TGF-Beta Signaling in Bone with Chronic Kidney Disease

Abstract
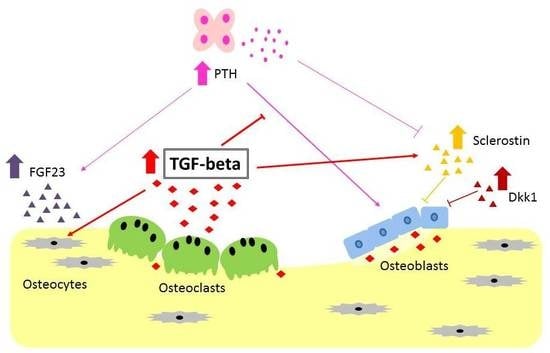
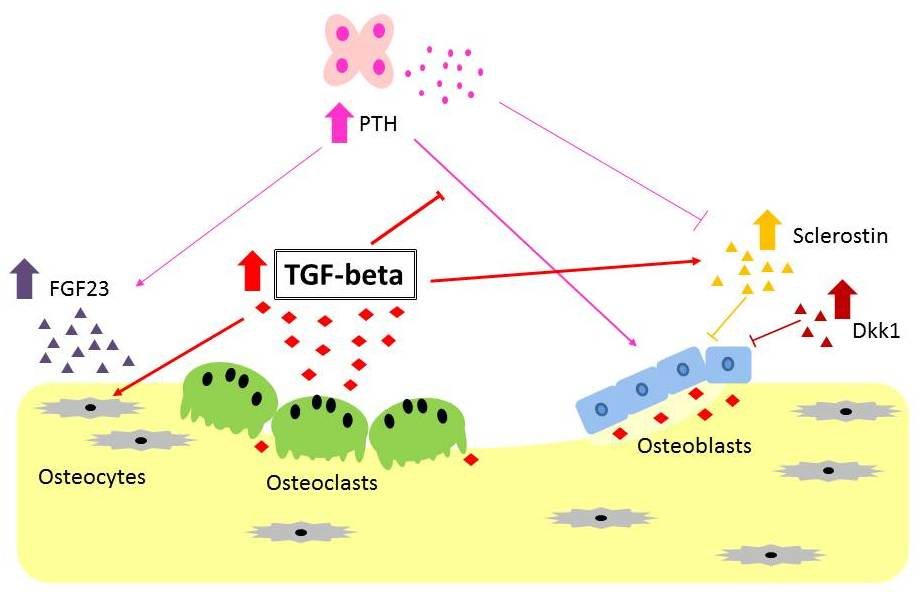
:
{kind=link}
{kind=link}
1. Introduction
2. TGF-β Exists in Bone Tissue and Is Implicated in Bone Metabolism
2.1. TGF-β Acts on Osteoblasts
2.2. TGF-β Acts on Osteoclasts
2.3. TGF-β Regulates Bone Homeostasis Mediated by Osteocytes
3. Abnormalities of Bone and Mineral Metabolism in Chronic Kidney Disease
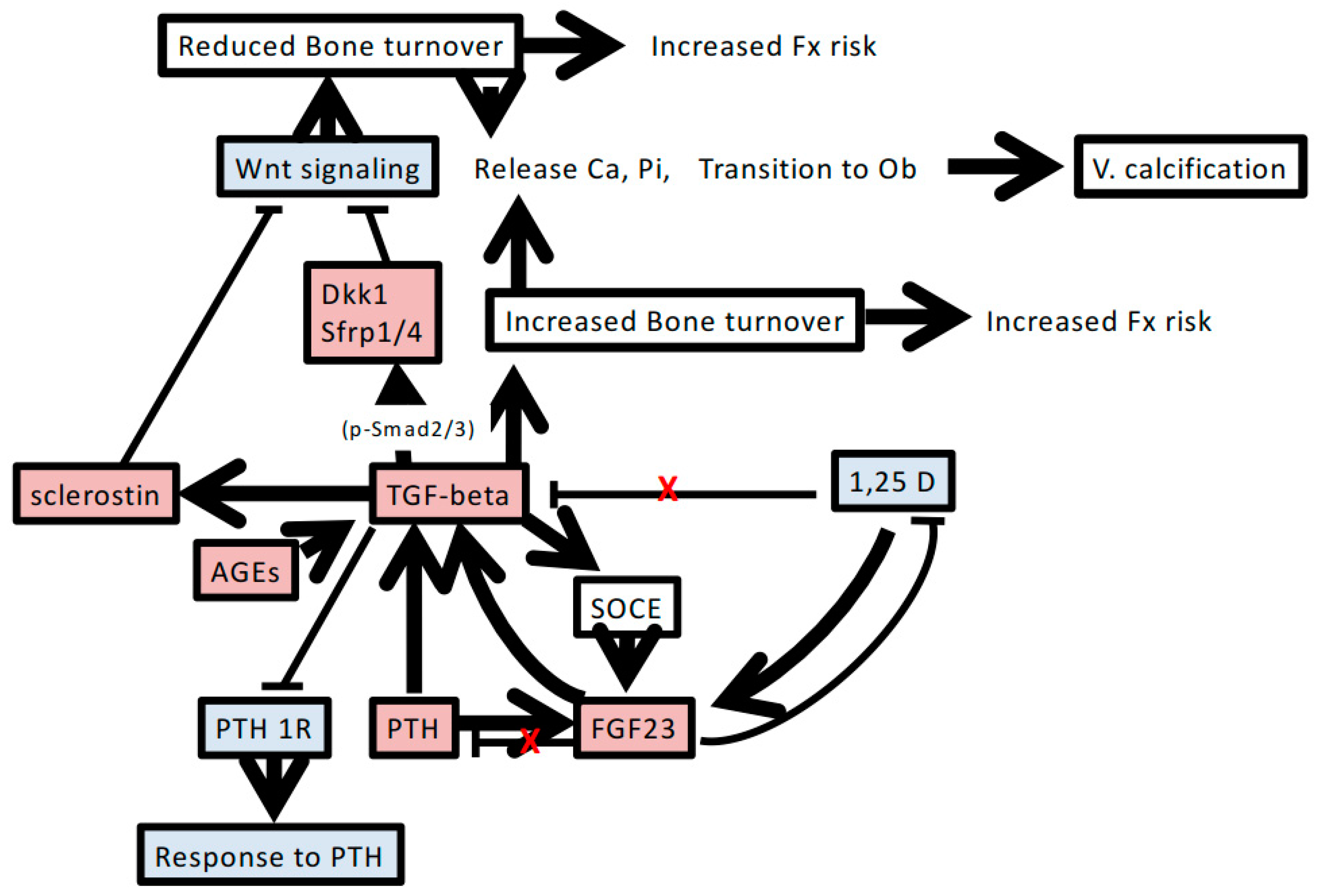
4. Possible Role of TGF-β in Chronic Kidney Disease-Mineral and Bone Disorder
4.1. Renal Osteodystrophy and Resistance to PTH
4.2. High Serum FGF23 Level
4.3. Wnt Inhibitors
5. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| Runx2 | Runt-related transcription factor 2 |
| MMP | matrix metalloprotease |
| RANKL | receptor activator of nuclear factor-kappa B ligand |
| M-CSF | macrophage-colony stimulating factor |
| OPG | osteoprotegerin |
| ROD | renal osteodystrophy |
| CKD-MBD | chronic kidney disease-mineral and bone disorder |
| GFR | glomerular filtration rate |
| 1,25D | 1,25 dihydroxyvitamin D3 |
| BMD | bone mineral density |
| Dkk1 | Dickkopf-1 |
References
- Fleisch, M.C.; Maxwell, C.A.; Barcellos-Hoff, M.-H. The pleiotropic roles of transforming growth factor beta in homeostasis and carcinogenesis of endocrine organs. Endocr. Relat. Cancer 2006, 13, 379–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnert, S.A.; Akhurst, R.J. Embryonic expression pattern of TGF beta type-1 RNA suggests both paracrine and autocrine mechanisms of action. Development 1988, 104, 263–273. [Google Scholar] [PubMed]
- Horner, A.; Kemp, P.; Summers, C.; Bord, S.; Bishop, N.J.; Kelsall, A.W.; Coleman, N.; Compston, J.E. Expression and distribution of transforming growth factor-β isoforms and their signaling receptors in growing human bone. Bone 1998, 23, 5–102. [Google Scholar] [CrossRef]
- Berendsen, A.D.; Olsen, B.R. Bone development. Bone 2015, 80, 14–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, F.; Ornitz, D.M. Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 2013, 5, a008334. [Google Scholar] [CrossRef] [PubMed]
- Geiser, A.G.; Hummel, C.W.; Draper, M.W.; Henck, J.W.; Cohen, I.R.; Rudmann, D.G.; Donnelly, K.B.; Adrian, M.D.; Shepherd, T.A.; Wallace, O.B.; et al. A new selective estrogen receptor modulator with potent uterine antagonist activity, agonist activity in bone, and minimal ovarian stimulation. Endocrinology 2005, 46, 4524–4535. [Google Scholar] [CrossRef] [PubMed]
- Dunker, N.; Krieglstein, K. Tgfβ2−/− Tgfβ3−/− double knockout mice display severe midline fusion defects and early embryonic lethality. Anat. Embryol. 2002, 206, 73–83. [Google Scholar] [PubMed]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGF beta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [PubMed]
- Crane, J.L.; Xian, L.; Cao, X. Role of TGF-β signaling in coupling bone remodeling. Methods Mol. Biol. 2016, 1344, 287–300. [Google Scholar] [PubMed]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.H.; Derynck, R. Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [PubMed]
- Sakou, T.; Onishi, T.; Yamamoto, T.; Nagamine, T.; Sampath, T.K.; Ten Dijke, P. Localization of Smads, the TGF-β family intracellular signaling components during endochondral ossification. J. Bone Miner. Res. 1999, 14, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Serra, R.; Karaplis, A.; Sohn, P. Parathyroid hormone-related peptide (PTHrP)-dependent and -independent effects of transforming growth factor β (TGF-β) on endochondral bone formation. J. Cell Biol. 1999, 145, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Li, T.F.; O’Keefe, R.J.; Chen, D. TGF-β signaling in chondrocytes. Front. Biosci. 2005, 10, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Gentry, L.E.; Nash, B.W. The pro domain of pre-pro-transforming growth factor beta 1 when independently expressed is a functional binding protein for the mature growth factor. Biochemistry 1990, 29, 6851–6857. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Jarrett, J.A.; Chen, E.Y.; Eaton, D.H.; Bell, J.R.; Assoian, R.K.; Roberts, A.B.; Sporn, M.B.; Goeddel, D.V. Human transforming growth factor-β complementary DNA sequence and expression in normal and transformed cells. Nature 1985, 316, 701–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, S.S.; Cain, S.A.; Morgan, A.; Dallas, S.L.; Shuttleworth, C.A.; Kielty, C.M. Fibrillin-1 regulates the bioavailability of TGFβ1. J. Cell Biol. 2007, 176, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taipale, J.; Miyazono, K.; Heldin, C.H.; Keski-Oja, J. Latent transforming growth factor-β1 associates to fibroblast extracellular matrix via latent TGF-β binding protein. J. Cell Biol. 1994, 124, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Nunes, I.; Gleizes, P.E.; Metz, C.N.; Rifkin, D.B. Latent transforming growth factor-β binding protein domains involved in activation and transglutaminase-dependent cross-linking of latent transforming growth factor-β. J. Cell Biol. 1997, 136, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Unsöld, C.; Hyytiäinen, M.; Bruckner-Tuderman, L.; Keski-Oja, J. Latent TGF-β binding protein LTBP-1 contains three potential extracellular matrix interacting domains. J. Cell Sci. 2001, 114, 187–197. [Google Scholar] [PubMed]
- Isogai, Z.; Ono, R.N.; Ushiro, S.; Keene, D.R.; Chen, Y.; Mazzieri, R.; Charbonneau, N.L.; Reinhardt, D.P.; Rifkin, D.B.; Sakai, L.Y. Latent transforming growth factor β-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J. Biol. Chem. 2003, 278, 2750–2757. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Rosser, J.L.; Mundy, G.R.; Bonewald, L.F. Proteolysis of latent transforming growth factor-β (TGF-β)-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-β from bone matrix. J. Biol. Chem. 2002, 277, 21352–21360. [Google Scholar] [CrossRef] [PubMed]
- Oreffo, R.O.; Mundy, G.R.; Seyedin, S.M.; Bonewald, L.F. Activation of the bone-derived latent TGF β complex by isolated osteoclasts. Biochem. Biophys. Res. Commun. 1989, 158, 817–823. [Google Scholar] [CrossRef]
- Van der Kraan, P.M.; Goumans, M.J.; Blaney Davidson, E.; Ten Dijke, P. Age-dependent alteration of TGF-β signalling in osteoarthritis. Cell Tissue Res. 2012, 347, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Van den Bosch, M.H.; Blom, A.B.; van Lent, P.L.; van Beuningen, H.M.; Davidson, E.N.B.; van der Kraan, P.M.; van den Berg, W.B. Canonical Wnt signaling skews TGF-β signaling in chondrocytes towards signaling via ALK1 and Smad 1/5/8. Cell. Signal. 2014, 26, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Choi, K.Y.; Cho, J.Y.; Jung, S.H.; Song, K.B.; Park, E.K.; Choi, J.-Y.; Shin, H.-I.; Kim, S.-Y.; Woo, K.-M.; et al. TGF-β2 stimulates cranial suture closure through activation of the Erk-MAPK pathway. J. Cell Biochem. 2006, 98, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Urano, T.; Yashiroda, H.; Muraoka, M.; Tanaka, K.; Hosoi, T.; Inoue, S.; Ouchi, Y.; Tanaka, K.; Toyoshima, H. p57(Kip2) is degraded through the proteasome in osteoblasts stimulated to proliferation by transforming growth factor β1. J. Biol. Chem. 1999, 74, 12197–12200. [Google Scholar] [CrossRef]
- Sowa, H.; Kaji, H.; Yamaguchi, T.; Sugimoto, T.; Chihara, K. Smad3 promotes alkaline phosphatase activity and mineralization of osteoblastic MC3T3-E1 cells. J. Bone Miner. Res. 2002, 17, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Kwak, J.H.; Zachariah, M.; He, Y.; Wang, L.; Choi, M.E. TGF-β-activated kinase 1 and TAK1-binding protein 1 cooperate to mediate TGF-β1-induced MKK3-p38 MAPK activation and stimulation of type I collagen. Am. J. Physiol. Renal Physiol. 2007, 292, F1471–F1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.F.; Cheng, S.L. Signal transductions induced by bone morphogenetic protein-2 and transforming growth factor-beta in normal human osteoblastic cells. J. Biol. Chem. 2002, 277, 15514–15522. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S. TGF-β regulates β-catenin signaling and osteoblast differentiation in human mesenchymal stem cells. J. Cell Biochem. 2011, 112, 1651–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, T.; Sundar, R.; Widmark, A.; Landström, M.; Persson, E. Osteoblast-derived factors promote metastatic potential in human prostate cancer cells, in part via non-canonical transforming growth factor β (TGFβ) signaling. Prostate 2018, 78, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Hong, S.H.; Bae, S.C. Both the Smad and p38 MAPK pathways play a crucial role in Runx2 expression following induction by transforming growth factor-β and bone morphogenetic protein. Oncogene 2002, 21, 7156–7163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, F.; Thornell, A.P.; Crompton, T.; Denzel, A.; Gilmour, K.C.; Rosewell, I.R.; Stamp, G.W.H.; Beddington, R.S.P.; Mundlos, S.; Olsen, B.R.; et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997, 89, 765–771. [Google Scholar] [CrossRef]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef]
- Koga, T.; Matsui, Y.; Asagiri, M.; Kodama, T.; de Crombrugghe, B.; Nakashima, K.; Takayanagi, H. NFAT and Osterix cooperatively regulate bone formation. Nat. Med. 2005, 11, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Tuckermann, J.P.; Pittois, K.; Partridge, N.C.; Merregaert, J.; Angel, P. Collagenase-3 (MMP-13) and integral membrane protein 2a (Itm2a) are marker genes of chondrogenic/osteoblastic cells in bone formation: Sequential temporal, and spatial expression of Itm2a, alkaline phosphatase, MMP-13, and osteocalcin in the mouse. J. Bone Miner. Res. 2000, 15, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Alliston, T.; Choy, L.; Ducy, P.; Karsenty, G.; Derynck, R. TGF-β-induced repression of CBFA1 by Smad3 decreases cbfa1 and osteocalcin expression and inhibits osteoblast differentiation. EMBO J. 2001, 20, 2254–2272. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Larsen, L.; Engsig, M.T.; Lou, H.; Ferreras, M.; Lochter, A.; Delaissé, J.M.; Foged, N.T. Matrix metalloproteinase-dependent activation of latent transforming growth factor-β controls the conversion of osteoblasts into osteocytes by blocking osteoblast apoptosis. J. Biol. Chem. 2002, 277, 44061–44067. [Google Scholar] [CrossRef] [PubMed]
- Borton, A.J.; Frederick, J.P.; Datto, M.B.; Wang, X.F.; Weinstein, R.S. The loss of Smad 3 results in lower rate of bone formation and osteopenia through dysregulation of osteoblast differentiation and apoptosis. J. Bone Miner. Res. 2001, 16, 1754–1764. [Google Scholar] [CrossRef] [PubMed]
- Pilkington, M.F.; Sims, S.M.; Dixon, S.J. Transforming growth factor-beta induces osteoclast ruffling and chemotaxis: Potential role in osteoclast recruitment. J. Bone Miner. Res. 2001, 16, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Takahashi, N.; Udagawa, N.; Jimi, E.; Gillespie, M.T.; Martin, T.J. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr. Rev. 1999, 20, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pixley, F.J.; Stanley, E.R. CSF-1 regulation of the wandering macrophage: Complexity in action. Trends Cell Biol. 2004, 4, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, T.; Nojima, T.; Nakagawa, M.; Ogasawara, A.; Kaneko, H.; Sato, T.; Mano, H.; Kumegawa, M.; Hakeda, Y. Endogenous production of TGF-β is essential for osteoclastogenesis induced by a combination of receptor activator of NF-κB ligand and macrophage-colony-stimulating factor. J. Immunol. 2000, 165, 4254–4263. [Google Scholar] [CrossRef] [PubMed]
- Galvin, R.J.S.; Gatlin, C.L.; Horn, J.W.; Fuson, T.R. TGF-β enhances osteoclast differentiation in hematopoietic cell cultures stimulated with RANKL and M-CSF. Biochem. Biophys. Res. Commun. 1999, 265, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Fuller, K.; Lean, J.M.; Bayley, K.E.; Wani, M.R.; Chambers, T.J. A role for TGFbeta(1) in osteoclast differentiation and survival. J. Cell Sci. 2000, 113, 2445–2453. [Google Scholar] [PubMed]
- Yasui, T.; Kadono, Y.; Nakamura, M.; Oshima, Y.; Matsumoto, T.; Masuda, H.; Hirose, J.; Omata, Y.; Yasuda, H.; Imamura, T.; et al. Regulation of RANKL-induced osteoclastogenesis by TGF-β through molecular interaction between Smad3 and Traf6. J. Bone Miner. Res. 2011, 26, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, K.S.; Chen, C.G.; Balooch, G.; Stebbins, E.; McKenna, C.R.; Davis, H.; Niewolna, M.; Peng, X.H.; Nguyen, D.H.N.; Ionova-Martin, S.S.; et al. Pharmacologic inhibition of the TGF-β type I receptor kinase has anabolic and anti-catabolic effects on bone. PLoS ONE 2009, 4, e5275. [Google Scholar] [CrossRef] [PubMed]
- Karst, M.; Gorny, G.; Galvin, R.J.S.; Oursler, M.J. Roles of stromal cell RANKL, OPG, and M-CSF expression in biphasic TGF-β regulation of osteoclast differentiation. J. Cell Physiol. 2004, 200, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Yamamoto, M.; Yamamoto, M.; Ono, K.; Nishikawa, M.; Nagata, N.; Motoyoshi, K.; Akatsu, T. Transforming growth factor-β1 increases mRNA levels of osteoclastogenesis inhibitory factor in osteoblastic/stromal cells and inhibits the survival of murine osteoclast-like cells. Biochem. Biophys. Res. Commun. 1998, 252, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukkarasu, K. Stimulation of osteoprotegerin (OPG) gene expression by transforming growth factor-β (TGF-β): Mapping of the OPG promoter region that mediates TGF-β effects. J. Biol. Chem. 2001, 276, 36241–36250. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, M.; Hawse, J.R.; Bruinsma, E.S.; Grygo, S.B.; Cicek, M.; Oursler, M.J.; Spelsberg, T.C. TGFβ inducible early gene-1 directly binds to, and represses, the OPG promoter in osteoblasts. Biochem. Biophys. Res. Commun. 2010, 392, 72–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.; Yamaguchi, Y.; Hakeda, Y. Isolated chick osteocytes stimulate formation and bone-resorbing activity of osteoclast-like cells. J. Bone Miner. Metab. 1995, 13, 61–70. [Google Scholar] [CrossRef]
- Qing, H.; Bonewald, L.F. Osteocyte remodeling of the perilacunar and pericanalicular matrix. Int. J. Oral Sci. 2009, 1, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Kogawa, M.; Wijenayaka, A.R.; Ormsby, R.T.; Thomas, G.P.; Anderson, P.H.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Sclerostin regulates release of bone mineral by osteocytes by induction of carbonic anhydrase 2. J. Bone Miner. Res. 2013, 28, 2436–2448. [Google Scholar] [CrossRef] [PubMed]
- Qing, H.; Ardeshirpour, L.; Pajevic, P.D.; Dusevich, V.; Jahn, K.; Kato, S.; Wysolmerski, J.; Bonewald, L.F. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J. Bone Miner. Res. 2012, 27, 1018–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wysolmerski, J.J. Osteocytes remove and replace perilacunar mineral during reproductive cycles. Bone 2013, 54, 230–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, A.E.; Stoddart, M.J.; Davies, C.M.; Jähn, K.; Furlong, P.I.; Gasser, J.A.; Jones, D.B.; Noble, B.S.; Richards, R.G. TGF β3 and loading increases osteocyte survival in human cancellous bone cultured ex vivo. Cell Biochem. Funct. 2009, 27, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Verborgt, O.; Tatton, N.A.; Majeska, R.J.; Schaffler, M.B. Spatial distribution of Bax and Bcl-2 in osteocytes after bone fatigue: Complementary roles in bone remodeling regulation? J. Bone Miner. Res. 2002, 17, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, S.; Ishii, K.; Amizuka, N.; Li, M.; Kobayashi, T.; Kohno, K.; Ito, M.; Takeshita, S.; Ikeda, K. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007, 5, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Moe, S.; Drüeke, T.; Cunningham, J.; Goodman, W.; Martin, K.; Olgaard, K.; Ott, S.; Sprague, S.; Lameire, N.; Eknoyan, G. Kidney Disease: Improving Global Outcomes (KDIGO). Definition, evaluation, and classification of renal osteodystrophy: A position statement from kidney disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006, 69, 1945–1953. [Google Scholar] [CrossRef] [PubMed]
- Evenepoel, P.; Meijers, B.; Viaene, L.; Bammens, B.; Claes, K.; Kuypers, D.; Vanderschueren, D.; Vanrenterghem, Y. Fibroblast growth factor-23 in early chronic kidney disease: Additional support in favor of a phosphate-centric paradigm for the pathogenesis of secondary hyperparathyroidism. Clin. J. Am. Soc. Nephrol. 2010, 5, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Wahl, P.; Vargas, G.S.; Gutiérrez, O.M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; Chen, J.; et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickolas, T.L.; Stein, E.M.; Dworakowski, E.; Nishiyama, K.K.; Komandah-Kosseh, M.; Zhang, C.A.; McMahon, D.J.; Liu, X.S.; Boutroy, S.; Cremers, S.; et al. Rapid cortical bone loss in patients with chronic kidney disease. J. Bone Miner. Res. 2013, 28, 1811–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, C.R.; Taccetti, G.; Caneva, P.; Mannarino, A.; Maranghi, P.; Ricca, M. Volumetric bone density and geometry assessed by peripheral quantitative computed tomography in uremic patients on maintenance hemodialysis. Osteoporos. Int. 1998, 8, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Paranhos-Neto, F.P.; Lima, G.A.C.; Silva, L.C.; Madeira, M.; Vieira, L.N.; Mendonça, L.M.C.; Lima, I.C.B.; Delgado, A.G.; Leite, M., Jr.; Gomes, C.P.; et al. HR-pQCT detects alterations in bone microstructure in men with CKD stages 3 and 4, which are influenced by hormonal changes and body composition. Clin. Nephrol. 2018, 89, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Fukagawa, M.; Kazama, J.J.; Shigematsu, T. Skeletal resistance to PTH as a basic abnormality underlying uremic bone disease. Am. J. Kidney Dis. 2001, 38 (Suppl. 1), S152–S155. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Yamato, H.; Nii-Kono, T.; Fujieda, A.; Uchida, M.; Hosokawa, A.; Motojima, M.; Fukagawa, M. Insufficiency of PTH action on bone in uremia. Kidney Int. 2006, 70, S34–S36. [Google Scholar] [CrossRef] [PubMed]
- Bover, J.; Ureña, P.; Brandenburg, V.; Goldsmith, D.; Ruiz, C.; DaSilva, I.; Bosch, R.J. Adynamic bone disease: From bone to vessels in chronic kidney disease. Semin. Nephrol. 2014, 34, 626–640. [Google Scholar] [CrossRef] [PubMed]
- Rudser, K.D.; de Boer, I.H.; Dooley, A.; Young, B.; Kestenbaum, B. Fracture risk after parathyroidectomy among chronic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 2401–2407. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, Y.; Taniguchi, M.; Kazama, J.J.; Yokoyama, K.; Hosoya, T.; Yokoo, T.; Shigematsu, T.; Iseki, K.; Tsubakihara, Y. A higher serum alkaline phosphatase is associated with the incidence of hip fracture and mortality among patients receiving hemodialysis in Japan. Nephrol. Dial. Transplant. 2014, 29, 1532–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atsumi, K.; Kushida, K.; Yamazaki, K.; Shimizu, S.; Ohmura, A.; Inoue, T. Risk factors for vertebral fracture in renal osteodystrophy. Am. J. Kidney Dis. 1999, 33, 287–293. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Kazama, J.J.; Yamato, H.; Fukagawa, M. Changes in chemical composition of cortical bone associated with bone fragility in rat model with chronic kidney disease. Bone 2011, 48, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, V.M.; D’Haese, P.; Deck, A.; Mekahli, D.; Meijers, B.; Neven, E.; Evenepoel, P. From skeletal to cardiovascular disease in 12 steps-the evolution of sclerostin as a major player in CKD-MBD. Pediatr. Nephrol. 2016, 31, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Seifert, M.E.; de las Fuentes, L.; Rothstein, M.; Dietzen, D.J.; Bierhals, A.J.; Cheng, S.C.; Ross, W.; Windus, D.; Dávila-Román, V.G.; Hruska, K.A. Effects of phosphate binder therapy on vascular stiffness in early-stage chronic kidney disease. Am. J. Nephrol. 2013, 38, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, I.; Wolf, G. Transforming growth factor-β and the progression of renal disease. Nephrol. Dial. Transplant. 2014, 29 (Suppl. 1), i37–i45. [Google Scholar] [CrossRef] [PubMed]
- Seifert, M.E.; de las Fuentes, L.; Rothstein, M.; Dietzen, D.J.; Bierhals, A.J.; Cheng, S.C.; Ross, W.; Windus, D.; Dávila-Román, V.G.; Hruska, K.A. Circulating transforming growth factor-β1 levels and the risk for kidney disease in African Americans. Kidney Int. 2009, 76, 72–80. [Google Scholar] [Green Version]
- Mathew, S.; Davies, M.; Lund, R.; Saab, G.; Hruska, K.A. Function and effect of bone morphogenetic protein-7 in kidney bone and the bone-vascular links in chronic kidney disease. Eur. J. Clin. Investig. 2006, 36 (Suppl. 2), 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Kanai, H.; Shigehara, T.; Maezawa, A.; Yano, S.; Naruse, T. Metabolism of transforming growth factor-beta in patients receiving hemodialysis especially those with renal osteodystrophy. Ren. Fail. 1998, 20, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Duarte, M.E.; Carvalho, E.F.; Cruz, E.A.; Lucena, S.B.; Andress, D.L. Cytokine accumulation in osteitis fibrosa of renal osteodystrophy. Braz. J. Med. Biol. Res. 2002, 35, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, F.R.; Moysés, R.M.; Montenegro, F.L.; Jorgetti, V.; Noronha, I.L. IL-1beta, TNF-alpha, TGF-beta, and bFGF expression in bone biopsies before and after parathyroidectomy. Kidney Int. 2003, 63, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Hoyland, J.A.; Picton, M.L. Cellular mechanisms of renal osteodystrophy. Kidney Int. 1999, 56, S8–S13. [Google Scholar] [CrossRef]
- Hughes, D.E.; Boyce, B.F. Apoptosis in bone physiology and disease. Mol. Pathol. 1997, 50, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Song, W.; Boulanger, J.H.; Tang, W.; Sabbagh, Y.; Kelley, B.; Gotschall, R.; Ryan, S.; Phillips, L.; Malley, K.; et al. Role of TGF-β in a mouse model of high turnover renal osteodystrophy. J. Bone Miner. Res. 2014, 29, 1141–1157. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, T.L.; Centrella, M. Novel links among Wnt and TGF-β signaling and Runx2. Mol. Endocrinol. 2010, 24, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Massry, S.G.; Coburn, J.W.; Lee, D.B.M.; Towsey, J.; Kleeman, C. Skeletal resistance to parathyroid hormone in renal failure. Study in 105 human subjects. Ann. Intern. Med. 1973, 78, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Martin-Malo, A.; Martinez, M.E.; Torres, A.; Felsenfeld, A.J.; Llach, F. Calcemic response to parathyroid hormone in renal failure: Role of phosphorus and its effect on calcitriol. Kidney Int. 1991, 40, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Bover, J.; Jara, A.; Trinidad, P.; Rodriguez, M.; Martin Malo, A.; Felsenfeld, A.J. The calcemic response to PTH in the rat: Effect of elevated PTH levels and uremia. Kidney Int. 1994, 46, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Felsenfeld, A.J.; Llach, F. Calcemic response to parathyroid hormone in renal failure: Role of calcitriol and the effect parathyroidectomy. Kidney Int. 1991, 40, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Urena, P.; Kubrusly, M.; Mannstradt, M.; Hruby, M.; Tan, M.T.T.; Silve, C.; Lacour, B.; Abou-Samra, A.; Segre, G.; Drüeke, T. The renal PTH/PTHrP receptor is downregulated in rats with chronic renal failure. Kidney Int. 1994, 45, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Urena, P.; Mannstadt, M.; Hruby, M.; Ferreira, A.; Schmitt, F.; Silve, C.; Ardaillou, R.; Lacour, B.; Abou-Samra, A.B.; Segre, G.V.; et al. Parathyroidectomy does not prevent the renal PTH/PTHrP receptor down regulation in uremic rats. Kidney Int. 1995, 47, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Jongen, J.W.; Willemstein-Van Hove, E.C.; Van der Meer, J.M.; Bos, M.P.; Juppner, H.; Segre, G.V.; Abou-Samra, A.B.; Feyen, J.H.M.; Herrmann-Erlee, M.P.M. Down-regulation of the receptor for parathyroid hormone (PTH) and PTH-related peptide by transforming growth factor-beta in primary fetal rat osteoblasts. Endocrinology 1995, 136, 3260–3266. [Google Scholar] [CrossRef] [PubMed]
- Qiu, T.; Wu, X.; Zhang, F.; Clemens, T.L.; Wan, M.; Cao, X. TGF-beta type II receptor phosphorylates PTH receptor to integrate bone remodelling signalling. Nat. Cell Biol. 2010, 12, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Nii-Kono, T.; Iwasaki, Y.; Uchida, M.; Fujieda, A.; Hosokawa, A.; Motojima, M.; Yamato, H.; Kurokawa, K.; Fukagawa, M. Indoxyl sulfate induces skeletal resistance to parathyroid hormone in cultured osteoblastic cells. Kidney Int. 2007, 71, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Iwasaki, Y.; Yamato, H.; Mori, Y.; Komaba, H.; Watanabe, H.; Maruyama, T.; Fukagawa, M. p-Cresyl sulfate induces osteoblast dysfunction through activating JNK and p38 MAPK pathways. Bone 2013, 56, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Yamaguchi, T.; Kanazawa, I.; Ogawa, N.; Hayashi, K.; Yamauchi, M.; Sugimoto, T. The uraemic toxin phenylacetic acid inhibits osteoblastic proliferation and differentiation: An implication for the pathogenesis of low turnover bone in chronic renal failure. Nephrol. Dial. Transplant. 2007, 22, 3160–3165. [Google Scholar] [CrossRef] [PubMed]
- Westerberg, P.A.; Linde, T.; Wikström, B.; Ljunggren, O.; Stridsberg, M.; Larsson, T.E. Regulation of fibroblast growth factor-23 in chronic kidney disease. Nephrol. Dial. Transplant. 2007, 22, 3202–3207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isakova, T.; Xie, H.; Yang, W.; Xie, D.; Anderson, A.H.; Scialla, J.; Wahl, P.; Gutiérrez, O.M.; Steigerwalt, S.; He, J.; et al. Chronic Renal Insufficiency Cohort (CRIC) Study Group. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 2011, 305, 2432–2439. [Google Scholar] [CrossRef] [PubMed]
- Lavi-Moshayoff, V.; Wasserman, G.; Meir, T.; Silver, J.; Naveh-Many, T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: A bone parathyroid feedback loop. Am. J. Physiol. Ren. Physiol. 2010, 299, F882–F889. [Google Scholar] [CrossRef] [PubMed]
- Feger, M.; Hase, P.; Zhang, B.; Hirche, F.; Glosse, P.; Lang, F.; Föller, M. The production of fibroblast growth factor 23 is controlled by TGF-β2. Sci. Rep. 2017, 7, 4982. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, M.; Komaba, H.; Nakanishi, S.; Fujimori, A.; Fukagawa, M. Cinacalcet treatment and serum FGF23 levels in haemodialysis patients with secondary hyperparathyroidism. Nephrol. Dial. Transplant. 2012, 27, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Shiizaki, K.; Kuro-o, M.; Moe, O.W. Fibroblast growth factor 23 and Klotho: Physiology and pathophysiology of an endocrine network of mineral metabolism. Annu. Rev. Physiol. 2013, 75, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Ben-Dov, I.Z.; Galitzer, H.; Lavi-Moshayoff, V.; Goetz, R.; Kuro-o, M.; Mohammadi, M.; Sirkis, R.; Naveh-Many, T.; Silver, J. The parathyroid is a target organ for FGF23 in rats. J. Clin. Investig. 2007, 117, 4003–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galitzer, H.; Ben-Dov, I.Z.; Silver, J.; Naveh-Many, T. Parathyroid cell resistance to fibroblast growth factor 23 in secondary hyperparathyroidism of chronic kidney disease. Kidney Int. 2010, 77, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canalejo, R.; Canalejo, A.; Martinez-Moreno, J.M.; Rodriguez-Ortiz, M.E.; Estepa, J.C.; Mendoza, F.J.; Munoz-Castaneda, J.R.; Shalhoub, V.; Almaden, Y.; Rodriguez, M. FGF23 fails to inhibit uremic parathyroid glands. J. Am. Soc. Nephrol. 2010, 21, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Komaba, H.; Goto, S.; Fujii, H.; Hamada, Y.; Kobayashi, A.; Shibuya, K.; Tominaga, Y.; Otsuki, N.; Nibu, K.; Nakagawa, K.; et al. Depressed expression of Klotho and FGF receptor 1 in hyperplastic parathyroid glands from uremic patients. Kidney Int. 2010, 77, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Shi, M.; Zhang, J.; Quiñones, H.; Kuro-o, M.; Moe, O.W. Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int. 2010, 78, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Meir, T.; Durlacher, K.; Pan, Z.; Amir, G.; Richards, W.G.; Silver, J.; Naveh-Many, T. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int. 2014, 86, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.K.; Kaneko, I.; Jurutka, P.W.; Forster, R.; Hsieh, A.; Hsieh, J.C.; Haussler, M.R.; Whitfield, G.K. 1,25-dihydroxyvitamin D-3 regulation of fibroblast growth factor-23 expression in bone cells: Evidence for primary and secondary mechanisms modulated by leptin and interleukin-6. Calcif. Tissue Int. 2013, 92, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yan, J.; Umbach, A.T.; Fakhri, H.; Fajol, A.; Schmidt, S.; Salker, M.S.; Chen, H.; Alexander, D.; Spichtig, D.; et al. NFκB-sensitive Orai1 expression in the regulation of FGF23 release. J. Mol. Med. 2016, 94, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Mace, M.L.; Gravesen, E.; Nordholm, A.; Hofman-Bang, J.; Secher, T.; Olgaard, K.; Lewin, E. Kidney fibroblast growth factor 23 does not contribute to elevation of its circulating levels in uremia. Kidney Int. 2017, 92, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Holt, S.G.; Hewitson, T.D. FGF23 activates injury-primed renal fibroblasts via FGFR4-dependent signalling and enhancement of TGF-β autoinduction. Int. J. Biochem. Cell Biol. 2017, 92, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Komaba, H.; Kaludjerovic, J.; Hu, D.Z.; Nagano, K.; Amano, K.; Ide, N.; Sato, T.; Densmore, M.J.; Hanai, J.I.; Olauson, H.; et al. Klotho expression in osteocytes regulates bone metabolism and controls bone formation. Kidney Int. 2017, 92, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Cool, S.; Jackson, R.; Pincus, P.; Dickinson, I.; Nurcombe, V. Fibroblast growth factor receptor 4 (FGFR4) expression in newborn murine calvaria and primary osteoblast cultures. Int. J. Dev. Biol. 2002, 46, 519–523. [Google Scholar] [PubMed]
- Zerr, P.; Vollath, S.; Palumbo-Zerr, K.; Tomcik, M.; Huang, J.; Distler, A.; Beyer, C.; Dees, C.; Gela, K.; Distler, O.; et al. Vitamin D receptor regulates TGF-β signalling in systemic sclerosis. Ann. Rheum. Dis. 2015, 74, e20. [Google Scholar] [CrossRef] [PubMed]
- Cejka, D.; Jäger-Lansky, A.; Kieweg, H.; Weber, M.; Bieglmayer, C.; Haider, D.G.; Diarra, D.; Patsch, J.M.; Kainberger, F.; Bohle, B.; et al. Sclerostin serum levels correlate positively with bone mineral density and microarchitecture in haemodialysis patients. Nephrol. Dial. Transplant. 2012, 27, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Thambiah, S.; Roplekar, R.; Manghat, P.; Fogelman, I.; Fraser, W.D.; Goldsmith, D.; Hampson, G. Circulating sclerostin and Dickkopf-1 (DKK1) in predialysis chronic kidney disease (CKD): Relationship with bone density and arterial stiffness. Calcif. Tissue Int. 2012, 90, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, Y.; Graciolli, F.G.; O’Brien, S.; Tang, W.; dos Reis, L.M.; Ryan, S.; Phillips, L.; Boulanger, J.; Song, W.; Bracken, C.; et al. Repression of osteocyte Wnt/β-catenin signaling is an early event in the progression of renal osteodystrophy. J. Bone Miner. Res. 2012, 27, 1757–1772. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, R.B.; Graciolli, F.G.; dos Reis, L.M.; Cancela, A.L.; Cuppari, L.; Canziani, M.E.; Carvalho, A.B.; Jorgetti, V.; Moysés, R.M. Disturbances of Wnt/β-catenin pathway and energy metabolism in early CKD: Effect of phosphate binders. Nephrol. Dial. Transplant. 2013, 28, 2510–2517. [Google Scholar] [CrossRef] [PubMed]
- Graciolli, F.G.; Neves, K.R.; Barreto, F.; Barreto, D.V.; Dos Reis, L.M.; Canziani, M.E.; Sabbagh, Y.; Carvalho, A.B.; Jorgetti, V.; Elias, R.M.; et al. The complexity of chronic kidney disease-mineral and bone disorder across stages of chronic kidney disease. Kidney Int. 2017, 91, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Mödder, U.I.; Hoey, K.A.; Amin, S.; McCready, L.K.; Achenbach, S.J.; Riggs, B.L.; Melton, L.J., III; Khosla, S. Relation of age, gender, and bone mass to circulating sclerostin levels in women and men. J. Bone Miner. Res. 2011, 26, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishimura, E.; Okuno, S.; Ichii, M.; Norimine, K.; Yamakawa, T.; Shoji, S.; Nishizawa, Y.; Inaba, M. Relationship between serum sclerostin, bone metabolism markers, and bone mineral density in maintenance hemodialysis patients. J. Clin. Endocrinol. Metab. 2014, 99, 4315–4320. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, R.A.; Barreto, F.C.; Mendes, M.; dos Reis, L.M.; Castro, J.H.; Britto, Z.M.; Marques, I.D.; Carvalho, A.B.; Moysés, R.M.; Jorgetti, V. Peritoneal dialysis per se is a risk factor for sclerostin-associated adynamic bone disease. Kidney Int. 2015, 87, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Cejka, D.; Herberth, J.; Branscum, A.J.; Fardo, D.W.; Monier-Faugere, M.C.; Diarra, D.; Haas, M.; Malluche, H.H. Sclerostin and Dickkopf-1 in renal osteodystrophy. Clin. J. Am. Soc. Nephrol. 2011, 6, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Viaene, L.; Behets, G.J.; Claes, K.; Meijers, B.; Blocki, F.; Brandenburg, V.; Evenepoel, P.; D’Haese, P.C. Sclerostin: Another bone-related protein related to all-cause mortality in haemodialysis? Nephrol. Dial. Transplant. 2013, 28, 3024–3030. [Google Scholar] [CrossRef] [PubMed]
- Keller, H.; Kneissel, M. SOST is a target gene for PTH in bone. Bone 2005, 37, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Leupin, O.; Kramer, I.; Collette, N.M.; Loots, G.G.; Natt, F.; Kneissel, M.; Keller, H. Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J. Bone Miner. Res. 2007, 22, 1957–1967. [Google Scholar] [CrossRef] [PubMed]
- Loots, G.G.; Keller, H.; Leupin, O.; Murugesh, D.; Collette, N.M.; Genetos, D.C. TGF-β regulates sclerostin expression via the ECR5 enhancer. Bone 2012, 50, 663–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notsu, M.; Kanazawa, I.; Takeno, A.; Yokomoto-Umakoshi, M.; Tanaka, K.I.; Yamaguchi, T.; Sugimoto, T. Advanced glycation end product 3 (AGE3) increases apoptosis and the expression of sclerostin by stimulating TGF-β expression and secretion in osteocyte-like MLO-Y4-A2 cells. Calcif. Tissue Int. 2017, 100, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Wada, Y.; Cai, Z.; Iida, Y.; Horie, K.; Yasuda, Y.; Maeda, K.; Kurokawa, K.; van Ypersele de Strihou, C. Implication of an increased oxidative stress in the formation of advanced glycation end products in patients with end-stage renal failure. Kidney Int. 1997, 51, 1170–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.Z.; Richards, W.G.; Li, X.; Ominsky, M.S. Sclerostin and Dickkopf-1 as therapeutic targets in bone diseases. Endocr. Rev. 2012, 33, 747–783. [Google Scholar] [CrossRef] [PubMed]
- Kramer, I.; Loots, G.G.; Studer, A.; Keller, H.; Kneissel, M. Parathyroid hormone (PTH)-induced bone gain is blunted in SOST overexpressing and deficient mice. J. Bone Miner. Res. 2010, 25, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Roforth, M.M.; Demaray, S.; McGregor, U.; Kirmani, S.; McCready, L.K.; Peterson, J.M.; Drake, M.T.; Monroe, D.G.; Khosla, S. Effects of estrogen on bone mRNA levels of sclerostin and other genes relevant to bone metabolism in postmenopausal women. J. Clin. Endocrinol. Metab. 2014, 99, E81–E88. [Google Scholar] [CrossRef] [PubMed]
- Register, T.C.; Hruska, K.A.; Divers, J.; Bowden, D.W.; Palmer, N.D.; Carr, J.J.; Wagenknecht, L.E.; Hightower, R.C.; Xu, J.; Smith, S.C.; et al. Plasma Dickkopf1 (DKK1) concentrations negatively associate with atherosclerotic calcified plaque in African-Americans with type 2 diabetes. J. Clin. Endocrinol. Metab. 2013, 98, E60–E65. [Google Scholar] [CrossRef] [PubMed]
- Szulc, P.; Schoppet, M.; Rachner, T.D.; Chapurlat, R.; Hofbauer, L.C. Severe abdominal aortic calcification in older men is negatively associated with DKK1 serum levels: The STRAMBO study. J. Clin. Endocrinol. Metab. 2014, 99, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Hampson, G.; Edwards, S.; Conroy, S.; Blake, G.M.; Fogelman, I.; Frost, M.L. The relationship between inhibitors of the Wnt signalling pathway (Dickkopf-1(DKK1) and sclerostin), bone mineral density, vascular calcification and arterial stiffness in post-menopausal women. Bone 2013, 56, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Ginsberg, C.; Seifert, M.; Agapova, O.; Sugatani, T.; Register, T.C.; Freedman, B.I.; Monier-Faugere, M.C.; Malluche, H.; Hruska, K.A. CKD-induced wingless/integration1 inhibitors and phosphorus cause the CKD-mineral and bone disorder. J. Am. Soc. Nephrol. 2014, 25, 1760–1763. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, K.; Olauson, H.; Amin, R.; Ponnusamy, A.; Goetz, R.; Taylor, R.F.; Mohammadi, M.; Canfield, A.; Kublickiene, K.; Larsson, T.E. Arterial klotho expression and FGF23 effects on vascular calcification and function. PLoS ONE 2013, 8, e60658. [Google Scholar] [CrossRef] [PubMed]
- Agapova, O.A.; Fang, Y.; Sugatani, T.; Seifert, M.E.; Hruska, K.A. Ligand trap for the activin type IIA receptor protects against vascular disease and renal fibrosis in mice with chronic kidney disease. Kidney Int. 2016, 89, 1231–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeshima, A.; Nojima, Y.; Kojima, I. The role of the activin-follistatin system in the developmental and regeneration processes of the kidney. Cytokine Growth Factor Rev. 2001, 12, 289–298. [Google Scholar] [CrossRef]
- Ying, S.Y. Inhibins, activins, and follistatins: Gonadal proteins modulating the secretion of follicle-stimulating hormone. Endocr. Rev. 1988, 9, 267–293. [Google Scholar] [CrossRef] [PubMed]
- Sugatani, T.; Agapova, O.A.; Fang, Y.; Berman, A.G.; Wallace, J.M.; Malluche, H.H.; Faugere, M.C.; Smith, W.; Sung, V.; Hruska, K.A. Ligand trap of the activin receptor type IIA inhibits osteoclast stimulation of bone remodeling in diabetic mice with chronic kidney disease. Kidney Int. 2017, 91, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.J.; Sugatani, T.; Agapova, O.A.; Fang, Y.; Gaut, J.P.; Faugere, M.C.; Malluche, H.H.; Hruska, K.A. The activin receptor is stimulated in the skeleton, vasculature, heart, and kidney during chronic kidney disease. Kidney Int. 2018, 93, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-López, N.; Panizo, S.; Alonso-Montes, C.; Román-García, P.; Rodríguez, I.; Martínez-Salgado, C.; Dusso, A.S.; Naves, M.; Cannata-Andía, J.B. Direct inhibition of osteoblastic Wnt pathway by fibroblast growth factor 23 contributes to bone loss in chronic kidney disease. Kidney Int. 2016, 90, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Murali, S.K.; Roschger, P.; Zeitz, U.; Klaushofer, K.; Andrukhova, O.; Erben, R.G. FGF23 regulates bone mineralization in a 1,25(OH)2D3 and Klotho-independent manner. J. Bone Miner. Res. 2016, 31, 129–142. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwasaki, Y.; Yamato, H.; Fukagawa, M. TGF-Beta Signaling in Bone with Chronic Kidney Disease. Int. J. Mol. Sci. 2018, 19, 2352. https://doi.org/10.3390/ijms19082352
Iwasaki Y, Yamato H, Fukagawa M. TGF-Beta Signaling in Bone with Chronic Kidney Disease. International Journal of Molecular Sciences. 2018; 19(8):2352. https://doi.org/10.3390/ijms19082352
Chicago/Turabian StyleIwasaki, Yoshiko, Hideyuki Yamato, and Masafumi Fukagawa. 2018. "TGF-Beta Signaling in Bone with Chronic Kidney Disease" International Journal of Molecular Sciences 19, no. 8: 2352. https://doi.org/10.3390/ijms19082352
APA StyleIwasaki, Y., Yamato, H., & Fukagawa, M. (2018). TGF-Beta Signaling in Bone with Chronic Kidney Disease. International Journal of Molecular Sciences, 19(8), 2352. https://doi.org/10.3390/ijms19082352