The Therapeutic Strategy of HDAC6 Inhibitors in Lymphoproliferative Disease

Abstract
:
1. Introduction
2. HDAC Classification and Their Physiological Roles
2.1. HDAC Classification and Their Physiological Roles in Lymphoid Lineage
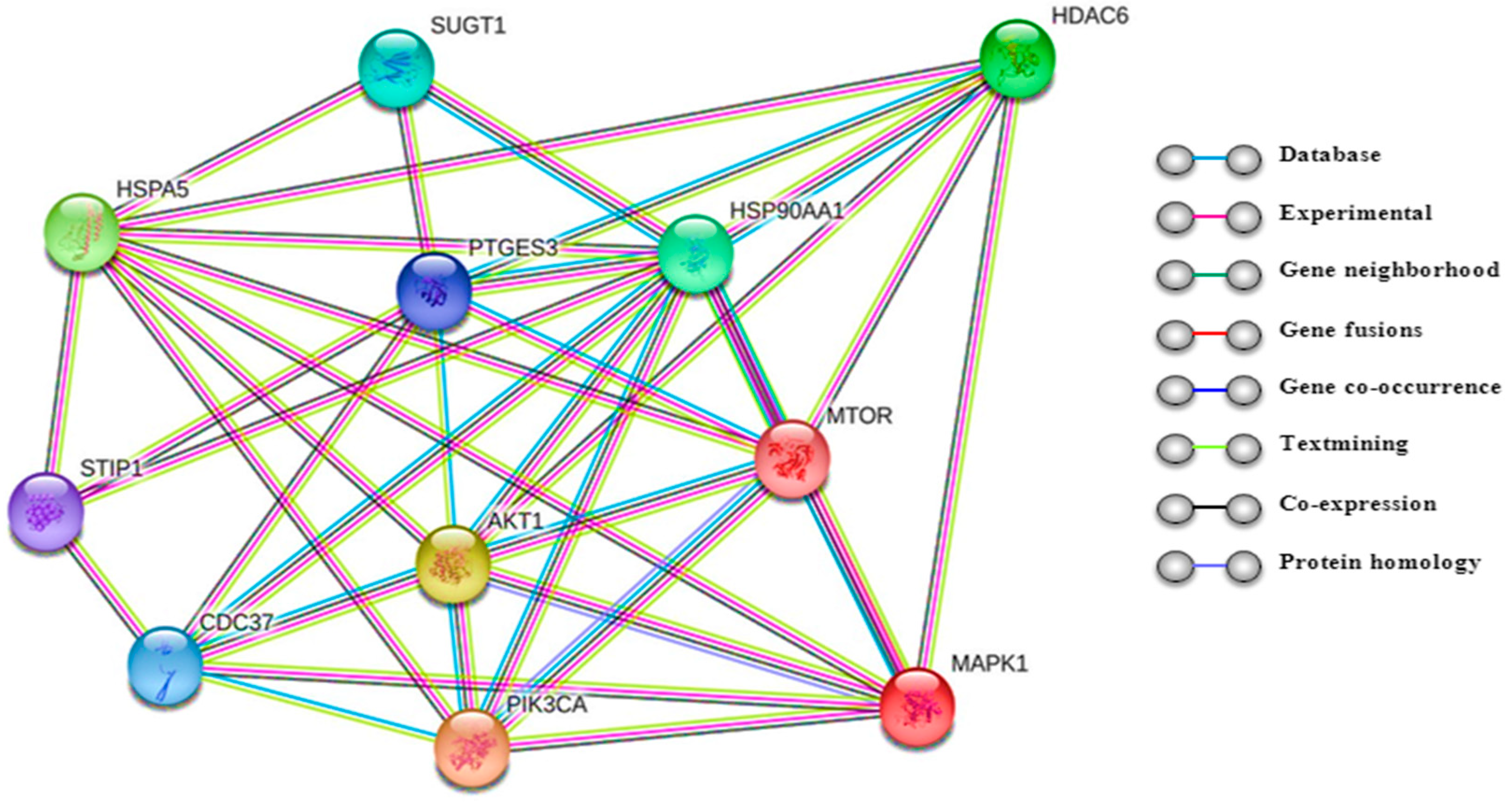
2.2. Biological Roles of Histone Deacetylase 6 (HDAC6)
3. Abnormal Expression HDAC in Lymphoproliferative Disease
4. Anticancer Effects of HDAC Inhibitors and Their Roles in Lymphoproliferative Disease
4.1. Classification of HDAC Inhibitors: Specific and Non-Specific HDACis
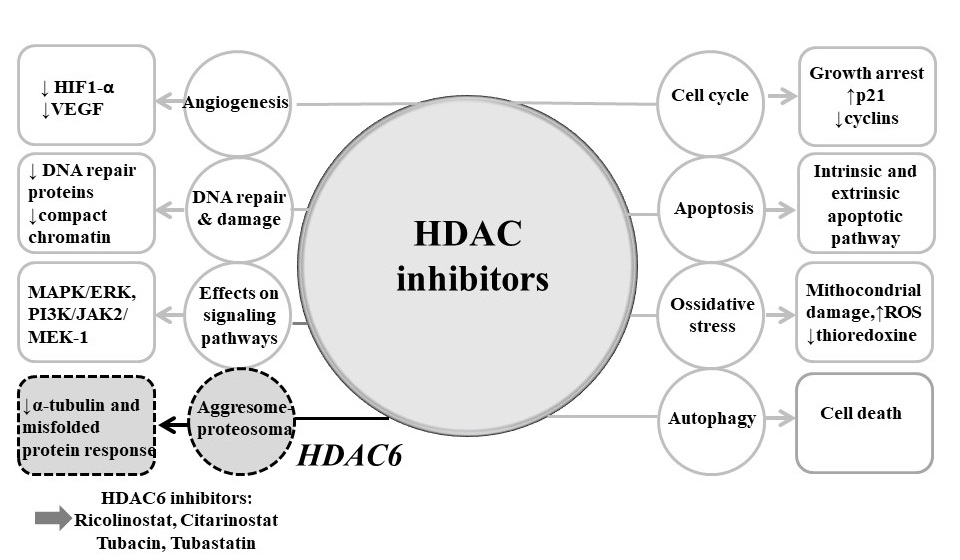
4.2. Mechanisms of Actions of HDAC Inhibitors
4.2.1. Cell Cycle Arrest
4.2.2. Apoptosis
4.2.3. Autophagy
4.2.4. Angiogenesis
4.2.5. Migration
4.2.6. Protein–Protein Interactions
4.3. Pan-HDAC Inhibitors in Lymphoproliferative Disorders: Preclinical and Clinical Data
4.4. Selective HDAC6 Inhibitors in Lymphoproliferative Diseases: Pre-Clinical and Clinical Data
4.4.1. Tubacin
4.4.2. Tubastatin
4.4.3. Ricolinostat
4.4.4. Citarinostat
5. Future Developments of HDAC Inhibitors
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perri, F.; Longo, F.; Giuliano, M.; Sabbatino, F.; Favia, G.; Ionna, F.; Addeo, R.; Della Vittoria Scarpati, G.; Di Lorenzo, G.; Pisconti, S. Epigenetic control of gene expression: Potential implications for cancer treatment. Crit. Rev. Oncol. Hematol. 2017, 111, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazirehi, A.R. Regulation of apoptosis-associated genes by histone deacetylase inhibitors: Implications in cancer therapy. Anticancer Drugs 2010, 21, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Schrump, D.S. Cytotoxicity mediated by histone deacetylase inhibitors in cancer cells: Mechanisms and potential clinical implications. Clin. Cancer Res. 2009, 15, 3947–3957. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Richon, V.M.; Rifkind, R.A. Histone deacetylase inhibitors: Inducers of differentiation or apoptosis of transformed cells. J. Natl. Cancer Inst. 2000, 92, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Bassett, S.A.; Barnett, M.P.G. The role of dietary histone deacetylases (HDACs) inhibitors in health and disease. Nutrients 2014, 6, 4273–4301. [Google Scholar] [CrossRef] [PubMed]
- Villagra, A.; Cheng, F.; Wang, H.-W.; Suarez, I.; Glozak, M.; Maurin, M.; Nguyen, D.; Wright, K.L.; Atadja, P.W.; Bhalla, K.; et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2009, 10, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Dovey, O.M.; Foster, C.T.; Conte, N.; Edwards, S.A.; Edwards, J.M.; Singh, R.; Vassiliou, G.; Bradley, A.; Cowley, S.M. Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice. Blood 2013, 121, 1335–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, R.D.W.; Cowley, S.M. The physiological roles of histone deacetylase (HDAC) 1 and 2: Complex co-stars with multiple leading parts. Biochem. Soc. Trans. 2013, 41, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Seigneurin-Berny, D.; Verdel, A.; Curtet, S.; Lemercier, C.; Garin, J.; Rousseaux, S.; Khochbin, S. Identification of components of the murine histone deacetylase 6 complex: Link between acetylation and ubiquitination signaling pathways. Mol. Cell. Biol. 2001, 21, 8035–8044. [Google Scholar] [CrossRef] [PubMed]
- Fischle, W.; Kiermer, V.; Dequiedt, F.; Verdin, E. The emerging role of class II histone deacetylases. Biochem. Cell Biol. 2001, 79, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Fischle, W.; Dequiedt, F.; Hendzel, M.J.; Guenther, M.G.; Lazar, M.A.; Voelter, W.; Verdin, E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 2002, 9, 45–57. [Google Scholar] [CrossRef]
- Grausenburger, R.; Bilic, I.; Boucheron, N.; Zupkovitz, G.; El-Housseiny, L.; Tschismarov, R.; Zhang, Y.; Rembold, M.; Gaisberger, M.; Hartl, A.; et al. Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. J. Immunol. 2010, 185, 3489–3497. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.-C.; Belmonte, P.J.; Constans, M.M.; Chen, M.W.; McWilliams, D.C.; Hiebert, S.W.; Shapiro, V.S. Histone Deacetylase 3 Is Required for T Cell Maturation. J. Immunol. 2015, 195, 1578–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, J.W.; Pagiatakis, C.; Salma, J.; Du, M.; Andreucci, J.J.; Zhao, J.; Hou, G.; Perry, R.L.; Dan, Q.; Courtman, D.; et al. Protein kinase A-regulated assembly of a MEF2{middle dot}HDAC4 repressor complex controls c-Jun expression in vascular smooth muscle cells. J. Biol. Chem. 2009, 284, 19027–19042. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, S.K.; Volinia, S.; Costinean, S.; Galasso, M.; Neinast, R.; Santhanam, R.; Parthun, M.R.; Perrotti, D.; Marcucci, G.; Garzon, R.; et al. miR-155 targets histone deacetylase 4 (HDAC4) and impairs transcriptional activity of B-cell lymphoma 6 (BCL6) in the Eμ-miR-155 transgenic mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, 20047–20052. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.H.; Bertos, N.R.; Vezmar, M.; Pelletier, N.; Crosato, M.; Heng, H.H.; Th’ng, J.; Han, J.; Yang, X.J. HDAC4, a human histone deacetylase related to yeast HDA1, is a transcriptional corepressor. Mol. Cell. Biol. 1999, 19, 7816–7827. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, X.; Yin, C.; Chen, X.; Zhang, Z.; Brown, S.; Xie, H.; Zhou, L.; Mi, Q.-S. HDAC4 is expressed on multiple T cell lineages but dispensable for their development and function. Oncotarget 2017, 8, 17562–17572. [Google Scholar] [CrossRef] [PubMed]
- Kasler, H.G.; Verdin, E. Histone deacetylase 7 functions as a key regulator of genes involved in both positive and negative selection of thymocytes. Mol. Cell. Biol. 2007, 27, 5184–5200. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.N.; Goebel, J.; Feijoo-Carnero, C.; Morrice, N.; Cantrell, D.A. Phosphoproteomic analysis reveals an intrinsic pathway for the regulation of histone deacetylase 7 that controls the function of cytotoxic T lymphocytes. Nat. Immunol. 2011, 12, 352–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azagra, A.; Román-González, L.; Collazo, O.; Rodríguez-Ubreva, J.; de Yébenes, V.G.; Barneda-Zahonero, B.; Rodríguez, J.; Castro de Moura, M.; Grego-Bessa, J.; Fernández-Duran, I.; et al. In vivo conditional deletion of HDAC7 reveals its requirement to establish proper B lymphocyte identity and development. J. Exp. Med. 2016, 213, 2591–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Zoeten, E.F.; Wang, L.; Sai, H.; Dillmann, W.H.; Hancock, W.W. Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology 2010, 138, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Beier, U.H.; Wang, L.; Han, R.; Akimova, T.; Liu, Y.; Hancock, W.W. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci. Signal. 2012, 5, ra45. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercurio, C.; Minucci, S.; Pelicci, P.G. Histone deacetylases and epigenetic therapies of hematological malignancies. Pharmacol. Res. 2010, 62, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-L.; Guan, Y.-J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Cherukuri, P.; Luo, J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J. Biol. Chem. 2005, 280, 11528–11534. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Aldana-Masangkay, G.I.; Sakamoto, K.M. The role of HDAC6 in cancer. J. Biomed. Biotechnol. 2011, 2011, 875824. [Google Scholar] [CrossRef] [PubMed]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.-F.; Yao, T.-P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giorgio, E.; Gagliostro, E.; Brancolini, C. Selective class IIa HDAC inhibitors: Myth or reality. Cell. Mol. Life Sci. 2015, 72, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Fernández, A.; Cabrero, J.R.; Serrador, J.M.; Sánchez-Madrid, F. HDAC6: A key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008, 18, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.-L.; Yang, W.-M. Beyond histone and deacetylase: An overview of cytoplasmic histone deacetylases and their nonhistone substrates. J. Biomed. Biotechnol. 2011, 2011, 146493. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.J.; Murphy, P.J.M.; Gaillard, S.; Zhao, X.; Wu, J.-T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.-P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Broemer, M.; Arslan, S.C.; Otto, A.; Mueller, E.-C.; Dettmer, R.; Scheidereit, C. Signal responsiveness of IkappaB kinases is determined by Cdc37-assisted transient interaction with Hsp90. J. Biol. Chem. 2007, 282, 32311–32319. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Boyault, C.; Zhang, Y.; Fritah, S.; Caron, C.; Gilquin, B.; Kwon, S.H.; Garrido, C.; Yao, T.-P.; Vourc’h, C.; Matthias, P.; Khochbin, S. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007, 21, 2172–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hideshima, T.; Bradner, J.E.; Wong, J.; Chauhan, D.; Richardson, P.; Schreiber, S.L.; Anderson, K.C. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. USA 2005, 102, 8567–8572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Gonzalez, A.; Lin, T.; Ikeda, A.K.; Simms-Waldrip, T.; Fu, C.; Sakamoto, K.M. Role of the aggresome pathway in cancer: Targeting histone deacetylase 6-dependent protein degradation. Cancer Res. 2008, 68, 2557–2560. [Google Scholar] [CrossRef] [PubMed]
- Boyault, C.; Sadoul, K.; Pabion, M.; Khochbin, S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 2007, 26, 5468–5476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, H.; Ali, Y.O.; Ravichandran, M.; Dong, A.; Qiu, W.; MacKenzie, F.; Dhe-Paganon, S.; Arrowsmith, C.H.; Zhai, R.G. Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J. Biol. Chem. 2012, 287, 2317–2327. [Google Scholar] [CrossRef] [PubMed]
- McConkey, D. Proteasome and HDAC: Who’s zooming who? Blood 2010, 116, 308–309. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Fiskus, W.; Yang, Y.; Lee, P.; Joshi, R.; Fernandez, P.; Mandawat, A.; Atadja, P.; Bradner, J.E.; Bhalla, K. HDAC6 inhibition enhances 17-AAG-mediated abrogation of hsp90 chaperone function in human leukemia cells. Blood 2008, 112, 1886–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.-P.; Zhou, D.; Ouyang, D.-Y.; Xu, L.-H.; Wang, Y.; Wang, L.-X.; Pan, H.; He, X.-H. LC3B-II deacetylation by histone deacetylase 6 is involved in serum-starvation-induced autophagic degradation. Biochem. Biophys. Res. Commun. 2013, 441, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.-W.; Shin, D.-H.; Lee, D.H.; Choi, J.; Han, G.; Lee, K.Y.; Kwon, S.H. HDAC6 deacetylates p53 at lysines 381/382 and differentially coordinates p53-induced apoptosis. Cancer Lett. 2017, 391, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Lim, K.-H.; Guo, X.; Kawaguchi, Y.; Gao, Y.; Barrientos, T.; Ordentlich, P.; Wang, X.-F.; Counter, C.M.; Yao, T.-P. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 2008, 68, 7561–7569. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-S.; Weng, S.-C.; Tseng, P.-H.; Lin, H.-P.; Chen, C.-S. Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J. Biol. Chem. 2005, 280, 38879–38887. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.A.; No, M.; Lee, J.M.; Shin, J.H.; Oh, J.S.; Choi, E.J.; Kim, I.H.; Atadja, P.; Bernhard, E.J. Epigenetic modulation of radiation response in human cancer cells with activated EGFR or HER-2 signaling: Potential role of histone deacetylase 6. Radiother. Oncol. 2009, 92, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Tien, S.-C.; Chang, Z.-F. Oncogenic Shp2 disturbs microtubule regulation to cause HDAC6-dependent ERK hyperactivation. Oncogene 2014, 33, 2938–2946. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Zhang, G.; Hwa, Y.L.; Li, J.; Dowdy, S.C.; Jiang, S.-W. Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther. 2010, 10, 935–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, A.A.; Chabner, B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Xu, W.-S. Histone deacetylase inhibitors: Potential in cancer therapy. J. Cell. Biochem. 2009, 107, 600–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Rao, R.; Fernandez, P.; Herger, B.; Yang, Y.; Chen, J.; Kolhe, R.; Mandawat, A.; Wang, Y.; Joshi, R.; et al. Molecular and biologic characterization and drug sensitivity of pan-histone deacetylase inhibitor-resistant acute myeloid leukemia cells. Blood 2008, 112, 2896–2905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquard, L.; Gjerdrum, L.M.; Christensen, I.J.; Jensen, P.B.; Sehested, M.; Ralfkiaer, E. Prognostic significance of the therapeutic targets histone deacetylase 1, 2, 6 and acetylated histone H4 in cutaneous T-cell lymphoma. Histopathology 2008, 53, 267–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mithraprabhu, S.; Kalff, A.; Chow, A.; Khong, T.; Spencer, A. Dysregulated Class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics 2014, 9, 1511–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquard, L.; Poulsen, C.B.; Gjerdrum, L.M.; de Nully Brown, P.; Christensen, I.J.; Jensen, P.B.; Sehested, M.; Johansen, P.; Ralfkiaer, E. Histone deacetylase 1, 2, 6 and acetylated histone H4 in B- and T-cell lymphomas. Histopathology 2009, 54, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Kafeel, M.I.; Avezbakiyev, B.; Chen, C.; Sun, Y.; Rathnasabapathy, C.; Kalavar, M.; He, Z.; Burton, J.; Lichter, S. Histone deacetylase in chronic lymphocytic leukemia. Oncology 2011, 81, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Gloghini, A.; Buglio, D.; Khaskhely, N.M.; Georgakis, G.; Orlowski, R.Z.; Neelapu, S.S.; Carbone, A.; Younes, A. Expression of histone deacetylases in lymphoma: Implication for the development of selective inhibitors. Br. J. Haematol. 2009, 147, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Adams, H.; Fritzsche, F.R.; Dirnhofer, S.; Kristiansen, G.; Tzankov, A. Class I histone deacetylases 1, 2 and 3 are highly expressed in classical Hodgkin’s lymphoma. Expert Opin. Ther. Targets 2010, 14, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Reid, R.C.; Iyer, A.; Sweet, M.J.; Fairlie, D.P. Towards isozyme-selective HDAC inhibitors for interrogating disease. Curr. Top. Med. Chem. 2012, 12, 1479–1499. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, A.L.; Rosenwald, A.; Staudt, L.M. Lymphoid malignancies: The dark side of B-cell differentiation. Nat. Rev. Immunol. 2002, 2, 920–932. [Google Scholar] [CrossRef] [PubMed]
- Lemercier, C.; Brocard, M.-P.; Puvion-Dutilleul, F.; Kao, H.-Y.; Albagli, O.; Khochbin, S. Class II histone deacetylases are directly recruited by BCL6 transcriptional repressor. J. Biol. Chem. 2002, 277, 22045–22052. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Maddipoti, S.; Quesada, A.; Bohannan, Z.; Cabrero Calvo, M.; Colla, S.; Wei, Y.; Estecio, M.; Wierda, W.; Bueso-Ramos, C.; et al. Analysis of class I and II histone deacetylase gene expression in human leukemia. Leuk. Lymphoma 2015, 56, 3426–3433. [Google Scholar] [CrossRef] [PubMed]
- Prince, H.M.; Bishton, M.J.; Harrison, S.J. Clinical studies of histone deacetylase inhibitors. Clin. Cancer Res. 2009, 15, 3958–3969. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.A. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 1999, 260, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef] [PubMed]
- Ai, T.; Cui, H.; Chen, L. Multi-targeted histone deacetylase inhibitors in cancer therapy. Curr. Med. Chem. 2012, 19, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Matthews, G.M.; Lefebure, M.; Doyle, M.A.; Shortt, J.; Ellul, J.; Chesi, M.; Banks, K.M.; Vidacs, E.; Faulkner, D.; Atadja, P.; et al. Preclinical screening of histone deacetylase inhibitors combined with ABT-737, rhTRAIL/MD5-1 or 5-azacytidine using syngeneic Vk*MYC multiple myeloma. Cell Death Dis. 2013, 4, e798. [Google Scholar] [CrossRef] [PubMed]
- Wanczyk, M.; Roszczenko, K.; Marcinkiewicz, K.; Bojarczuk, K.; Kowara, M.; Winiarska, M. HDACi—Going through the mechanisms. Front. Biosci. (Landmark Ed.) 2011, 16, 340–359. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Cottini, F.; Ohguchi, H.; Jakubikova, J.; Gorgun, G.; Mimura, N.; Tai, Y.-T.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Rational combination treatment with histone deacetylase inhibitors and immunomodulatory drugs in multiple myeloma. Blood Cancer J. 2015, 5, e312. [Google Scholar] [CrossRef] [PubMed]
- Heider, U.; von Metzler, I.; Kaiser, M.; Rosche, M.; Sterz, J.; Rötzer, S.; Rademacher, J.; Jakob, C.; Fleissner, C.; Kuckelkorn, U.; et al. Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in mantle cell lymphoma. Eur. J. Haematol. 2008, 80, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Gao, M.; Yang, G.; Tao, Y.; Kong, Y.; Yang, R.; Meng, X.; Ai, G.; Wei, R.; Wu, H.; et al. Synergistic Activity of Carfilzomib and Panobinostat in Multiple Myeloma Cells via Modulation of ROS Generation and ERK1/2. Biomed. Res. Int. 2015, 2015, 459052. [Google Scholar] [CrossRef] [PubMed]
- Yee, A.J.; Bensinger, W.I.; Supko, J.G.; Voorhees, P.M.; Berdeja, J.G.; Richardson, P.G.; Libby, E.N.; Wallace, E.E.; Birrer, N.E.; Burke, J.N.; et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: A multicentre phase 1b trial. Lancet Oncol. 2016, 17, 1569–1578. [Google Scholar] [CrossRef]
- Cyrenne, B.M.; Lewis, J.M.; Weed, J.G.; Carlson, K.R.; Mirza, F.N.; Foss, F.M.; Girardi, M. Synergy of BCL2 and histone deacetylase inhibition against leukemic cells from cutaneous T-cell lymphoma patients. Blood 2017, 130, 2073–2083. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Almeciga-Pinto, I.; Tamang, D.; Yang, M.; Jones, S.S.; Quayle, S.N. Enhancement of pomalidomide anti-tumor response with ACY-241, a selective HDAC6 inhibitor. PLoS ONE 2017, 12, e0173507. [Google Scholar] [CrossRef] [PubMed]
- Catley, L.; Weisberg, E.; Kiziltepe, T.; Tai, Y.-T.; Hideshima, T.; Neri, P.; Tassone, P.; Atadja, P.; Chauhan, D.; Munshi, N.C.; et al. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 2006, 108, 3441–3449. [Google Scholar] [CrossRef] [PubMed]
- Dasmahapatra, G.; Lembersky, D.; Kramer, L.; Fisher, R.I.; Friedberg, J.; Dent, P.; Grant, S. The pan-HDAC inhibitor vorinostat potentiates the activity of the proteasome inhibitor carfilzomib in human DLBCL cells in vitro and in vivo. Blood 2010, 115, 4478–4487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [PubMed]
- Bereshchenko, O.R.; Gu, W.; Dalla-Favera, R. Acetylation inactivates the transcriptional repressor BCL6. Nat. Genet. 2002, 32, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Kurland, J.F.; Tansey, W.P. Myc-mediated transcriptional repression by recruitment of histone deacetylase. Cancer Res. 2008, 68, 3624–3629. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Curet, I.; Perkins, R.S.; Bennett, R.; Feidler, K.L.; Dunn, S.P.; Krueger, L.J. c-Myc inhibition negatively impacts lymphoma growth. J. Pediatr. Surg. 2006, 41, 207–211, discussion 207–211. [Google Scholar] [CrossRef] [PubMed]
- Kretsovali, A.; Hadjimichael, C.; Charmpilas, N. Histone deacetylase inhibitors in cell pluripotency, differentiation, and reprogramming. Stem Cells Int. 2012, 2012, 184154. [Google Scholar] [CrossRef] [PubMed]
- Piekarz, R.L.; Bates, S.E. Epigenetic modifiers: Basic understanding and clinical development. Clin. Cancer Res. 2009, 15, 3918–3926. [Google Scholar] [CrossRef] [PubMed]
- Peart, M.J.; Smyth, G.K.; van Laar, R.K.; Bowtell, D.D.; Richon, V.M.; Marks, P.A.; Holloway, A.J.; Johnstone, R.W. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 3697–3702. [Google Scholar] [CrossRef] [PubMed]
- Peart, M.J.; Tainton, K.M.; Ruefli, A.A.; Dear, A.E.; Sedelies, K.A.; O’Reilly, L.A.; Waterhouse, N.J.; Trapani, J.A.; Johnstone, R.W. Novel mechanisms of apoptosis induced by histone deacetylase inhibitors. Cancer Res. 2003, 63, 4460–4471. [Google Scholar] [PubMed]
- Kawamata, N.; Chen, J.; Koeffler, H.P. Suberoylanilide hydroxamic acid (SAHA; vorinostat) suppresses translation of cyclin D1 in mantle cell lymphoma cells. Blood 2007, 110, 2667–2673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandor, V.; Senderowicz, A.; Mertins, S.; Sackett, D.; Sausville, E.; Blagosklonny, M.V.; Bates, S.E. P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br. J. Cancer 2000, 83, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Abukhdeir, A.M.; Park, B.H. P21 and p27: Roles in carcinogenesis and drug resistance. Expert Rev. Mol. Med. 2008, 10, e19. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; He, Q.; Tsai, C.; Lei, J.; Chen, J.; Vienna Makcey, L.; Coy, D.H. HDAC inhibitors suppressed small cell lung cancer cell growth and enhanced the suppressive effects of receptor-targeting cytotoxins via upregulating somatostatin receptor II. Am. J. Transl. Res. 2018, 10, 545–553. [Google Scholar] [PubMed]
- Zhou, H.; Cai, Y.; Liu, D.; Li, M.; Sha, Y.; Zhang, W.; Wang, K.; Gong, J.; Tang, N.; Huang, A.; et al. Pharmacological or transcriptional inhibition of both HDAC1 and 2 leads to cell cycle blockage and apoptosis via p21Waf1/Cip1 and p19INK4d upregulation in hepatocellular carcinoma. Cell Prolif. 2018, 51, e12447. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Yang, Y.; Liu, S.; Lu, J.; Huang, B.; Zhang, Y. HDAC inhibitor PAC-320 induces G2/M cell cycle arrest and apoptosis in human prostate cancer. Oncotarget 2018, 9, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Mensah, A.A.; Kwee, I.; Gaudio, E.; Rinaldi, A.; Ponzoni, M.; Cascione, L.; Fossati, G.; Stathis, A.; Zucca, E.; Caprini, G.; et al. Novel HDAC inhibitors exhibit pre-clinical efficacy in lymphoma models and point to the importance of CDKN1A expression levels in mediating their anti-tumor response. Oncotarget 2015, 6, 5059–5071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C.P.; Singh, M.M.; Rivera-Del Valle, N.; Manton, C.A.; Chandra, J. Therapeutic strategies to enhance the anticancer efficacy of histone deacetylase inhibitors. J. Biomed. Biotechnol. 2011, 2011, 514261. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Modulation of TRAIL-induced apoptosis by HDAC inhibitors. Curr. Cancer Drug Targets 2008, 8, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Matthews, G.M.; Newbold, A.; Johnstone, R.W. Intrinsic and extrinsic apoptotic pathway signaling as determinants of histone deacetylase inhibitor antitumor activity. Adv. Cancer Res. 2012, 116, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Ruefli, A.A.; Ausserlechner, M.J.; Bernhard, D.; Sutton, V.R.; Tainton, K.M.; Kofler, R.; Smyth, M.J.; Johnstone, R.W. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2001, 98, 10833–10838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powis, G.; Mustacich, D.; Coon, A. The role of the redox protein thioredoxin in cell growth and cancer. Free Radic. Biol. Med. 2000, 29, 312–322. [Google Scholar] [CrossRef]
- Hrebackova, J.; Hrabeta, J.; Eckschlager, T. Valproic acid in the complex therapy of malignant tumors. Curr. Drug Targets 2010, 11, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.; Choi, I.-K.; Kwon, H.J. Inhibition of histone deacetylase1 induces autophagy. Biochem. Biophys. Res. Commun. 2008, 369, 1179–1183. [Google Scholar] [CrossRef] [PubMed]
- Bánréti, A.; Sass, M.; Graba, Y. The emerging role of acetylation in the regulation of autophagy. Autophagy 2013, 9, 819–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohsumi, Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Deroanne, C.F.; Bonjean, K.; Servotte, S.; Devy, L.; Colige, A.; Clausse, N.; Blacher, S.; Verdin, E.; Foidart, J.-M.; Nusgens, B.V.; et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002, 21, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.-T.; Chang, H.-C.; Chiang, L.-C.; Hung, W.-C. Histone deacetylase inhibitor up-regulates RECK to inhibit MMP-2 activation and cancer cell invasion. Cancer Res. 2003, 63, 3069–3072. [Google Scholar] [PubMed]
- Khan, O.; La Thangue, N.B. HDAC inhibitors in cancer biology: Emerging mechanisms and clinical applications. Immunol. Cell Biol. 2012, 90, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Rosato, R.R.; Grant, S. Histone deacetylase inhibitors in cancer therapy. Cancer Biol. Ther. 2003, 2, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef]
- Zhang, X.D.; Gillespie, S.K.; Borrow, J.M.; Hersey, P. The histone deacetylase inhibitor suberic bishydroxamate: A potential sensitizer of melanoma to TNF-related apoptosis-inducing ligand (TRAIL) induced apoptosis. Biochem. Pharmacol. 2003, 66, 1537–1545. [Google Scholar] [CrossRef]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Bae, S.-C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar] [PubMed]
- Trüe, O.; Matthias, P. Interplay between histone deacetylases and autophagy--from cancer therapy to neurodegeneration. Immunol. Cell Biol. 2012, 90, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Batlevi, Y.; Baehrecke, E.H.; Taylor, J.P. HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system and neurodegeneration. Autophagy 2007, 3, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Zain, J.; O’Connor, O.A. Targeting histone deacetyalses in the treatment of B- and T-cell malignancies. Investig. New Drugs 2010, 28 (Suppl. 1), S58–S78. [Google Scholar] [CrossRef]
- Home—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 19 June 2018).
- Duvic, M.; Vu, J. Vorinostat: A new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin. Investig. Drugs 2007, 16, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Foss, F.; Duvic, M.; Lerner, A.; Waksman, J.; Whittaker, S. Clinical Efficacy of Romidepsin in Tumor Stage and Folliculotropic Mycosis Fungoides. Clin. Lymphoma Myeloma Leuk. 2016, 16, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.-H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San-Miguel, J.F.; Hungria, V.T.M.; Yoon, S.-S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Günther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751–2767. [Google Scholar] [CrossRef] [PubMed]
- Mackay, H.J.; Hirte, H.; Colgan, T.; Covens, A.; MacAlpine, K.; Grenci, P.; Wang, L.; Mason, J.; Pham, P.-A.; Tsao, M.-S.; et al. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. Eur. J. Cancer 2010, 46, 1573–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pili, R.; Salumbides, B.; Zhao, M.; Altiok, S.; Qian, D.; Zwiebel, J.; Carducci, M.A.; Rudek, M.A. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br. J. Cancer 2012, 106, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Thompson, L.A.; Wenger, S.D.; O’Bryant, C.L. Romidepsin: A histone deacetylase inhibitor for refractory cutaneous T-cell lymphoma. Ann. Pharmacother. 2012, 46, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Bishton, M.J.; Harrison, S.J.; Martin, B.P.; McLaughlin, N.; James, C.; Josefsson, E.C.; Henley, K.J.; Kile, B.T.; Prince, H.M.; Johnstone, R.W. Deciphering the molecular and biologic processes that mediate histone deacetylase inhibitor-induced thrombocytopenia. Blood 2011, 117, 3658–3668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, T.; Hideshima, T.; Anderson, K.C. Histone deacetylase inhibitors in multiple myeloma: From bench to bedside. Int. J. Hematol. 2016, 104, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Simms-Waldrip, T.; Rodriguez-Gonzalez, A.; Lin, T.; Ikeda, A.K.; Fu, C.; Sakamoto, K.M. The aggresome pathway as a target for therapy in hematologic malignancies. Mol. Genet. Metab. 2008, 94, 283–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.-C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amengual, J.E.; Johannet, P.; Lombardo, M.; Zullo, K.; Hoehn, D.; Bhagat, G.; Scotto, L.; Jirau-Serrano, X.; Radeski, D.; Heinen, J.; et al. Dual Targeting of Protein Degradation Pathways with the Selective HDAC6 Inhibitor ACY-1215 and Bortezomib Is Synergistic in Lymphoma. Clin. Cancer Res. 2015, 21, 4663–4675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.-X.; Wan, R.-Z.; Liu, Z.-P. Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur. J. Med. Chem. 2018, 143, 1406–1418. [Google Scholar] [CrossRef] [PubMed]
- Dallavalle, S.; Pisano, C.; Zunino, F. Development and therapeutic impact of HDAC6-selective inhibitors. Biochem. Pharmacol. 2012, 84, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Namdar, M.; Perez, G.; Ngo, L.; Marks, P.A. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. USA 2010, 107, 20003–20008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, C.; Jarzembowski, J.A.; Opipari, A.W.; Castle, V.P.; Kwok, R.P.S. HDAC6 deacetylates Ku70 and regulates Ku70-Bax binding in neuroblastoma. Neoplasia 2011, 13, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Kerr, E.; Holohan, C.; McLaughlin, K.M.; Majkut, J.; Dolan, S.; Redmond, K.; Riley, J.; McLaughlin, K.; Stasik, I.; Crudden, M.; et al. Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell Death Differ. 2012, 19, 1317–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrero, J.R.; Serrador, J.M.; Barreiro, O.; Mittelbrunn, M.; Naranjo-Suárez, S.; Martín-Cófreces, N.; Vicente-Manzanares, M.; Mazitschek, R.; Bradner, J.E.; Avila, J.; et al. Lymphocyte chemotaxis is regulated by histone deacetylase 6, independently of its deacetylase activity. Mol. Biol. Cell 2006, 17, 3435–3445. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Ping, L.; Feng, L.; Zheng, X.; Song, Y.; Zhu, J. Histone deacetylase 6 activity is critical for the metastasis of Burkitt’s lymphoma cells. Cancer Cell Int. 2014, 14, 139. [Google Scholar] [CrossRef] [PubMed]
- Mithraprabhu, S.; Khong, T.; Jones, S.S.; Spencer, A. Histone deacetylase (HDAC) inhibitors as single agents induce multiple myeloma cell death principally through the inhibition of class I HDAC. Br. J. Haematol. 2013, 162, 559–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishima, Y.; Santo, L.; Eda, H.; Cirstea, D.; Nemani, N.; Yee, A.J.; O’Donnell, E.; Selig, M.K.; Quayle, S.N.; Arastu-Kapur, S.; et al. Ricolinostat (ACY-1215) induced inhibition of aggresome formation accelerates carfilzomib-induced multiple myeloma cell death. Br. J. Haematol. 2015, 169, 423–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogl, D.T.; Raje, N.; Jagannath, S.; Richardson, P.; Hari, P.; Orlowski, R.; Supko, J.G.; Tamang, D.; Yang, M.; Jones, S.S.; et al. Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2017, 23, 3307–3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasmahapatra, G.; Patel, H.; Friedberg, J.; Quayle, S.N.; Jones, S.S.; Grant, S. In vitro and in vivo interactions between the HDAC6 inhibitor ricolinostat (ACY1215) and the irreversible proteasome inhibitor carfilzomib in non-Hodgkin lymphoma cells. Mol. Cancer Ther. 2014, 13, 2886–2897. [Google Scholar] [CrossRef] [PubMed]
- Amengual, J.E.; Prabhu, S.A.; Lombardo, M.; Zullo, K.; Johannet, P.M.; Gonzalez, Y.; Scotto, L.; Serrano, X.J.; Wei, Y.; Duong, J.; et al. Mechanisms of Acquired Drug Resistance to the HDAC6 Selective Inhibitor Ricolinostat Reveals Rational Drug-Drug Combination with Ibrutinib. Clin. Cancer Res. 2017, 23, 3084–3096. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Cai, Y.; Yang, Y.; Li, A.; Bi, R.; Wang, L.; Shen, X.; Wang, W.; Jia, Y.; Yu, B.; et al. Activation of MET signaling by HDAC6 offers a rationale for a novel ricolinostat and crizotinib combinatorial therapeutic strategy in diffuse large B-cell lymphoma. J. Pathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cosenza, M.; Civallero, M.; Marcheselli, L.; Sacchi, S.; Pozzi, S. Ricolinostat, a selective HDAC6 inhibitor, shows anti-lymphoma cell activity alone and in combination with bendamustine. Apoptosis 2017, 22, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Das, D.S.; Song, Y.; Hideshima, T.; Tai, Y.-T.; Chauhan, D.; Anderson, K.C. Combination of a novel HDAC6 inhibitor ACY-241 and anti-PD-L1 antibody enhances anti-tumor immunity and cytotoxicity in multiple myeloma. Leukemia 2018, 32, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Qi, J.; Paranal, R.M.; Tang, W.; Greenberg, E.; West, N.; Colling, M.E.; Estiu, G.; Mazitschek, R.; Perry, J.A.; et al. Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc. Natl. Acad. Sci. USA 2016, 113, 13162–13167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawada, J.; Zou, P.; Mazitschek, R.; Bradner, J.E.; Cohen, J.I. Tubacin kills Epstein-Barr virus (EBV)-Burkitt lymphoma cells by inducing reactive oxygen species and EBV lymphoblastoid cells by inducing apoptosis. J. Biol. Chem. 2009, 284, 17102–17109. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [PubMed]
- Simões-Pires, C.A.; Zwick, V.; Cretton, S.; Cuendet, M. Simultaneous Measurement of HDAC1 and HDAC6 Activity in HeLa Cells Using UHPLC-MS. J. Vis. Exp. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Mazitschek, R.; Santo, L.; Mimura, N.; Gorgun, G.; Richardson, P.G.; Raje, N.; Anderson, K.C. Induction of differential apoptotic pathways in multiple myeloma cells by class-selective histone deacetylase inhibitors. Leukemia 2014, 28, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Lwin, T.; Zhao, X.; Cheng, F.; Zhang, X.; Huang, A.; Shah, B.; Zhang, Y.; Moscinski, L.C.; Choi, Y.S.; Kozikowski, A.P.; et al. A microenvironment-mediated c-Myc/miR-548m/HDAC6 amplification loop in non-Hodgkin B cell lymphomas. J. Clin. Investig. 2013, 123, 4612–4626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raje, N.S.; Bensinger, W.; Cole, C.E.; Lonial, S.; Jagannath, S.; Arce-Lara, C.E.; Valent, J.; Rosko, A.E.; Harb, W.A.; Sandhu, I.; et al. Ricolinostat (ACY-1215), the First Selective HDAC6 Inhibitor, Combines Safely with Pomalidomide and Dexamethasone and Shows Promosing Early Results in Relapsed-and-Refractory Myeloma (ACE-MM-102 Study). Blood 2015, 126, 4228. [Google Scholar]
- Niesvizky, R.; Richardson, P.G.; Gabrail, N.Y.; Madan, S.; Yee, A.J.; Quayle, S.N.; Almeciga-Pinto, I.; Jones, S.S.; Houston, L.; Hayes, D.; et al. ACY-241, a Novel, HDAC6 Selective Inhibitor: Synergy with Immunomodulatory (IMiD®) Drugs in Multiple Myeloma (MM) Cells and Early Clinical Results (ACE-MM-200 Study). Blood 2015, 126, 3040. [Google Scholar]
- Yee, A.J.; Voorhees, P.M.; Bensinger, W.; Berdeja, J.G.; Supko, J.G.; Richardson, P.G.; Tamang, D.; Jones, S.S.; Patrick, G.; Wheeler, C.; et al. Ricolinostat (ACY-1215), a Selective HDAC6 Inhibitor, in Combination with Lenalidomide and Dexamethasone: Results of a Phase 1b Trial in Relapsed and Relapsed Refractory Multiple Myeloma. Blood 2014, 124, 4772. [Google Scholar]
- Vogl, D.T.; Raje, N.; Hari, P.; Jones, S.S.; Supko, J.G.; Leone, G.; Wheeler, C.; Orlowski, R.Z.; Richardson, P.G.; Lonial, S.; et al. Phase 1B Results of Ricolinostat (ACY-1215) Combination Therapy with Bortezomib and Dexamethasone in Patients with Relapsed or Relapsed and Refractory Multiple Myeloma (MM). Blood 2014, 124, 4764. [Google Scholar]
- Hideshima, T.; Richardson, P.G.; Anderson, K.C. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol. Cancer Ther. 2011, 10, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Guerrero, E.; Danhof, S.; Schreder, M.; Pérez-Simón, J.A.; Einsele, H.; Hudecek, M. Upregulation of CD38 Expression on Multiple Myeloma Cells By the HDAC Inhibitor Ricolinostat Augments the Efficacy of Daratumumab. Blood 2017, 130, 1803. [Google Scholar]
- Vogl, D.T.; Hari, P.N.; Jagannath, S.; Jones, S.S.; Supko, J.G.; Leone, G.; Wheeler, C.; Orlowski, R.Z.; Richardson, P.G.; Lonial, S. ACY-1215, a Selective Histone Deacetylase (HDAC) 6 Inhibitor: Interim Results Of Combination Therapy With Bortezomib In Patients With Multiple Myeloma (MM). Blood 2013, 122, 759. [Google Scholar]
- Vorhees, P.; Bensinger, W.I.; Berdeja, J.; Supko, J.G.; Richardson, P.G.; Jones, S.S.; Patrick, G.; Wheeler, C.; Raje, N. ACY-1215, a Selective Histone Deacetylase (HDAC) 6 Inhibitor, In Combination With Lenalidomide and Dexamethasone (dex), Is Well Tolerated Without Dose Limiting Toxicity (DLT) In Patients (Pts) With Multiple Myeloma (MM) At Doses Demonstrating Biologic Activity: Interim Results Of a Phase 1b Trial. Blood 2013, 122, 3190. [Google Scholar]
- Huang, P.; Almeciga-Pinto, I.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Yang, M.; Jones, S.S.; Quayle, S.N. Selective HDAC inhibition by ACY-241 enhances the activity of paclitaxel in solid tumor models. Oncotarget 2017, 8, 2694–2707. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Hideshima, T.; Tai, Y.-T.; Song, Y.; Richardson, P.; Raje, N.; Munshi, N.C.; Anderson, K.C. Histone deacetylase (HDAC) inhibitor ACY241 enhances anti-tumor activities of antigen-specific central memory cytotoxic T lymphocytes against multiple myeloma and solid tumors. Leukemia 2018. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.S. ACY-1215, a First-In-Class Selective Inhibitor of HDAC6, Demonstrates Significant Synergy With Immunomodulatory Drugs (IMiDs) In Preclinical Models Of Multiple Myeloma (MM). Blood 2013, 122, 1952. [Google Scholar]
- Cosenza, M.; Civallero, M.; Sacchi, S.; Pozzi, S. Momelotinib and Citarinostat: Co-Targeting JAK2/STAT3 and HDAC6 in Lymphoid Malignancies, a New Potential Therapeutic Combination. Blood 2017, 130, 5201. [Google Scholar]
- Mehrling, T.; Chen, Y. The Alkylating-HDAC Inhibition Fusion Principle: Taking Chemotherapy to the Next Level with the First in Class Molecule EDO-S101. Anticancer Agents Med. Chem. 2016, 16, 20–28. [Google Scholar] [CrossRef] [PubMed]
- López-Iglesias, A.A.; San-Segundo, L.; González-Méndez, L.; Hernández-García, S.; Primo, D.; Garayoa, M.; Hernández, A.B.; Paíno, T.; Mateos, M.-V.; Chen, Y.; et al. The Alkylating Histone Deacetylase Inhibitor Fusion Molecule Edo-S101 Displays Full Bi-Functional Properties in Preclinical Models of Hematological Malignancies. Blood 2014, 124, 2100. [Google Scholar]
- López-Iglesias, A.-A.; Herrero, A.B.; Chesi, M.; San-Segundo, L.; González-Méndez, L.; Hernández-García, S.; Misiewicz-Krzeminska, I.; Quwaider, D.; Martín-Sánchez, M.; Primo, D.; et al. Preclinical anti-myeloma activity of EDO-S101, a new bendamustine-derived molecule with added HDACi activity, through potent DNA damage induction and impairment of DNA repair. J. Hematol. Oncol. 2017, 10, 127. [Google Scholar] [CrossRef] [PubMed]
- Besse, L.; Kraus, M.; Besse, A.; Bader, J.; Silzle, T.; Mehrling, T.; Driessen, C. The first-in-class alkylating HDAC inhibitor EDO-S101 is highly synergistic with proteasome inhibition against multiple myeloma through activation of multiple pathways. Blood Cancer J. 2017, 7, e589. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Xu, K.; Lin, J.; Jayachandran, G.; Wang, B.; Watanabe, Y.; Ge, Q.; Wu, Y.; Guo, D.; Chen, Y.; et al. Abstract 2741: Synergistic inhibition of tumor growth and overcoming chemo-resistance by simultaneously targeting key components in DNA damage/repair, epigenetic, and putative cancer stem cell signaling pathways using novel dual-functional DNA-alkylating/HDAC inhibitor and tumor suppressor gene nanoparticles in lung cancer. Cancer Res. 2012, 72, 2741. [Google Scholar] [CrossRef]
- Shuttleworth, S.J. Abstract 3996: KA2237 and KA2507: Novel, oral cancer immunotherapeutics targeting PI3K-p110β/p110δ and HDAC6 with single-agent and combination activity. Cancer Res. 2016, 76, 3996. [Google Scholar] [CrossRef]
- Dallavalle, S.; Cincinelli, R.; Nannei, R.; Merlini, L.; Morini, G.; Penco, S.; Pisano, C.; Vesci, L.; Barbarino, M.; Zuco, V.; et al. Design, synthesis, and evaluation of biphenyl-4-yl-acrylohydroxamic acid derivatives as histone deacetylase (HDAC) inhibitors. Eur. J. Med. Chem. 2009, 44, 1900–1912. [Google Scholar] [CrossRef] [PubMed]
- Zuco, V.; De Cesare, M.; Cincinelli, R.; Nannei, R.; Pisano, C.; Zaffaroni, N.; Zunino, F. Synergistic antitumor effects of novel HDAC inhibitors and paclitaxel in vitro and in vivo. PLoS ONE 2011, 6, e29085. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Class | Members | Cellular Localization | Biological Functions |
|---|---|---|---|
| I | HDAC1 | Nucleus | Proliferation control, apoptosis; p21 and p27 CDK (cyclin-dependent kinase) inhibitor repression; Represses transcription; Binds to transcription factors; Resistance to chemotherapy; Suppresses cytokine production in activated T cells and during T effector cell differentiation |
| HDAC2 | Nucleus | Negatively regulates transcription by being recruited to DNA as a corepressor; Proliferation control; Apoptosis | |
| HDAC3 | Nucleus | Proliferation; Differentiation, represses transcription; Binds to transcription factors; Deacetylates FOXP3 (forkhead box P3) that reduces Treg development and suppressive function | |
| HDAC8 | Nucleus | Proliferation; Differentiation | |
| IIA | HDAC4 | Nucleus/Cytoplasm | Differentiation, angiogenesis; Deacetylates BCL6 (B-cell lymphoma 6) which activates genes for lymphocyte activation |
| HDAC5 | Nucleus/Cytoplasm | Differentiation; Deacetylates BCL6 which activates genes for lymphocyte activation | |
| HDAC7 | Nucleus/Cytoplasm | Angiogenesis; Suppresses Nur77 expression during TCR (T-cell receptor) negative selection; Regulates gene expression during TCR positive selection; Deacetylates BCL6 which activates genes for lymphocyte activation | |
| HDAC9 | Nucleus/Cytoplasm | Deacetylates FOXP3, which reduces Treg development and immunosuppressive activity | |
| IIB | HDAC6 | Cytoplasm | Regulation of protein degradation both via aggresome and the regulation of Hsp90 chaperone activity; Migration; Angiogenesis; Controls IgM and IgG levels upon antigen stimulation; T-cell migration; Immune synapse formation; Deacetylates FOXP3 that decreases Treg development and immunosuppressive activity |
| HDAC10 | Cytoplasm | Angiogenesis | |
| III | SIRT 1 | Nucleus, Cytoplasm | DNA repair/genome stability; Chromatin organization; Stress; Cancer |
| SIRT 2 | Nucleus | Mitosis; DNA repair; Chromatin condensation | |
| SIRT 3 | Mitochondria | Cancer, chromatin silencing; DNA repair; Cellular stress | |
| SIRT 4 | Mitochondria | have not yet been fully determined | |
| SIRT 5 | Mitochondria | have not yet been fully determined | |
| SIRT6 | Nucleus | DNA repair/genome stability; Telomeric chromatin/senescence | |
| SIRT7 | Nucleus | Cellular transformation | |
| IV | HDAC11 | Nucleus | Regulates the protein stability of DNA replication factor CDT1 (chromatin licensing and DNA replication factor 1) and the expression of IL-10; Suppresses IL10 expression in APCs (antigen presenting cells) |
| Class | Members | Expression of HDACs Increased in Lymphoproliferative Disease (Cell Lines and Primary Cell) | Reference |
|---|---|---|---|
| I | HDAC1 | MM, HL, MCL, DLBCL, ALCL, CLL PTCL, | [59,60,61,62,63,64] |
| HDAC2 | MM, HL, MCL, DLBCL, ALCL, PTCL | [59,60,61,63,64] | |
| HDAC3 | MCL, CLL, DLBCL, HL; MM | [59,60,61,62,63,64,65] | |
| HDAC8 | MM | [60] | |
| IIA | HDAC4 | DLBCL, PTCL | [61,63] |
| HDAC5 | MM | [60] | |
| HDAC7 | CLL, MCL | [59,61,62,63] | |
| HDAC9 | CLL, MCL | [59,62,63] | |
| IIB | HDAC6 | MM, MCL, DLBCL, PTCL, CTCL, CLL, | [59,60,61,62,63,64] |
| HDAC10 | CLL, MCL, HL | [59,61,62,63] | |
| III | SIRT 1 | CLL | [62] |
| SIRT 2 | |||
| SIRT 3 | |||
| SIRT 4 | |||
| SIRT 5 | |||
| SIRT6 | |||
| SIRT7 | CLL | [59,62,63] | |
| IV | HDAC11 | MCL, HL | [63] |
| Class | HDACis | Target HDAC | Clinical Trial Active in Lymphoproliferative Disease (clinicaltrials.gov) |
|---|---|---|---|
| Hidroxamic acids | Trichostatin A | Pan | Preclinical |
| Vorinostat/SAHA | Pan | * Phase I/II/III MM and lymphoma | |
| Belinostat | Pan | ** Phase I/II Lymphoma | |
| Panobinostat | Pan | *** Phase I/II MM and lymphoma | |
| Givinostat | Pan | Phase I/II completed for MM and lymphoma | |
| Resminostat | Pan | Phase II CTCL | |
| Abexinostat | Pan | Phase I/II completed for MM and lymphoma | |
| Quisinostat | Pan | Phase I/II completed for MM and lymphoma | |
| Ricolinostat/Acy-1215 | II selective | Phase I/II clinical trials for MM and lymphoma | |
| Citarinostat/Acy-241 | II selective | Phase I MM | |
| Practilinostat | I, II, IV | / | |
| CHR-3996 | I | / | |
| Aliphatic acid | Valproic acid | I, IIa | Phase I/II completed for lymphoma |
| Butyric acid | I, IIa | Phase I/II completed for lymphoma | |
| Phenylbutyric acid | I, IIa | Phase I/II completed for MM and lymphoma | |
| Benzamides | Entinostat | I | Phase I/II completed—MM. Phase I/II—lymphoma |
| Tacedinaline | I | Phase II completed—MM. | |
| 4SC202 | I | Phase I completed—Advanced Hematologic | |
| Malignancies | |||
| Mocetinostat | I, IV | Phase I/II clinical trials—lymphoma | |
| Cyclic tetrapepides | Romidepsin | I | Approved for * CTCL and ** PTCL |
| Several studies of phase I/II lymphoma | |||
| Phase I/II clinical trials—MM. | |||
| Sirtuins inhibitors | Nicotinamide | Class III | Phase I/II MM. Phase I lymphoma |
| Sirtinol | SIRT 1 and 2 | Preclinical | |
| Cambinol | SIRT 1 and 2 | Preclinical | |
| Ex-527 | SIRT 1 and 2 | Preclinical |
| HDAC6 Inhibitors | Lymphoproliferative Disease | Preclinical and Clinical Study (Ref.) | Clinical Trials State |
|---|---|---|---|
| Ricolinostat (Acy-1215) | MM cell | Alone [145] | Phase 1/2 combo poma and dex in MM (NCT01997840) (active) |
| + Bortezomib [135] | |||
| + Carfilzomib [146] | |||
| + Lenalidomide [78] | |||
| +Dexamethasone [78,147] | |||
| Non-NHL | + Carfilzomib [148] | Phase 1/2 combo lena e dex in MM (NCT01583283) (active) | |
| DLBCL, MCL, TCL | + Bortezomib [136] | Phase 1 combo poma and low-dose dex in relapsed-and-refractory MM (NCT02189343) (active) | |
| DLBCL | + Ibrutinib [149] | Phase 1/2 combo bort and dex in relapsed and refractory MM (NCT01323751) (termined) | |
| + Crizotinib [150] | |||
| FL, MCL, TCL | + Bendamustine [151] | Phase 1 /2 relapsed or refractory lymphoid malignancies (NCT02091063) (recruiting) | |
| Citarinostat (Acy-241) | MM and MCL | + Pomalidomide [80] | Phase 1 combo poma and dex in MM (NCT02400242) (active) |
| + Lenalidomide [80] | |||
| MM | + anti-PD-L1 [152] | ||
| Tubacin | MM and lymphoma | + Bortezomib [42,153] | Preclinical studies. Compound not tested in clinical trials: it is not optimized for oral delivery |
| Burkitt’s lymphoma | [144,154] | ||
| Tubastatin A | Lymphoma | [155,156] | Preclinical studies compound not tested in clinical trials: It is not optimized for oral delivery |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cosenza, M.; Pozzi, S. The Therapeutic Strategy of HDAC6 Inhibitors in Lymphoproliferative Disease. Int. J. Mol. Sci. 2018, 19, 2337. https://doi.org/10.3390/ijms19082337
Cosenza M, Pozzi S. The Therapeutic Strategy of HDAC6 Inhibitors in Lymphoproliferative Disease. International Journal of Molecular Sciences. 2018; 19(8):2337. https://doi.org/10.3390/ijms19082337
Chicago/Turabian StyleCosenza, Maria, and Samantha Pozzi. 2018. "The Therapeutic Strategy of HDAC6 Inhibitors in Lymphoproliferative Disease" International Journal of Molecular Sciences 19, no. 8: 2337. https://doi.org/10.3390/ijms19082337
APA StyleCosenza, M., & Pozzi, S. (2018). The Therapeutic Strategy of HDAC6 Inhibitors in Lymphoproliferative Disease. International Journal of Molecular Sciences, 19(8), 2337. https://doi.org/10.3390/ijms19082337