Modeling Endometrial Cancer: Past, Present, and Future


Abstract
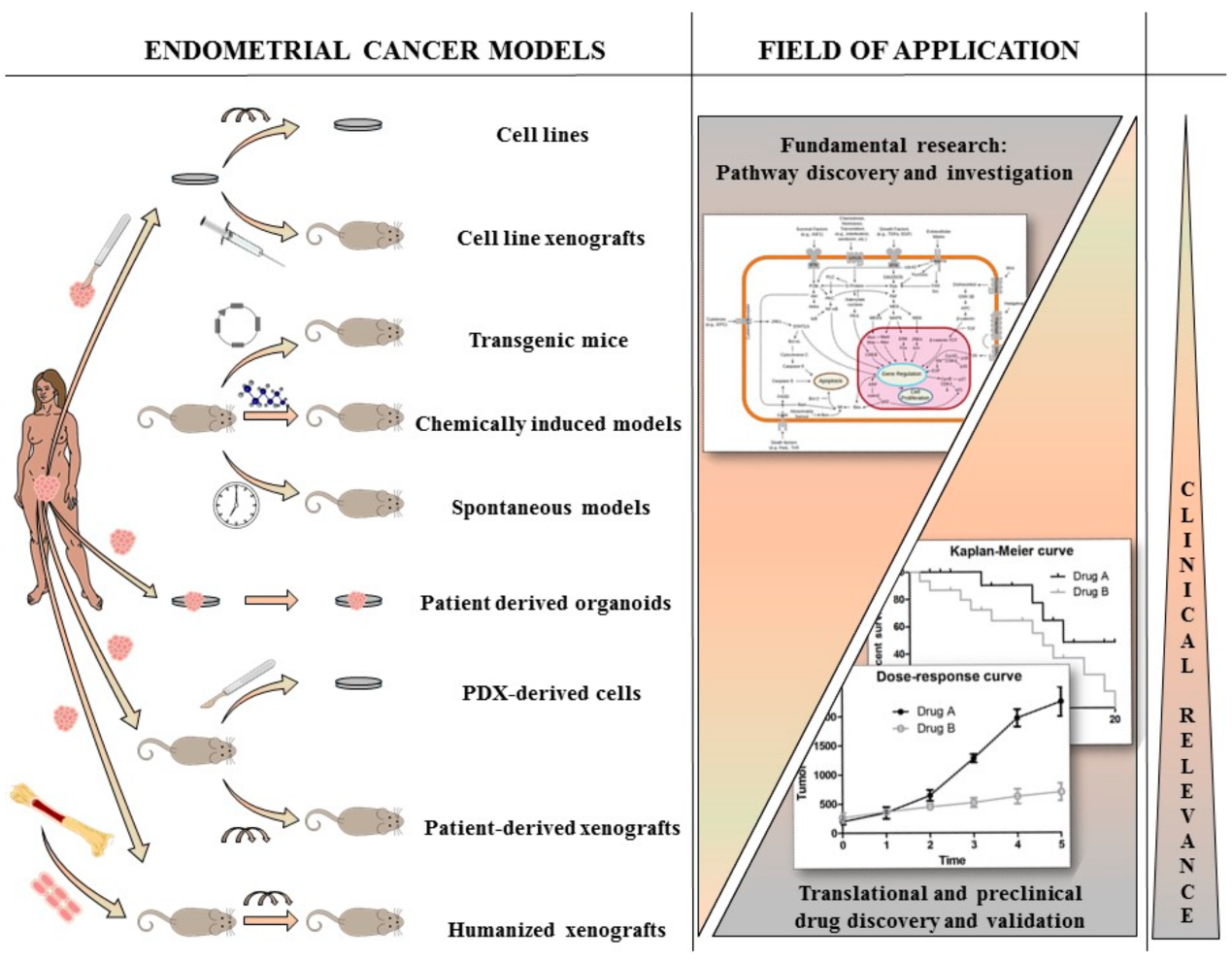
:1. Introduction
2. Cell Lines and Cell Line-Derived Xenograft Models
3. Organoids and Organs-on-a-Chip Models
3.1. EC Organoids Models
3.2. Organs-on-Chip Models
4. In Vivo Models
4.1. Spontaneous EC Rodent Models
4.2. Chemically Induced EC Rodent Models
4.3. Transgenic Mouse Models
4.3.1. PTEN Knock-Out Mouse Models
4.3.2. TP53 Knock-Out Models
4.3.3. The Mitogen Inducible Gene 6 (MIG-6) Knock-Out Model
4.3.4. Transgenic Models: Remarks
4.4. Patient-Derived Xenografts (PDXs) and Humanized Mice
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3D | Three-dimensional |
| Akt | Protein kinase B |
| AMPK | Adenosine monophosphate-activated protein kinase |
| ARID1A | AT-rich interaction domain 1A |
| BDII/Han | Berlin-Druckrey II/Hannover |
| CCLE | Cancer Cell Line Encyclopedia |
| CDK4,6 | Cyclin-dependent kinase 4,6 |
| Cdk6 | Cyclin-dependent kinase 6 |
| CMT1000 | Center for Molecular Therapeutics 1000 |
| Cre-lox | Cyclization recombinase-locus of X-over P1 |
| CTNNB1 | Catenin beta 1 |
| DA/Han | Dark Agouti/Hannover |
| E:P | Estrogen:progesterone |
| EC | Endometrial cancer |
| EEC | Endometrioid endometrial cancer |
| EGFR | Epidermal growth factor receptor |
| ENNG | N-ethyl-N-nitro-N-nitrosoguanidine |
| ER | Estrogen receptor |
| ERBB2 | Erb-b2 receptor tyrosine kinase 2 |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| F344 | Fischer 344 |
| FBXW7 | F-box and WD repeat domain containing protein 7 |
| FGFR2 | Fibroblast growth factor receptor 2 |
| FIGO | International Federation of Gynecology and Obstetrics |
| Han:Wistar | Hannover Wistar |
| HOTAIR | Homeobox transcript antisense RNA |
| ICR | Institute of Cancer Research |
| JFCR-39 | Japanese Foundation of Cancer Research 39 |
| KRAS | KRAS proto-oncogene, GTPase |
| LKB1 | Serine/threonine kinase 11 |
| loxP | Locus of X-over P1 |
| MDM4 | Double minute 4 protein |
| MIG-6 | Mitogen Inducible Gene 6 |
| MLH | MutL homolog 1 |
| MNU | N-methyl-N-nitrosourea |
| mTOR | Mammalian target of rapamycin |
| NCI60 | National Cancer Institute 60 |
| OOC | Organ-on-chip |
| PARP | Poly (ADP-ribose) polymerase |
| PI3K | Phosphoinositide 3-kinase |
| PIK3CA | Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha |
| PDX | Patient-derived xenograft |
| PDC | Patient-derived cell culture |
| PMS2 | Mismatch repair endonuclease PMS2 |
| POLE | Polymerase ε |
| POT1A | Protection of telomeres protein 1A |
| PR | Progesterone receptor |
| PTEN | Phosphatase and tensin homolog |
| STAT3STR | Signal transducer and activator of transcription 3Short tandem repeat |
| TP53 | Tumor protein p53 |
| Wnt | Wingless/integration-1 |
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in Globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Morice, P.; Leary, A.; Creutzberg, C.; Abu-Rustum, N.; Darai, E. Endometrial cancer. Lancet 2016, 387, 1094–1108. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Amant, F.; Moerman, P.; Neven, P.; Timmerman, D.; van Limbergen, E.; Vergote, I. Endometrial cancer. Lancet 2005, 366, 491–505. [Google Scholar] [CrossRef]
- Saso, S.; Chatterjee, J.; Georgiou, E.; Ditri, A.M.; Smith, J.R.; Ghaem-Maghami, S. Endometrial cancer. BMJ 2011, 343, d3954. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Watanabe, M.; Amikura, T.; Obata, H.; Sekine, M.; Yahata, T.; Fujita, K.; Tanaka, K. Adjuvant chemotherapy as treatment of high-risk stage I and II endometrial cancer. Gynecol. Oncol. 2004, 94, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Dizon, D.S. Treatment options for advanced endometrial carcinoma. Gynecol. Oncol. 2010, 117, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Nogami, Y.; Banno, K.; Kisu, I.; Yanokura, M.; Umene, K.; Masuda, K.; Kobayashi, Y.; Yamagami, W.; Nomura, H.; Tominaga, E.; et al. Current status of molecular-targeted drugs for endometrial cancer (review). Mol. Clin. Oncol. 2013, 1, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Oza, A.M. Endometrial cancer-targeted therapies myth or reality? Review of current targeted treatments. Eur. J. Cancer 2016, 59, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Brasseur, K.; Gevry, N.; Asselin, E. Chemoresistance and targeted therapies in ovarian and endometrial cancers. Oncotarget 2017, 8, 4008–4042. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Bokhman, J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983, 15, 10–17. [Google Scholar] [CrossRef]
- Lax, S.F.; Kurman, R.J. A dualistic model for endometrial carcinogenesis based on immunohistochemical and molecular genetic analyses. Verh. Dtsch. Ges. Pathol. 1997, 81, 228–232. [Google Scholar] [PubMed]
- Buhtoiarova, T.N.; Brenner, C.A.; Singh, M. Endometrial carcinoma: Role of current and emerging biomarkers in resolving persistent clinical dilemmas. Am. J. Clin. Pathol. 2016, 145, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; Benz, C.C.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [PubMed] [Green Version]
- Ellenson, L.H.; Wu, T.C. Focus on endometrial and cervical cancer. Cancer Cell 2004, 5, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, C.A.; Cheung, M.K.; Osann, K.; Chen, L.; Teng, N.N.; Longacre, T.A.; Powell, M.A.; Hendrickson, M.R.; Kapp, D.S.; Chan, J.K. Uterine papillary serous and clear cell carcinomas predict for poorer survival compared to grade 3 endometrioid corpus cancers. Br. J. Cancer 2006, 94, 642–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirisano, F.D.; Robboy, S.J.; Dodge, R.K.; Bentley, R.C.; Krigman, H.R.; Synan, I.S.; Soper, J.T.; Clarke-Pearson, D.L. Epidemiologic and surgicopathologic findings of papillary serous and clear cell endometrial cancers when compared to endometrioid carcinoma. Gynecol. Oncol. 1999, 74, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Depreeuw, J.; Stelloo, E.; Osse, E.; Creutzberg, C.; Nout, R.; Moisse, M.; Garcia-Dios, D.; Dewaele, M.; Willekens, K.; Marine, J.; et al. Amplification of 1q32.1 refines the molecular classification of endometrial carcinoma. Clin. Cancer Res. 2017, 23, 7232–7241. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Haber, D.A.; Settleman, J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat. Rev. Cancer 2010, 10, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Korch, C.; Spillman, M.A.; Jackson, T.A.; Jacobsen, B.M.; Murphy, S.K.; Lessey, B.A.; Jordan, V.C.; Bradford, A.P. DNA profiling analysis of endometrial and ovarian cell lines reveals misidentification, redundancy and contamination. Gynecol. Oncol. 2012, 127, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaab, W.E.; Risinger, J.I.; Umar, A.; Kunkel, T.A.; Barrett, J.C.; Tindall, K.R. Characterization of distinct human endometrial carcinoma cell lines deficient in mismatch repair that originated from a single tumor. J. Biol. Chem. 1998, 273, 26662–26669. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Warne, P.H.; Lambros, M.B.; Reis-Filho, J.S.; Downward, J. PI3K pathway dependencies in endometrioid endometrial cancer cell lines. Clin. Cancer Res. 2013, 19, 3533–3544. [Google Scholar] [CrossRef] [PubMed]
- McConechy, M.K.; Ding, J.; Cheang, M.C.; Wiegand, K.; Senz, J.; Tone, A.; Yang, W.; Prentice, L.; Tse, K.; Zeng, T.; et al. Use of mutation profiles to refine the classification of endometrial carcinomas. J. Pathol. 2012, 228, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Stokoe, D.; Taketani, Y.; McCormick, F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 2005, 65, 10669–10673. [Google Scholar] [CrossRef] [PubMed]
- Cheung, L.W.; Hennessy, B.T.; Li, J.; Yu, S.; Myers, A.P.; Djordjevic, B.; Lu, Y.; Stemke-Hale, K.; Dyer, M.D.; Zhang, F.; et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011, 1, 170–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, D.; Cogdell, D.; Hu, L.; Xue, F.; Broaddus, R.; Zhang, W. Genomic characterization of gene copy-number aberrations in endometrial carcinoma cell lines derived from endometrioid-type endometrial adenocarcinoma. Technol. Cancer Res. Treat. 2010, 9, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Philip, C.A.; Laskov, I.; Beauchamp, M.C.; Marques, M.; Amin, O.; Bitharas, J.; Kessous, R.; Kogan, L.; Baloch, T.; Gotlieb, W.H.; et al. Inhibition of PI3K-Akt-mTOR pathway sensitizes endometrial cancer cell lines to PARP inhibitors. BMC Cancer 2017, 17, 638. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Wang, Y.; Chen, D.; Sheng, X.; Liu, J.; Xiong, H. Cisplatin regulates cell autophagy in endometrial cancer cells via the PI3K/Akt/mTOR signalling pathway. Oncol. Lett. 2017, 13, 3567–3571. [Google Scholar] [CrossRef] [PubMed]
- Dedes, K.J.; Wetterskog, D.; Ashworth, A.; Kaye, S.B.; Reis-Filho, J.S. Emerging therapeutic targets in endometrial cancer. Nat. Rev. Clin. Oncol. 2011, 8, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Vallone, C.; Rigon, G.; Gulia, C.; Baffa, A.; Votino, R.; Morosetti, G.; Zaami, S.; Briganti, V.; Catania, F.; Gaffi, M.; et al. Non-coding RNAs and endometrial cancer. Genes (Basel) 2018, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.Y.; Zhu, J.Y.; Zhang, C.Y.; Zhang, M.; Song, Y.N.; Rahman, K.; Zhang, L.J.; Zhang, H. Autophagy regulated by lncRNA HOTAIR contributes to the cisplatin-induced resistance in endometrial cancer cells. Biotechnol. Lett. 2017, 39, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Z.; Yu, H. MiR-205 inhibits cell growth by targeting Akt-mTOR signaling in progesterone-resistant endometrial cancer Ishikawa cells. Oncotarget 2017, 8, 28042–28051. [Google Scholar] [CrossRef] [PubMed]
- Daley-Brown, D.; Oprea-Iles, G.; Vann, K.T.; Lanier, V.; Lee, R.; Candelaria, P.V.; Quarshie, A.; Pattillo, R.; Gonzalez-Perez, R.R. Type II endometrial cancer overexpresses NILCO: A preliminary evaluation. Dis. Mark. 2017, 2017, 8248175. [Google Scholar] [CrossRef] [PubMed]
- Cali, G.; Insabato, L.; Conza, D.; Bifulco, G.; Parrillo, L.; Mirra, P.; Fiory, F.; Miele, C.; Raciti, G.A.; Di Jeso, B.; et al. Grp78 mediates cell growth and invasiveness in endometrial cancer. J. Cell Physiol. 2014, 229, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Zhang, D.; Shi, H.; Bian, Y.; Guo, R. MiR-143 inhibits endometrial cancer cell proliferation and metastasis by targeting MAPK1. Oncotarget 2017, 8, 84384–84395. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, C.; Jiang, Y.; Wan, Y.; Zhou, S.; Cheng, W. Tumor-suppressor role of miR-139-5p in endometrial cancer. Cancer Cell Int. 2018, 18, 51. [Google Scholar] [CrossRef] [PubMed]
- Oki, S.; Sone, K.; Oda, K.; Hamamoto, R.; Ikemura, M.; Maeda, D.; Takeuchi, M.; Tanikawa, M.; Mori-Uchino, M.; Nagasaka, K.; et al. Oncogenic histone methyltransferase EZH2: A novel prognostic marker with therapeutic potential in endometrial cancer. Oncotarget 2017, 8, 40402–40411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartosch, C.; Lopes, J.M.; Jeronimo, C. Epigenetics in endometrial carcinogenesis—Part 2: Histone modifications, chromatin remodeling and noncoding RNAs. Epigenomics 2017, 9, 873–892. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, C.; Lopes, J.M.; Jeronimo, C. Epigenetics in endometrial carcinogenesis—Part 1: DNA methylation. Epigenomics 2017, 9, 737–755. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Fan, Y.; Xu, W.; Chen, J.; Meng, Y.; Fang, D.; Wang, J. Exploration of miR-1202 and miR-196a in human endometrial cancer based on high throughout gene screening analysis. Oncol. Rep. 2017, 37, 3493–3501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brasseur, K.; Fabi, F.; Adam, P.; Parent, S.; Lessard, L.; Asselin, E. Post-translational regulation of the cleaved fragment of Par-4 in ovarian and endometrial cancer cells. Oncotarget 2016, 7, 36971–36987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hevir-Kene, N.; Rizner, T.L. The endometrial cancer cell lines Ishikawa and HEC-1A, and the control cell line HIEEC, differ in expression of estrogen biosynthetic and metabolic genes, and in androstenedione and estrone-sulfate metabolism. Chem. Biol. Interact. 2015, 234, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Bai, H.; Liu, S.; Cao, D.; Wu, H.; Shen, K.; Tai, Y.; Yang, J. Targeting stearoyl-Coa desaturase 1 to repress endometrial cancer progression. Oncotarget 2018, 9, 12064–12078. [Google Scholar] [CrossRef] [PubMed]
- Byrne, F.L.; Poon, I.K.; Modesitt, S.C.; Tomsig, J.L.; Chow, J.D.; Healy, M.E.; Baker, W.D.; Atkins, K.A.; Lancaster, J.M.; Marchion, D.C.; et al. Metabolic vulnerabilities in endometrial cancer. Cancer Res. 2014, 74, 5832–5845. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.D.; van Geldermalsen, M.; Otte, N.J.; Lum, T.; Vellozzi, M.; Thoeng, A.; Pang, A.; Nagarajah, R.; Zhang, B.; Wang, Q.; et al. ASCT2 regulates glutamine uptake and cell growth in endometrial carcinoma. Oncogenesis 2017, 6, e367. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Ishikawa, K.; Kawai, S.; Torii, Y.; Kawamura, K.; Kato, R.; Tsukada, K.; Udagawa, Y. Overcoming paclitaxel resistance in uterine endometrial cancer using a COX-2 inhibitor. Oncol. Rep. 2013, 30, 2937–2944. [Google Scholar] [CrossRef] [PubMed]
- Kozak, J.; Wdowiak, P.; Maciejewski, R.; Torres, A. A guide for endometrial cancer cell lines functional assays using the measurements of electronic impedance. Cytotechnology 2018, 70, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, T.; Yogosawa, S.; Yamada, T.; Kitawaki, J.; Sakai, T. Combination of a novel HDAC inhibitor OBP-801/YM753 and a PI3K inhibitor LY294002 synergistically induces apoptosis in human endometrial carcinoma cells due to increase of Bim with accumulation of ROS. Gynecol. Oncol. 2013, 129, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Aslan, O.; Cremona, M.; Morgan, C.; Cheung, L.W.; Mills, G.B.; Hennessy, B.T. Preclinical evaluation and reverse phase protein array-based profiling of PI3K and MEK inhibitors in endometrial carcinoma in vitro. BMC Cancer 2018, 18, 168. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.I.; Schultz, C.R.; Buras, A.L.; Friedman, E.; Fedorko, A.; Seamon, L.; Chandramouli, G.V.R.; Maxwell, G.L.; Bachmann, A.S.; Risinger, J.I. Ornithine decarboxylase as a therapeutic target for endometrial cancer. PLoS ONE 2017, 12, e0189044. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, R.H. The nci60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Yamori, T. JFCR39, a panel of 39 human cancer cell lines, and its application in the discovery and development of anticancer drugs. Bioorg. Med. Chem. 2012, 20, 1947–1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, U.; Sharma, S.V.; Dowell, L.; Greninger, P.; Montagut, C.; Lamb, J.; Archibald, H.; Raudales, R.; Tam, A.; Lee, D.; et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc. Natl. Acad. Sci. USA 2007, 104, 19936–19941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boretto, M.; Cox, B.; Noben, M.; Hendriks, N.; Fassbender, A.; Roose, H.; Amant, F.; Timmerman, D.; Tomassetti, C.; Vanhie, A.; et al. Development of organoids from mouse and human endometrium showing endometrial epithelium physiology and long-term expandability. Development 2017, 144, 1775–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turco, M.Y.; Gardner, L.; Hughes, J.; Cindrova-Davies, T.; Gomez, M.J.; Farrell, L.; Hollinshead, M.; Marsh, S.G.E.; Brosens, J.J.; Critchley, H.O.; et al. Long-term, hormone-responsive organoid cultures of human endometrium in a chemically defined medium. Nat. Cell Biol. 2017, 19, 568–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.P.T.; Xiao, L.; Deane, J.A.; Tan, K.S.; Cousins, F.L.; Masuda, H.; Sprung, C.N.; Rosamilia, A.; Gargett, C.E. N-cadherin identifies human endometrial epithelial progenitor cells by in vitro stem cell assays. Hum. Reprod. 2017, 32, 2254–2268. [Google Scholar] [CrossRef] [PubMed]
- Van der Zee, M.; Sacchetti, A.; Cansoy, M.; Joosten, R.; Teeuwssen, M.; Heijmans-Antonissen, C.; Ewing-Graham, P.C.; Burger, C.W.; Blok, L.J.; Fodde, R. IL6/JAK1/STAT3 signaling blockade in endometrial cancer affects the ALDHhi/CD126+ stem-like component and reduces tumor burden. Cancer Res. 2015, 75, 3608–3622. [Google Scholar] [CrossRef] [PubMed]
- Kiyohara, Y.; Yoshino, K.; Kubota, S.; Okuyama, H.; Endo, H.; Ueda, Y.; Kimura, T.; Kimura, T.; Kamiura, S.; Inoue, M. Drug screening and grouping by sensitivity with a panel of primary cultured cancer spheroids derived from endometrial cancer. Cancer Sci. 2016, 107, 452–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girda, E.; Huang, E.C.; Leiserowitz, G.S.; Smith, L.H. The use of endometrial cancer patient-derived organoid culture for drug sensitivity testing is feasible. Int. J. Gynecol. Cancer 2017, 27, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Dasari, V.R.; Mazack, V.; Feng, W.; Nash, J.; Carey, D.J.; Gogoi, R. Verteporfin exhibits yap-independent anti-proliferative and cytotoxic effects in endometrial cancer cells. Oncotarget 2017, 8, 28628–28640. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Hassell, B.A.; Goyal, G.; Lee, E.; Sontheimer-Phelps, A.; Levy, O.; Chen, C.S.; Ingber, D.E. Human organ chip models recapitulate orthotopic lung cancer growth, therapeutic responses, and tumor dormancy in vitro. Cell Rep. 2017, 21, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.E.; Yang, N.; Pehlke, C.; Keely, P.J.; Eliceiri, K.W.; Friedl, A.; Beebe, D.J. Transition to invasion in breast cancer: A microfluidic in vitro model enables examination of spatial and temporal effects. Integr. Biol. (Camb.) 2011, 3, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Coppeta, J.R.; Rogers, H.B.; Isenberg, B.C.; Zhu, J.; Olalekan, S.A.; McKinnon, K.E.; Dokic, D.; Rashedi, A.S.; Haisenleder, D.J.; et al. A microfluidic culture model of the human reproductive tract and 28-day menstrual cycle. Nat. Commun. 2017, 8, 14584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Rangarajan, A.; Weinberg, R.A. Opinion: Comparative biology of mouse versus human cells: Modelling human cancer in mice. Nat. Rev. Cancer 2003, 3, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N.; Ukraintseva, S.V.; Yashin, A.I. Cancer in rodents: Does it tell us about cancer in humans? Nat. Rev. Cancer 2005, 5, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Jia, Y.; Chen, L.; Ezeogu, L.; Yu, B.; Xu, N.; Liao, D.J. Weaknesses and pitfalls of using mice and rats in cancer chemoprevention studies. J. Cancer 2015, 6, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Deerberg, F.; Rehm, S.; Pittermann, W. Uncommon frequency of adenocarcinomas of the uterus in virgin Han: Wistar rats. Vet. Pathol. 1981, 18, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, G. Endometrial cancer: Experimental models useful for studies on molecular aspects of endometrial cancer and carcinogenesis. Endocr. Relat. Cancer 2003, 10, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, T.; Onodera, H.; Matsushima, Y.; Todate, A.; Shibutani, M.; Ogasawara, H.; Maekawa, A. Spontaneous uterine adenocarcinomas in aged rats and their relation to endocrine imbalance. J. Cancer Res. Clin. Oncol. 1990, 116, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Tanoguchi, K.; Yaegashi, N.; Jiko, K.; Maekawa, A.; Sato, S.; Yajima, A. K-ras point mutations in spontaneously occurring endometrial adenocarcinomas in the Donryu rat. Tohoku J. Exp. Med. 1999, 189, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, T.; Takeuchi, M.; Onodera, H.; Matsushima, Y.; Ando-Lu, J.; Maekawa, A. Sequential observation of spontaneous endometrial adenocarcinoma development in Donryu rats. Toxicol. Pathol. 1994, 22, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Watanabe, G.; Suzuki, T.; Inoue, K.; Takahashi, M.; Maekawa, A.; Taya, K.; Nishikawa, A. Long-term treatment with bromocriptine inhibits endometrial adenocarcinoma development in rats. J. Reprod. Dev. 2009, 55, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Tanaka, T.; Mori, H. Chemoprevention of spontaneous endometrial cancer in female Donryu rats by dietary indole-3-carbinol. Cancer Res. 1994, 54, 1446–1449. [Google Scholar] [PubMed]
- Yoshida, M.; Katsuda, S.; Tanimoto, T.; Asai, S.; Nakae, D.; Kurokawa, Y.; Taya, K.; Maekawa, A. Induction of different types of uterine adenocarcinomas in Donryu rats due to neonatal exposure to high-dose p-t-octylphenol for different periods. Carcinogenesis 2002, 23, 1745–1750. [Google Scholar] [CrossRef] [PubMed]
- Kakehashi, A.; Tago, Y.; Yoshida, M.; Sokuza, Y.; Wei, M.; Fukushima, S.; Wanibuchi, H. Hormonally active doses of isoflavone aglycones promote mammary and endometrial carcinogenesis and alter the molecular tumor environment in Donryu rats. Toxicol. Sci. 2012, 126, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, T.; Onodera, H.; Hayashi, Y.; Maekawa, A. Influence of high-fat diets on the occurrence of spontaneous uterine endometrial adenocarcinomas in rats. Teratog. Carcinog. Mutagen. 1995, 15, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, T.; Takegawa, K.; Takeuchi, M.; Maekawa, A. Effects of reproduction on spontaneous development of endometrial adenocarcinomas and mammary tumors in Donryu rats. Jpn. J. Cancer Res. 2000, 91, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Kaspareit-Rittinghausen, J.; Deerberg, F.; Rapp, K. Mortality and incidence of spontaneous neoplasms in BDII/Han rats. Z. Versuchstierkd. 1987, 30, 209–216. [Google Scholar] [PubMed]
- Samuelson, E.; Hedberg, C.; Nilsson, S.; Behboudi, A. Molecular classification of spontaneous endometrial adenocarcinomas in BDII rats. Endocr. Relat. Cancer 2009, 16, 99–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deerberg, F.; Pohlmeyer, G.; Lorcher, K.; Petrow, V. Total suppression of spontaneous endometrial carcinoma in BDII/Han rats by melengestrol acetate. Oncology 1995, 52, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Adamovic, T.; Hamta, A.; Roshani, L.; Lu, X.; Rohme, D.; Helou, K.; Klinga-Levan, K.; Levan, G. Rearrangement and allelic imbalance on chromosome 5 leads to homozygous deletions in the CDKN2A/2B tumor suppressor gene region in rat endometrial cancer. Cancer Genet. Cytogenet. 2008, 184, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Nordlander, C.; Samuelson, E.; Klinga-Levan, K.; Behboudi, A. Recurrent chromosome 10 aberrations and Tp53 mutations in rat endometrial adenocarcinomas. Adv. Exp. Med. Biol. 2008, 617, 519–525. [Google Scholar] [PubMed]
- Samuelson, E.; Nordlander, C.; Levan, G.; Behboudi, A. Amplification studies of met and CDK6 in a rat endometrial tumor model and their correlation to human type I endometrial carcinoma tumors. Adv. Exp. Med. Biol. 2008, 617, 511–517. [Google Scholar] [PubMed]
- Karlsson, S.; Klinga-Levan, K. Expression analysis of human endometrial adenocarcinoma in an inbred rat model. Adv. Exp. Med. Biol. 2008, 617, 503–509. [Google Scholar] [PubMed]
- Falck, E.; Klinga-Levan, K. Expression patterns of Phf5a/PHF5A and Gja1/GJA1 in rat and human endometrial cancer. Cancer Cell Int. 2013, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Samuelson, E.; Levan, K.; Adamovic, T.; Levan, G.; Horvath, G. Recurrent gene amplifications in human type I endometrial adenocarcinoma detected by fluorescence in situ hybridization. Cancer Genet. Cytogenet. 2008, 181, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Walentinsson, A.; Helou, K.; Wallenius, V.; Hedrich, H.J.; Szpirer, C.; Levan, G. Independent amplification of two gene clusters on chromosome 4 in rat endometrial cancer: Identification and molecular characterization. Cancer Res. 2001, 61, 8263–8273. [Google Scholar] [PubMed]
- Roshani, L.; Mallon, P.; Sjostrand, E.; Wedekind, D.; Szpirer, J.; Szpirer, C.; Hedrich, H.J.; Klinga-Levan, K. Genetic analysis of susceptibility to endometrial adenocarcinoma in the BDII rat model. Cancer Genet. Cytogenet. 2005, 158, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Hamta, A.; Adamovic, T.; Heloua, K.; Levan, G. Cytogenetic aberrations in spontaneous endometrial adenocarcinomas in the BDII rat model as revealed by chromosome banding and comparative genome hybridization. Cancer Genet. Cytogenet. 2005, 159, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Willson, C.J.; Herbert, R.A.; Cline, J.M. Hormone receptor expression in spontaneous uterine adenocarcinoma in fischer 344 rats. Toxicol. Pathol. 2015, 43, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Onogi, K.; Niwa, K.; Tang, L.; Yun, W.; Mori, H.; Tamaya, T. Inhibitory effects of Hochu-ekki-to on endometrial carcinogenesis induced by N-methyl-N-nitrosourea and 17beta-estradiol in mice. Oncol. Rep. 2006, 16, 1343–1348. [Google Scholar] [PubMed]
- Niwa, K.; Hashimoto, M.; Morishita, S.; Yokoyama, Y.; Lian, Z.; Tagami, K.; Mori, H.; Tamaya, T. Preventive effects of danazol on endometrial carcinogenesis in mice. Cancer Lett. 2000, 158, 133–139. [Google Scholar] [CrossRef]
- Yoshida, M.; Katashima, S.; Ando, J.; Tanaka, T.; Uematsu, F.; Nakae, D.; Maekawa, A. Dietary indole-3-carbinol promotes endometrial adenocarcinoma development in rats initiated with N-ethyl-N′-nitro-n-nitrosoguanidine, with induction of cytochrome p450s in the liver and consequent modulation of estrogen metabolism. Carcinogenesis 2004, 25, 2257–2264. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Shimomoto, T.; Miyajima, K.; Yoshida, M.; Katashima, S.; Uematsu, F.; Maekawa, A.; Nakae, D. Effects of estrogens and metabolites on endometrial carcinogenesis in young adult mice initiated with N-ethyl-N′-nitro-N-nitrosoguanidine. Cancer Lett. 2004, 211, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Osterman-Golkar, S. Reaction kinetics of N-methyl-N′-nitro-N-nitrosoguanidine and N-ethyl-N′-nitro-N-nitrosoguanidine. Mutat. Res. 1974, 24, 219–226. [Google Scholar] [CrossRef]
- Niwa, K.; Murase, T.; Furui, T.; Morishita, S.; Mori, H.; Tanaka, T.; Tamaya, T. Enhancing effects of estrogens on endometrial carcinogenesis initiated by N-methyl-N-nitrosourea in ICR mice. Jpn. J. Cancer Res. 1993, 84, 951–955. [Google Scholar] [CrossRef] [PubMed]
- Niwa, K.; Tanaka, T.; Mori, H.; Yokoyama, Y.; Furui, T.; Tamaya, T. Rapid induction of endometrial carcinoma in ICR mice treated with N-methyl-N-nitrosourea and 17 beta-estradiol. Jpn. J. Cancer Res. 1991, 82, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Shimomoto, T.; Miyajima, K.; Iizuka, S.; Watanabe, T.; Yoshida, M.; Kurokawa, Y.; Maekawa, A. Promotion, but not progression, effects of tamoxifen on uterine carcinogenesis in mice initiated with N-ethyl-N′-nitro-N-nitrosoguanidine. Carcinogenesis 2002, 23, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- Daikoku, T.; Hirota, Y.; Tranguch, S.; Joshi, A.R.; DeMayo, F.J.; Lydon, J.P.; Ellenson, L.H.; Dey, S.K. Conditional loss of uterine PTEN unfailingly and rapidly induces endometrial cancer in mice. Cancer Res. 2008, 68, 5619–5627. [Google Scholar] [CrossRef] [PubMed]
- Mirantes, C.; Eritja, N.; Dosil, M.A.; Santacana, M.; Pallares, J.; Gatius, S.; Bergada, L.; Maiques, O.; Matias-Guiu, X.; Dolcet, X. An inducible knockout mouse to model the cell-autonomous role of PTEN in initiating endometrial, prostate and thyroid neoplasias. Dis. Model Mech. 2013, 6, 710–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Liu, P.; Zhang, F.; Xu, E.; Symonds, L.; Ohlson, C.E.; Bronson, R.T.; Maira, S.M.; Di Tomaso, E.; Li, J.; et al. A genetic mouse model of invasive endometrial cancer driven by concurrent loss of Pten and Lkb1 is highly responsive to mTOR inhibition. Cancer Res. 2014, 74, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Janzen, D.M.; Paik, D.Y.; Rosales, M.A.; Yep, B.; Cheng, D.; Witte, O.N.; Kayadibi, H.; Ryan, C.M.; Jung, M.E.; Faull, K.; et al. Low levels of circulating estrogen sensitize PTEN-null endometrial tumors to PARP inhibition in vivo. Mol. Cancer Ther. 2013, 12, 2917–2928. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Douglas, W.; Lia, M.; Edelmann, W.; Kucherlapati, R.; Podsypanina, K.; Parsons, R.; Ellenson, L.H. DNA mismatch repair deficiency accelerates endometrial tumorigenesis in PTEN heterozygous mice. Am. J. Pathol. 2002, 160, 1481–1486. [Google Scholar] [CrossRef]
- Contreras, C.M.; Akbay, E.A.; Gallardo, T.D.; Haynie, J.M.; Sharma, S.; Tagao, O.; Bardeesy, N.; Takahashi, M.; Settleman, J.; Wong, K.K.; et al. Lkb1 inactivation is sufficient to drive endometrial cancers that are aggressive yet highly responsive to mTOR inhibitor monotherapy. Dis. Model Mech. 2010, 3, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eritja, N.; Dolcet, X.; Matias-Guiu, X. Three-dimensional epithelial cultures: A tool to model cancer development and progression. Histol. Histopathol. 2013, 28, 1245–1256. [Google Scholar] [PubMed]
- Eritja, N.; Santacana, M.; Maiques, O.; Gonzalez-Tallada, X.; Dolcet, X.; Matias-Guiu, X. Modeling glands with PTEN deficient cells and microscopic methods for assessing PTEN loss: Endometrial cancer as a model. Methods 2015, 77–78, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Akbay, E.A.; Pena, C.G.; Ruder, D.; Michel, J.A.; Nakada, Y.; Pathak, S.; Multani, A.S.; Chang, S.; Castrillon, D.H. Cooperation between p53 and the telomere-protecting shelterin component pot1a in endometrial carcinogenesis. Oncogene 2013, 32, 2211–2219. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Yoo, J.Y.; Jeong, J.W. Mig-6 mouse model of endometrial cancer. Adv. Exp. Med. Biol. 2017, 943, 243–259. [Google Scholar] [PubMed]
- Kim, T.H.; Yoo, J.Y.; Kim, H.I.; Gilbert, J.; Ku, B.J.; Li, J.; Mills, G.B.; Broaddus, R.R.; Lydon, J.P.; Lim, J.M.; et al. MIG-6 suppresses endometrial cancer associated with PTEN deficiency and ERK activation. Cancer Res. 2014, 74, 7371–7382. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Wang, H.; Jiang, G.; Douglas, W.; Chan, J.S.; Korach, K.S.; Ellenson, L.H. Endometrial tumorigenesis in PTEN(+/−) mice is independent of coexistence of estrogen and estrogen receptor α. Am. J. Pathol. 2012, 180, 2536–2547. [Google Scholar] [CrossRef] [PubMed]
- Saito, F.; Tashiro, H.; To, Y.; Ohtake, H.; Ohba, T.; Suzuki, A.; Katabuchi, H. Mutual contribution of PTEN and estrogen to endometrial carcinogenesis in a PtenloxP/LoxP mouse model. Int. J. Gynecol. Cancer 2011, 21, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Saito, F.; Tashiro, H.; Yamaguchi, M.; Honda, R.; Ohba, T.; Suzuki, A.; Katabuchi, H. Development of a mouse model for testing therapeutic agents: The anticancer effect of dienogest on endometrial neoplasms. Gynecol. Endocrinol. 2016, 32, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Dosil, M.A.; Mirantes, C.; Eritja, N.; Felip, I.; Navaridas, R.; Gatius, S.; Santacana, M.; Colas, E.; Moiola, C.; Schoenenberger, J.A.; et al. Palbociclib has antitumour effects on Pten-deficient endometrial neoplasias. J. Pathol. 2017, 242, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Cheng, L.; Bi, X.; Zhang, X.; Liu, S.; Bai, X.; Li, F.; Zhao, A.Z. Elevation of ω-3 polyunsaturated fatty acids attenuates PTEN-deficiency induced endometrial cancer development through regulation of COX-2 and PGE2 production. Sci. Rep. 2015, 5, 14958. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Evans, D.B.; Wang, H.; Abbruzzese, J.L.; Fleming, J.B.; Gallick, G.E. Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat. Protoc. 2009, 4, 1670–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Bruna, A.; Rueda, O.M.; Greenwood, W.; Batra, A.S.; Callari, M.; Batra, R.N.; Pogrebniak, K.; Sandoval, J.; Cassidy, J.W.; Tufegdzic-Vidakovic, A.; et al. A biobank of breast cancer explants with preserved intra-tumor heterogeneity to screen anticancer compounds. Cell 2016, 167, 260–274 e222. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.T.; Alférez, D.G.; Amant, F.; Annibali, D.; Arribas, J.; Biankin, A.V.; Bruna, A.; Budinská, E.; Caldas, C.; Chang, D.K.; et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer 2017, 17, 254–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siolas, D.; Hannon, G.J. Patient-derived tumor xenografts: Transforming clinical samples into mouse models. Cancer Res. 2013, 73, 5315–5319. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Brehm, M.A.; Garcia-Martinez, J.V.; Greiner, D.L. Humanized mice for immune system investigation: Progress, promise and challenges. Nat. Rev. Immunol. 2012, 12, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Arora, E.; Masab, M.; Mittar, P.; Jindal, V.; Gupta, S.; Dourado, C. Role of immune checkpoint inhibitors in advanced or recurrent endometrial cancer. Cureus 2018, 10, e2521. [Google Scholar] [CrossRef] [PubMed]
- Moiola, C.P.; Lopez-Gil, C.; Cabrera, S.; Garcia, A.; van Nyen, T.; Annibali, D.; Fonnes, T.; Vidal, A.; Villanueva, A.; Matias-Guiu, X.; et al. Patient-derived xenograft models for endometrial cancer research. Int. J. Mol. Sci. 2018. Under revision. [Google Scholar]
- Girotti, M.R.; Gremel, G.; Lee, R.; Galvani, E.; Rothwell, D.; Viros, A.; Mandal, A.K.; Lim, K.H.; Saturno, G.; Furney, S.J.; et al. Application of sequencing, liquid biopsies, and patient-derived xenografts for personalized medicine in melanoma. Cancer Discov. 2016, 6, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Galuschka, C.; Proynova, R.; Roth, B.; Augustin, H.G.; Muller-Decker, K. Models in translational oncology: A public resource database for preclinical cancer research. Cancer Res. 2017, 77, 2557–2563. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cell Line | Tumor Location | Type | PTEN | KRAS | TP53 | PI3K/Akt Pathways Alteration(s) | Microsatellite Instability |
|---|---|---|---|---|---|---|---|
| AN3CA | Metastasis | I | Deletion | wt | Missense mutation | Yes | High |
| ARK1 | Primary | II | n/a | n/a | n/a | Yes | n/a |
| ARK2 | Primary | II | n/a | n/a | n/a | n/a | n/a |
| ECC-1 1 | Primary | I | Missense mutation | wt | Missense mutation | Yes | High |
| HEC1A | Primary | I | wt | Missense mutation | Missense mutation | Yes | High |
| HEC1B | Primary | I | wt | Missense mutation | Missense mutation | Yes | Low |
| HEC50co | Metastasis | n/a | wt | Missense mutation | Deletion | n/a | n/a |
| Ishikawa | Primary | I | Deletion | wt | Missense mutation | Yes | High |
| KLE | Metastasis | n/a | wt | wt | Missense mutation | No | Low |
| MFE-280 | Primary | I | wt | wt | Splice site mutation | Yes | Low |
| RL-95-2 | Primary | I | Missense mutation | wt | Deletion | Yes | High |
| SPEC2 | Primary | II | Not expressed | n/a | n/a | n/a | n/a |
| Research Category | Field of Application | References |
|---|---|---|
| Fundamental Research | Molecular Biology | |
| - Proliferation and migration | [23,25,29,36,37] | |
| - Tumorigenesis and dissemination mechanisms | ||
| - Therapy resistance mechanisms | ||
| - Pathways analysis and identification | ||
| Epigenetics | ||
| - DNA/histones modifications | [33,34,35,38,39,40,41,42,43,44] | |
| - Post-translational protein modification | ||
| - Non-coding RNAs | ||
| Metabolism | [45,46,47,48,49] | |
| - Hormone metabolism | ||
| - Glucose/glutamine metabolism | ||
| - Fatty acid metabolism | ||
| - Other | ||
| Functional analysis | [50] | |
| - New technologies development | ||
| Translational Research | Drug discovery and validation | |
| - Targeted therapies | ||
| - Overcoming therapy resistance | [25,30,36,49,51,52] | |
| Biomarkers discovery | ||
| - Distinguish different EC types | [36,43,53] | |
| - Identification of signatures linked to treatment response |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Nyen, T.; Moiola, C.P.; Colas, E.; Annibali, D.; Amant, F. Modeling Endometrial Cancer: Past, Present, and Future. Int. J. Mol. Sci. 2018, 19, 2348. https://doi.org/10.3390/ijms19082348
Van Nyen T, Moiola CP, Colas E, Annibali D, Amant F. Modeling Endometrial Cancer: Past, Present, and Future. International Journal of Molecular Sciences. 2018; 19(8):2348. https://doi.org/10.3390/ijms19082348
Chicago/Turabian StyleVan Nyen, Tom, Cristian P. Moiola, Eva Colas, Daniela Annibali, and Frédéric Amant. 2018. "Modeling Endometrial Cancer: Past, Present, and Future" International Journal of Molecular Sciences 19, no. 8: 2348. https://doi.org/10.3390/ijms19082348
APA StyleVan Nyen, T., Moiola, C. P., Colas, E., Annibali, D., & Amant, F. (2018). Modeling Endometrial Cancer: Past, Present, and Future. International Journal of Molecular Sciences, 19(8), 2348. https://doi.org/10.3390/ijms19082348