogenous ouabain-like factor in bipolar subjects. Psychiatry Res. 2010, 178, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Levy, T.; Galili, D.; Ovadia, H.; Yirmiya, R.; Rosen, H.; Lichtstein, D. Involvement of Na+,K+-ATPase and endogenous digitalis-like compounds in depressive disorders. Biol. Psychiatry 2006, 60, 491–499. [Google Scholar] [CrossRef] [PubMed]
- El-Mallakh, R.S.; El-Masri, M.A.; Huff, M.O.; Li, X.P.; Decker, S.; Levy, R.S. Intracerebroventricular administration of ouabain as a model of mania in rats. Bipolar Disord. 2003, 5, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Riegel, R.E.; Valvassori, S.S.; Elias, G.; Réus, G.Z.; Steckert, A.V.; de Souza, B.; Petronilho, F.; Gavioli, E.C.; Dal-Pizzol, F.; Quevedo, J. Animal model of mania induced by ouabain: Evidence of oxidative stress in submitochondrial particles of the rat brain. Neurochem. Int. 2009, 55, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Varela, R.B.; Valvassori, S.S.; Lopes-Borges, J.; Mariot, E.; Dal-Pont, G.C.; Amboni, R.T.; Bianchini, G.; Quevedo, J. Sodium butyrate and mood stabilizers block ouabain-induced hyperlocomotion and increase BDNF, NGF and GDNF levels in brain of Wistar rats. J. Psychiatr. Res. 2015, 61, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Valvassori, S.S.; Resende, W.R.; Lopes-Borges, J.; Mariot, E.; Dal-Pont, G.C.; Vitto, M.F.; Luz, G.; de Souza, C.T.; Quevedo, J. Effects of mood stabilizers on oxidative stress-induced cell death signaling pathways in the brains of rats subjected to the ouabain-induced animal model of mania: Mood stabilizers exert protective effects against ouabain-induced activation of the cell death pathway. J. Psychiatr. Res. 2015, 65, 63–70. [Google Scholar] [PubMed]
- Logan, R.W.; McClung, C.A. Animal models of bipolar mania: The past, present and future. Neuroscience 2016, 321, 163–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brocardo, P.S.; Budni, J.; Pavesi, E.; Franco, J.L.; Uliano-Silva, M.; Trevisan, R.; Terenzi, M.G.; Dafre, A.L.; Rodrigues, A.L. Folic acid administration prevents ouabain-induced hyperlocomotion and alterations in oxidative stress markers in the rat brain. Bipolar Disord. 2010, 12, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Lax, E.; Gispan-Herman, I.; Ovadia, H.; Rosen, H.; Yadid, G.; Lichtstein, D. Neutralization of endogenous digitalis-like compounds alters catecholamines metabolism in the brain and elicits anti-depressive behavior. Eur. Neuropsychopharmacol. 2012, 22, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Hodes, A.; Rosen, H.; Deutsch, J.; Lifschytz, T.; Einat, H.; Lichtstein, D. Endogenous cardiac steroids in animal models of mania. Bipolar Disord. 2016, 18, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Hodes, A.; Lifschytz, T.; Rosen, H.; Cohen, B.A.H.; Lichtstein, D. Reduction in endogenous cardiac steroids protects the brain from oxidative stress in a mouse model of mania induced by amphetamine. Brain Res. Bull. 2018, 137, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Valvassori, S.S.; Dal-Pont, G.C.; Resende, W.R.; Varela, R.B.; Peterle, B.R.; Gava, F.F.; Mina, F.G.; Cararo, J.H.; Carvalho, A.F.; Quevedo, J. Lithium and Tamoxifen Modulate Behavior and Protein Kinase C Activity in the Animal Model of Mania Induced by Ouabain. Int. J. Neuropsychopharmacol. 2017, 20, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.S.; Kim, S.H.; Park, H.G.; Kim, Y.S.; Ahn, Y.M. Activation of Akt signaling in rat brain by intracerebroventricular injection of ouabain: A rat model for mania. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2010, 34, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Yu, H.S.; Park, H.G.; Ha, K.; Kim, Y.S.; Shin, S.Y.; Ahn, Y.M. Intracerebroventricular administration of ouabain, a Na/K-ATPase inhibitor, activates mTOR signal pathways and protein translation in the rat frontal cortex. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 45, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Yu, H.S.; Park, H.G.; Jeon, W.J.; Song, J.Y.; Kang, U.G.; Ahn, Y.M.; Lee, Y.H.; Kim, Y.S. Dose-dependent effect of intracerebroventricular injection of ouabain on the phosphorylation of the MEK1/2-ERK1/2-p90RSK pathway in the rat brain related to locomotor activity. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zeng, Z.; Bhardwaj, S.K.; Jamali, S.; Srivastava, L.K. Lithium normalizes amphetamine-induced changes in striatal FoxO1 phosphorylation and behaviors in rats. Neuroreport 2013, 24, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, B.; Raineri, M.; Cadet, J.L.; Garcia-Rill, E.; Urbano, F.J.; Bisagno, V. Modafinil improves methamphetamine-induced object recognition deficits and restores prefrontal cortex ERK signaling in mice. Neuropharmacology 2014, 87, 188–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvela, M.; Rosen, H.; Ben-Ami, H.C.; Lichtstein, D. Endogenous ouabain regulates cell viability. Am. J. Physiol. Cell Physiol. 2012, 302, C442–C452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Gong, X.; Xie, Z. Signal-transducing function of Na+-K+-ATPase is essential for ouabain’s effect on [Ca2+]i in rat cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1899–H1907. [Google Scholar] [CrossRef] [PubMed]
- Buzaglo, N.; Rosen, H.; Ben Ami, H.C.; Inbal, A.; Lichtstein, D. Essential Opposite Roles of ERK and Akt Signaling in Cardiac Steroid-Induced Increase in Heart Contractility. J. Pharmacol. Exp. Ther. 2016, 357, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvela-Levitt, M.; Cohen-Ben Ami, H.; Rosen, H.; Ornoy, A.; Hochner-Celnikier, D.; Granat, M.; Lichtstein, D. Reduction in maternal circulating ouabain impairs offspring growth and kidney development. J. Am. Soc. Nephrol. 2015, 26, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Jefferys, J.G. Nonsynaptic modulation of neuronal activity in the brain: Electric currents and extracellular ions. Physiol. Rev. 1995, 75, 689–723. [Google Scholar] [CrossRef] [PubMed]
- Vizi, E.S.; Oberfrank, F. Na+/K+-ATPase, its endogenous ligands and neurotransmitter release. Neurochem. Int. 1992, 20, 11–17. [Google Scholar] [CrossRef]
- Noda, M.; Hiyama, T.Y. Sodium sensing in the brain. Pflugers Arch. 2015, 467, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Akiyama, T.; Kawada, T.; Sata, Y.; Turner, M.J.; Fukumitsu, M.; Yamamoto, H.; Kamiya, A.; Shishido, T.; Sugimachi, M. Sodium ion transport participates in non-neuronal acetylcholine release in the renal cortex of anesthetized rabbits. J. Physiol. Sci. 2017, 67, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Cavalier, M.; Crouzin, N.; Ben Sedrine, A.; de Jesus Ferreira, M.C.; Guiramand, J.; Cohen-Solal, C.; Fehrentz, J.A.; Martinez, J.; Barbanel, G.; Vignes, M. Involvement of PKA and ERK pathways in ghrelin-induced long-lasting potentiation of excitatory synaptic transmission in the CA1 area of rat hippocampus. Eur. J. Neurosci. 2015, 42, 2568–2576. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.M.; Wang, H.H.; Wang, J.Q. Antagonism of Muscarinic Acetylcholine Receptors Alters Synaptic ERK Phosphorylation in the Rat Forebrain. Neurochem. Res. 2017, 42, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Mao, L.M.; Choe, E.S.; Wang, J.Q. Synaptic ERK2 Phosphorylates and Regulates Metabotropic Glutamate Receptor 1 In Vitro and in Neurons. Mol. Neurobiol. 2017, 54, 7156–7170. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, L.; Pei, L.; Ju, W.; Ahmadian, G.; Lu, J.; Wang, Y.; Liu, F.; Wang, Y.T. Control of synaptic strength, a novel function of Akt. Neuron 2003, 38, 915–928. [Google Scholar] [CrossRef]
- Liu, G.; Feng, D.; Wang, J.; Zhang, H.; Peng, Z.; Cai, M.; Yang, J.; Zhang, R.; Wang, H.; Wu, S.; et al. rTMS Ameliorates PTSD Symptoms in Rats by Enhancing Glutamate Transmission and Synaptic Plasticity in the ACC via the PTEN/Akt Signalling Pathway. Mol. Neurobiol. 2018, 55, 3946–3958. [Google Scholar] [CrossRef] [PubMed]
- Pen, Y.; Borovok, N.; Reichenstein, M.; Sheinin, A.; Michaelevski, I. Membrane-tethered AKT kinase regulates basal synaptic transmission and early phase LTP expression by modulation of post-synaptic AMPA receptor level. Hippocampus 2016, 26, 1149–1167. [Google Scholar] [CrossRef] [PubMed]
- Caviedes, A.; Lafourcade, C.; Soto, C.; Wyneken, U. BDNF/NF-κB Signaling in the Neurobiology of Depression. Curr. Pharm. Des. 2017, 23, 3154–3163. [Google Scholar] [CrossRef] [PubMed]
- Dresselhaus, E.C.; Boersma, M.C.H.; Meffert, M.K. Targeting of NF-κB to Dendritic Spines Is Required for Synaptic Signaling and Spine Development. J. Neurosci. 2018, 38, 4093–4103. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, C.; Haenold, R. Transcriptional Control of Synaptic Plasticity by Transcription Factor NF-κB. Neural Plast. 2016, 2016, 7027949. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lichtstein, D.; Ilani, A.; Rosen, H.; Horesh, N.; Singh, S.V.; Buzaglo, N.; Hodes, A. Na+, K+-ATPase Signaling and Bipolar Disorder. Int. J. Mol. Sci. 2018, 19, 2314. https://doi.org/10.3390/ijms19082314
Lichtstein D, Ilani A, Rosen H, Horesh N, Singh SV, Buzaglo N, Hodes A. Na+, K+-ATPase Signaling and Bipolar Disorder. International Journal of Molecular Sciences. 2018; 19(8):2314. https://doi.org/10.3390/ijms19082314
Chicago/Turabian StyleLichtstein, David, Asher Ilani, Haim Rosen, Noa Horesh, Shiv Vardan Singh, Nahum Buzaglo, and Anastasia Hodes. 2018. "Na+, K+-ATPase Signaling and Bipolar Disorder" International Journal of Molecular Sciences 19, no. 8: 2314. https://doi.org/10.3390/ijms19082314
APA StyleLichtstein, D., Ilani, A., Rosen, H., Horesh, N., Singh, S. V., Buzaglo, N., & Hodes, A. (2018). Na+, K+-ATPase Signaling and Bipolar Disorder. International Journal of Molecular Sciences, 19(8), 2314. https://doi.org/10.3390/ijms19082314