Diffuse Gastric Cancer: A Summary of Analogous Contributing Factors for Its Molecular Pathogenicity




Abstract
:1. Introduction
2. Factors Associated with Molecular Pathogenicity of DGC
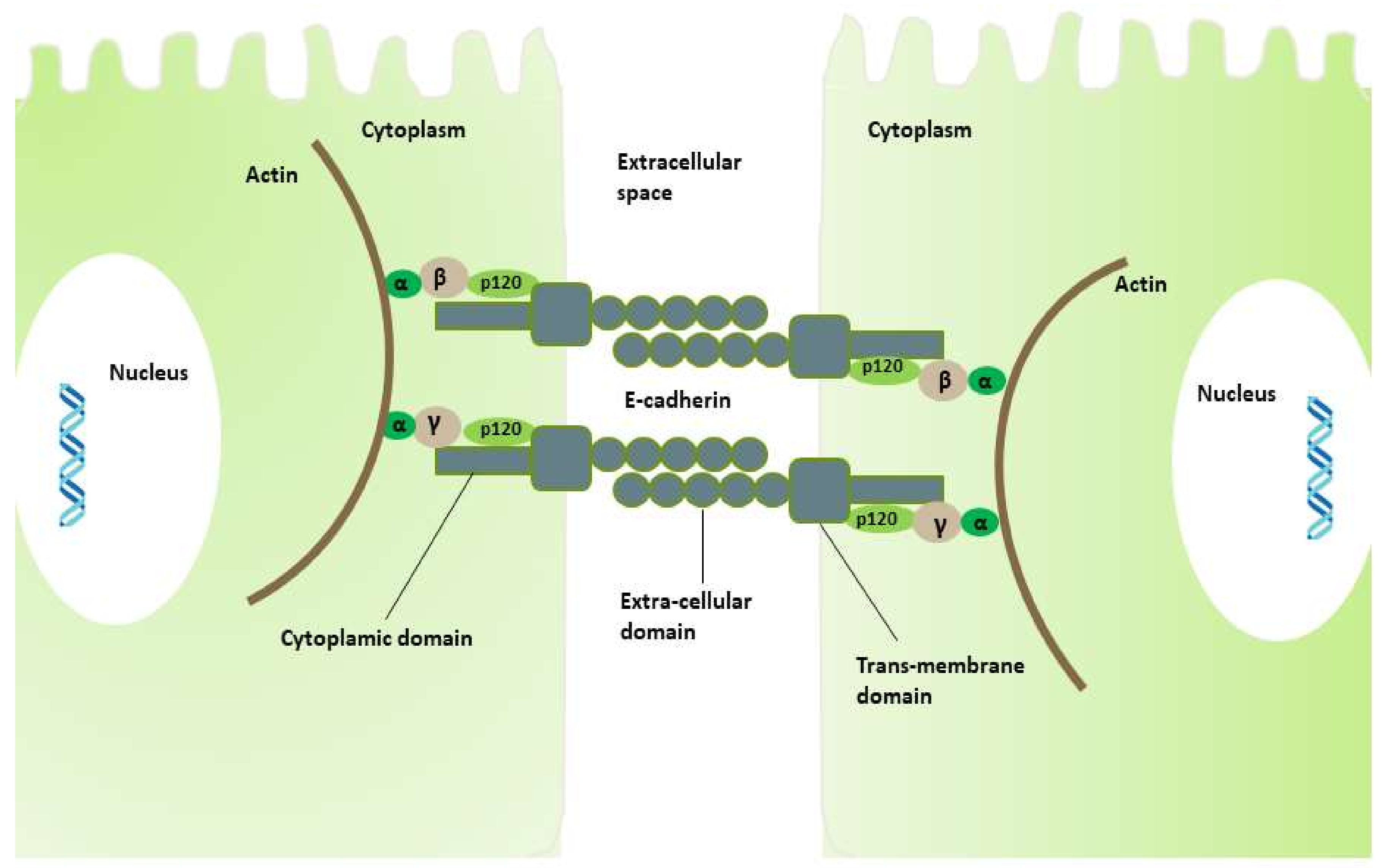
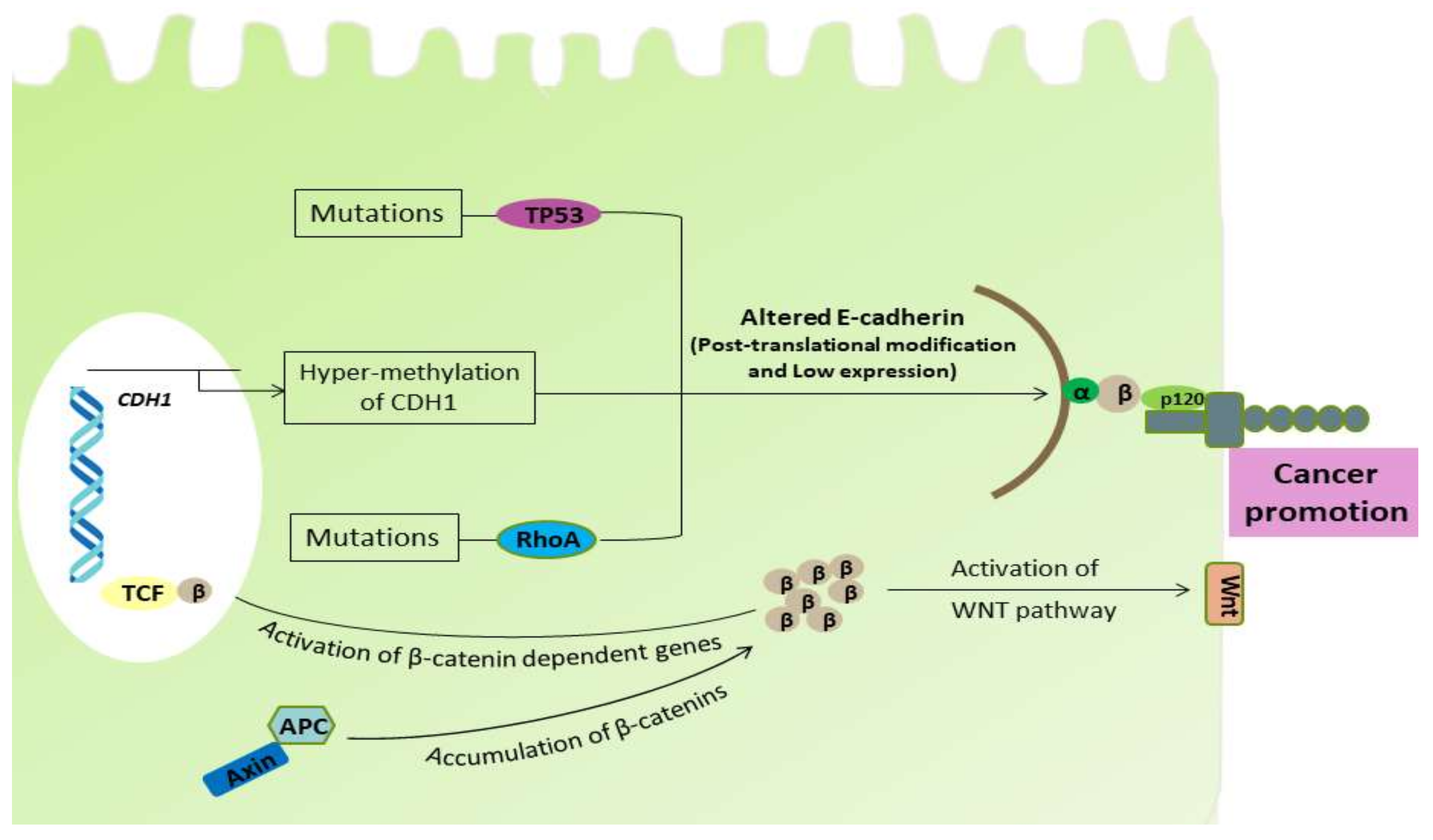
2.1. Role of E-Cadherin
2.2. Alterations in Ras Homolog Gene Family A (RhoA)
2.3. Role of Sphingosine-1-Phosphate
2.4. Role of Adenomatous Polyposis Coli
2.5. Role of Fibroblast Growth Factor Receptor (FGFR)
2.6. Role of Growth/Differentiation Factor 15 (GDF15)
2.7. Li-Fraumeni Syndrome with Germline Mutations in Tumor Protein 53 (TP53)
2.8. Role of Alteration in Other Genes
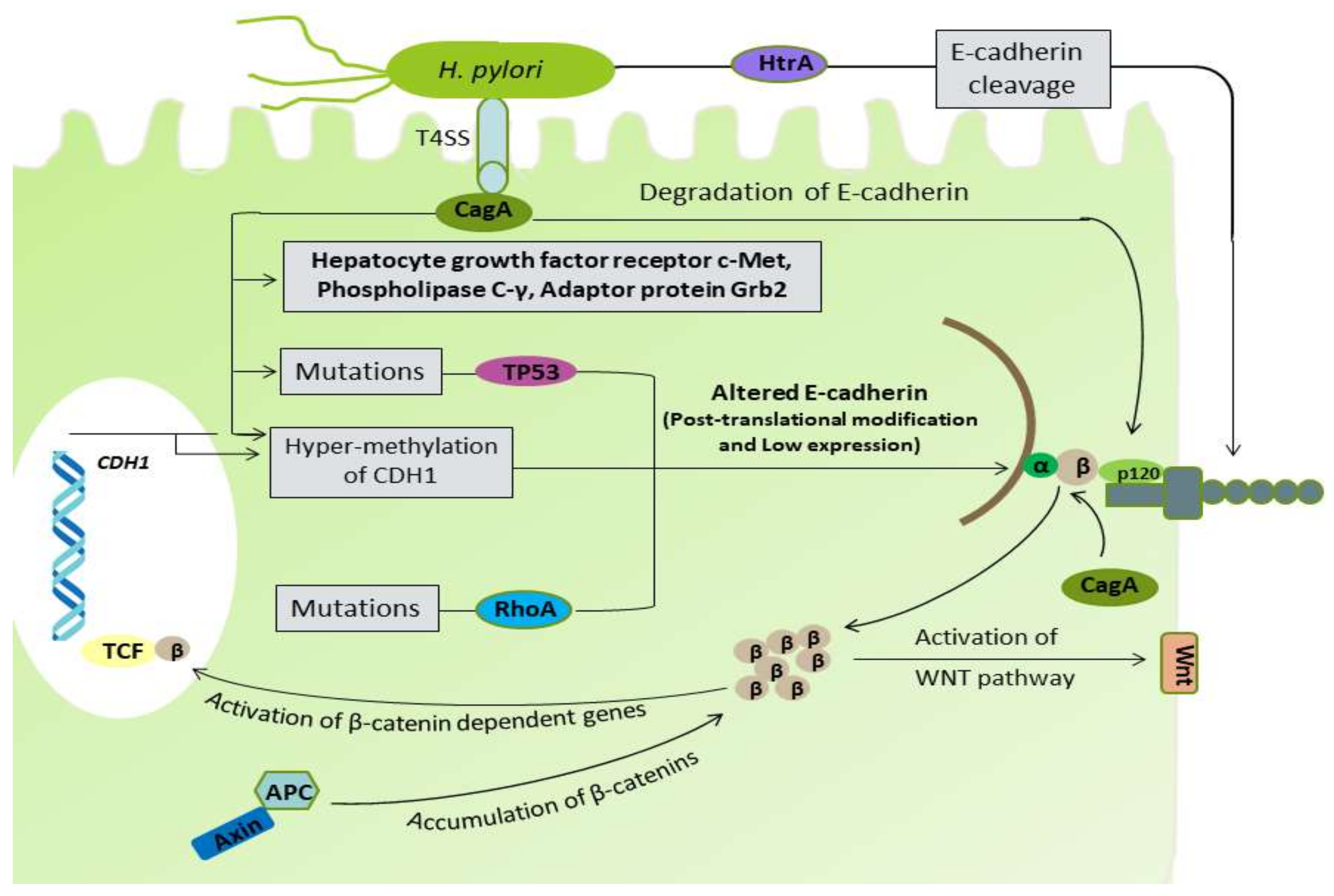
3. Helicobacter pylori Infection and DGC
4. Hereditary Diffuse Gastric Cancer (HDGC) and Germline Mutations
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, 359–386. [Google Scholar] [CrossRef] [PubMed]
- Hartgrink, H.H.; Jansen, E.P.; van Grieken, N.C.; van de Velde, C.J. Gastric cancer. Lancet 2009, 374, 477–490. [Google Scholar] [CrossRef] [Green Version]
- Hamashima, C.; Shibuya, D.; Yamazaki, H.; Inoue, K.; Fukao, A.; Saito, H.; Sobue, T. The Japanese guidelines for gastric cancer screening. Jpn. J. Clin. Oncol. 2008, 38, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.J. Endoscopic gastric cancer screening and surveillance in high-risk groups. Clin. Endosc. 2014, 47, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Fitzmaurice, C.; Dicker, D.; Pain, A.; Hamavid, H.; Moradi-Lakeh, M.; Maclntyre, M.F.; Allen, C.; Hansen, G.; Woodbrook, R.; Wolfe, C.; et al. The global burden of cancer 2013. JAMA Oncol. 2015, 1, 505–527. [Google Scholar] [CrossRef] [PubMed]
- Ajani, J.A.; Lee, J.; Sano, T.; Janjigian, Y.Y.; Fan, D.; Song, S. Gastric adenocarcinoma. Nat. Rev. Dis. Primer 2017, 3, 17036. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, P.F.; Zucca, E. Primary gastric lymphoma pathogenesis and treatment: What has changed over the past 10 years? Br. J. Haematol. 2007, 136, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, P.; Wu, G.Y.; Zhu, L. Primary gastrointestinal lymphoma. World J. Gastroenterol. 2011, 17, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Henson, D.E.; Dittus, C.; Younes, M.; Nguyen, H.; Albores-Saavedra, J. Differential trends in the intestinal and diffuse types of gastric carcinoma in the United States, 1973–2000: Increase in the signet ring cell type. Arch. Pathol. Lab. Med. 2004, 128, 765–770. [Google Scholar] [PubMed]
- Paredes, J.; Figueiredo, J.; Albergaria, A.; Oliveira, P.; Carvalho, J.; Ribeiro, A.S.; Caldeira, J.; Costa, A.M.; Simoes-Correia, J.; Oliveira, M.J.; et al. Epithelial E- and P-cadherins: Role and clinical significance in cancer. Biochim. Biophys. Acta 2012, 1826, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Flejou, J. WHO Classification of digestive tumors: The fourth edition. Ann. Pathol. 2011, 31, 27–31. [Google Scholar]
- Lauren, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta. Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, J.; Choi, I.J.; Kim, Y.W.; Ryu, K.W.; Kim, Y.I.; Oh, J.K.; Tran, B.T.; Kim, J. Dietary inflammatory index and the risk of gastric cancer in a Korean population. Oncotarget 2017, 8, 85452–85462. [Google Scholar] [CrossRef] [PubMed]
- Peleteiro, B.; Lopes, C.; Figueiredo, C.; Lunet, N. Salt intake and gastric cancer risk according to Helicobacter pylori infection, smoking, tumour site and histological type. Br. J. Cancer 2011, 104, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Rota, M.; Pelucchi, C.; Bertuccio, P.; Matsuo, K.; Zhang, Z.F.; Ito, H.; Hu, J.; Johnson, K.C.; Palli, D.; Ferraroni, M.; et al. Alcohol consumption and gastric cancer risk-A pooled analysis within the StoP project consortium. Int. J. Cancer 2017, 141, 1950–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binh, T.T.; Tuan, V.P.; Dung, H.D.Q.; Tung, P.H.; Tri, T.D.; Thuan, N.P.M.; Khien, V.V.; Hoan, P.Q.; Suzuki, R.; Uchida, T.; et al. Advanced non-cardia gastric cancer and Helicobacter pylori infection in Vietnam. Gut Pathog. 2017, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Ellison-Loschmann, L.; Sporle, A.; Corbin, M.; Cheng, S.; Harawira, P.; Gray, M.; Whaanga, T.; Guillford, P.; Koea, J.; Pearce, N. Risk of stomach cancer in Aotearoa/New Zealand: A Māori population based case-control study. PLoS ONE 2017, 12, E0181581. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Gong, E.J.; Chung, E.J.; Park, H.W.; Bae, S.E.; Kim, E.H.; Kim, J.; Do, Y.S.; Kim, T.H.; Chang, H.S.; et al. The characteristics and prognosis of diffuse-type early gastric cancer diagnosed during health check-ups. Gut Liver 2017, 11, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process-first American Cancer Society award lecture on cancer epidemiology and prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar] [PubMed]
- Crew, K.D.; Neugut, A.I. Epidemiology of gastric cancer. World J. Gastroenterol. 2006, 12, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Petrovchich, I.; Ford, J.M. Genetic predisposition to gastric cancer. Semin. Oncol. 2016, 43, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, F.; Charlton, A.; Huntsman, D.G. Hereditary diffuse gastric cancer. In WHO Classification of Tumours of the Digestive System, 4th ed.; Bosman, D.T., Carneiro, F., Hruban, R.H., Theise, N.D., Eds.; International Agency for Research on Cancer: Lyon, France, 2010; Volume 3, pp. 59–63. [Google Scholar]
- Guilford, P.; Hopkins, J.; Harraway, J.; McLeod, M.; McLeod, N.; Harawira, P.; Taite, H.; Scoular, R.; Miller, A.; Reeve, A.E. E-cadherin germline mutations in familial gastric cancer. Nature 1998, 392, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Kluijt, I.; Sijmons, R.H.; Hoogerbrugge, N.; Plukker, J.T.; de Jong, D.; van Krieken, J.H.; van Hillegersberg, R.; Ligtenberg, M.; Bleiker, E.; Cats, A.; et al. Familial gastric cancer: Guidelines for diagnosis, treatment and periodic surveillance. Fam. Cancer 2012, 11, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, R.C.; Hardwick, R.; Huntsman, D.; Carneiro, F.; Guilford, P.; Blair, V.; Chung, D.C.; Norton, J.; Ragunath, K.; Van Krieken, J.H.; et al. Hereditary diffuse gastric cancer: Updated consensus guidelines for clinical management and directions for future research. J. Med. Genet. 2010, 47, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Van der Post, R.S.; Vogelaar, I.P.; Carneiro, F.; Guilford, P.; Huntsman, D.; Hoogerbrugge, N.; Caldas, C.; Schreiber, K.E.; Hardwick, R.H.; Ausems, M.G.; et al. Hereditary diffuse gastric cancer: Updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J. Med. Genet. 2015, 52, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.; Yeoh, K.G. Genetics and Molecular Pathogenesis of Gastric Adenocarcinoma. Gastroenterology 2015, 149, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Jeanes, A.; Gottardi, C.J.; Yap, A.S. Cadherins and cancer: How does cadherin dysfunction promote tumor progression? Oncogene 2008, 27, 6920–6929. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Park, J.W.; Liu, Y.; Park, Y.S.; Kim, J.H.; Yang, H.; Um, H.; Ko, W.R.; Lee, B.I.; Kwon, S.Y.; et al. Sporadic early-onset diffuse gastric cancers have high frequency of somatic CDH1 alterations, but low frequency of somatic RHOA mutations compared with late-onset cancers. Gastroenterology 2017, 153, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, F.; Berx, G. The cell–cell adhesion molecule E-cadherin. Cell Mol. Life Sci. 2008, 65, 3756–3788. [Google Scholar] [CrossRef] [PubMed]
- Grady, W.M.; Willis, J.; Guilford, P.J.; Dunbier, A.K.; Toro, T.T.; Lynch, H.; Wiesner, G.; Ferguson, K.; Eng, C.; Park, J.G.; et al. Methylation of the CDH1 promoter as the second genetic hit in hereditary diffuse gastric cancer. Nat. Genet. 2000, 26, 16–17. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.; Catarino, T.A.; Dias, A.M.; Kato, M.; Almeida, A.; Hessling, B.; Figueiredo, J.; Gartner, F.; Sanches, J.M.; Ruppert, T.; et al. Preventing E-cadherin aberrant N-glycosylation at Asn-554 improves its critical function in gastric cancer. Oncogene 2016, 35, 1619–1631. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.S.; Seruca, R.; Gartner, F.; Yamaguchi, Y.; Gu, J.; Taniguchi, N.; Reis, C.A. Modulation of E-cadherin function and dysfunction by N-glycosylation. Cell Mol. Life Sci. 2011, 68, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Shen, C.Y.; Wu, H.S.; Hsieh, T.Y.; Chan, D.C.; Chen, C.J.; Yu, J.C.; Yu, C.P.; Ham, H.J.; Chen, P.J.; et al. Mechanisms inactivating the gene for E-cadherin in sporadic gastric carcinomas. World J. Gastroenterol. 2006, 12, 2168–2173. [Google Scholar] [CrossRef] [PubMed]
- Humar, B.; Graziano, F.; Cascinu, S.; Catalano, V.; Ruzzo, A.M.; Magnani, M.; Toro, T.; Burchill, T.; Futschik, M.E.; Merriman, T.; et al. Association of CDH1 haplotypes with susceptibility to sporadic diffuse gastric cancer. Oncogene 2002, 21, 8192–8195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakiuchi, M.; Nishizawa, T.; Ueda, H.; Gotoh, K.; Tanaka, A.; Hayashi, A.; Yamamoto, S.; Tatsuno, K.; Katoh, H.; Watanabe, Y.; et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat. Genet. 2014, 46, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Ramachandran, S.; Kim, E.Y.; Allegood, J.C.; Rashid, O.M.; Yamada, A.; Zhao, R.; Milstien, S.; Zhou, H.; Spiegel, S.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res. 2012, 72, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Hanyu, T.; Nagahashi, M.; Ichikawa, H.; Ishikawa, T.; Kobayashi, T.; Wakai, T. Expression of phosphorylated sphingosine kinase 1 is associated with diffuse type and lymphatic invasion in human gastric cancer. Surgery 2017. [Google Scholar] [CrossRef] [PubMed]
- Dumas, Y.R.; He, X. Wnt signaling: What the X@# is WTX? EMBO J. 2011, 30, 1415–1417. [Google Scholar]
- Ghatak, S.; Chakraborty, P.; Sarkar, S.R.; Chowdhury, B.; Bhaumik, A.; Kumar, N.S. Novel APC gene mutations associated with protein alteration in diffuse type gastric cancer. BMC Med. Genet. 2017, 18, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Itoh, H.; Uchino, S.; Hosokawa, K.; Ochiai, A.; Ino, Y.; Ishii, H.; Sakamoto, H.; Yamaguchi, N.; Yanagihara, K.; et al. Immunohistochemical detection of K-sam protein in stomach cancer. Clin. Cancer Res. 1996, 2, 1373–1381. [Google Scholar] [PubMed]
- Levine, A. The p53 tumor-suppressor gene. N. Engl. J. Med. 1992, 326, 1350–1352. [Google Scholar] [CrossRef] [PubMed]
- Kohno, Y.; Yamamoto, H.; Hirahashi, M.; Kumagae, Y.; Nakamura, M.; Oki, E.; Oda, Y. Reduced MUTYH, MTH1, and OGG1 expression and TP53 mutation in diffuse-type adenocarcinoma of gastric cardia. Hum. Pathol. 2016, 52, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Li, B.; Li, Y.; Li, Z.; Liu, Z.; Chen, Z.; Wu, J.; Gao, J.; Shen, L. Genomic alterations in advanced gastric cancer endoscopic biopsy samples using targeted next-generation sequencing. Am. J. Cancer Res. 2017, 7, 1540–1553. [Google Scholar] [PubMed]
- Choi, J.H.; Kim, Y.B.; Ahn, J.M.; Kim, M.J.; Bae, W.J.; Han, S.U.; Woo, H.G.; Lee, D. Identification of genomic aberrations associated with lymph node metastasis in diffuse-type gastric cancer. Experimen. Mol. Med. 2018, 50, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, M.J.; Costa, A.M.; Costa, A.C.; Ferreira, R.M.; Sampaio, P.; Machado, J.C.; Seruca, R.; Mareel, M.; Figueiredo, C. CagA associates with c-Met, E-cadherin, and p120-catenin in a multiproteic complex that suppresses Helicobacter pylori-induced cell-invasive phenotype. J. Infect. Dis. 2009, 200, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Meitner, P.; Konkin, T.; Cho, Y.; Resnick, M.; Moss, S. Altered expression of Skp2, c-Myc and p27 proteins but not mRNA after Helicobacter pylori eradication in chronic gastritis. Mod. Pathol. 2006, 19, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Andre, A.; Ferreira, M.; Mota, R.; Ferrasi, A.; Pardini, M.; Rabenhorst, S. Gastric adenocarcinoma and Helicobacter pylori: Correlation with p53 mutation and p27 immunoexpression. Cancer Epidemiol. 2010, 34, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, E.; Suzuki, H.; Takamaru, H.; Yamamoto, H.; Toyota, M.; Shinomura, Y. Role of DNA methylation in the development of diffuse-type gastric cancer. Digestion 2011, 83, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Perri, F.; Cotugno, R.; Piepoli, A.; Merla, A.; Quitadamo, M.; Gentile, A.; Pilotto, A.; Annese, V.; Andriulli, A. Aberrant DNA methylation in non-neoplastic gastric mucosa of Helicobacter pylori infected patients and effect of eradication. Am. J. Gastroenterol. 2007, 102, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Hoy, B.; Lower, M.; Weydig, C.; Carra, G.; Tegtmeyer, N.; Geppert, T.; Schroder, P.; Sewald, N.; Backert, S.; Schneider, G.; et al. Helicobacter pylori HtrA is a new secreted virulence factor that cleaves E-cadherin to disrupt intercellular adhesion. EMBO Rep. 2010, 11, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Tegtmeyer, N.; Moodley, Y.; Yamaoka, Y.; Pernitzsch, S.R.; Schmidt, V.; Traverso, F.R.; Schmidt, T.P.; Rad, R.; Yeoh, K.G.; Bow, H.; et al. Characterization of worldwide Helicobacter pylori strains reveals genetic conservation and essentiality of serine protease HtrA. Mol. Microbiol. 2016, 99, 925–944. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.P.; Goetz, C.; Huemer, M.; Schneider, G.; Wessler, S. Calcium binding protects E-cadherin from cleavage by Helicobacter pylori HtrA. Gut Pathog. 2016, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.P.; Pema, A.M.; Fugmann, T.; Bohm, M.; Jan, H.; Haller, S.; Gotz, C.; Tegtmeyer, N.; Hoy, B.; Rau, T.T.; et al. Identification of E-cadherin signature motifs functioning as cleavage sites for Helicobacter pylori HtrA. Sci. Rep. 2016, 6, 23264. [Google Scholar] [CrossRef] [PubMed]
- Hoy, B.; Geppert, T.; Boehm, M.; Reisen, F.; Plattner, P.; Gadermaier, G.; Sewald, N.; Ferreira, F.; Briza, P.; Schneider, G.; et al. Distinct roles of secreted HtrA proteases from gram-negative pathogens in cleaving the junctional protein and tumor suppressor E-cadherin. J. Biol. Chem. 2012, 287, 10115–10120. [Google Scholar] [CrossRef] [PubMed]
- Green, K.J.; Getsios, S.; Troyanovsky, S.; Godsel, L.M. Intercellular junction assembly, dynamics, and homeostasis. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Gumbiner, B.; Stevenson, B.; Grimaldi, A. The role of the cell adhesion molecule uvomorulin in the formation and maintenance of the epithelial junctional complex. J. Cell Biol. 1988, 107, 1575–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berx, G.; Nollet, F.; van Roy, F. Dysregulation of the E-cadherin/catenin complex by irreversible mutations in human carcinomas. Cell Adhes. Commun. 1998, 6, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Stemmler, M.P. Cadherins in development and cancer. Mol. Bio. Syst. 2008, 4, 835–850. [Google Scholar] [CrossRef] [PubMed]
- Berx, G.; van Roy, F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb. Perspect. Biol. 2009, 1. [Google Scholar] [CrossRef] [PubMed]
- Wheelock, M.J.; Johnson, K.R. Cadherins as modulators of cellular phenotype. Ann. Rev. Cell. Dev. Biol. 2003, 19, 207–325. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chu, K.-M. E-cadherin and gastric cancer: Cause, consequence, and applications. Biomed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Moridnia, A.; Tabatabaiefar, M.A.; Zeinalian, M.; Minakari, M.; Kheirollahi, M.; Moghaddam, N.A. Novel variants and copy number variation in CDH1 gene in Iranian patients with sporadic diffuse gastric cancer. J. Gastrointest. Cancer 2018. (ahead of print). [Google Scholar] [CrossRef] [PubMed]
- Moran, C.J.J.M.; McAnena, O.J. CDH1 associated gastric cancer: A report of a family and review of the literature. Eur. J. Surg. Oncol. 2005, 31, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, H.B.-C.R.; Seixas, S.; Carvalho, J.; Senz, J.; Oliveira, P.; Inácio, P.; Gusmao, L.; Rocha, J.; Huntsman, D.; Seruca, R.; et al. Allele-specific CDH1 down-regulation and hereditary diffuse gastric cancer. Hum. Mol. Genet. 2010, 19, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Tamura, G. Alterations of tumor suppressor and tumor-related genes in the development and progression of gastric cancer. World J. Gastroenterol. 2006, 12, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, S.; Rafati, M.; Sabokbar, T.; Dastan, J. A novel truncating mutation in the E-cadherin gene in the first Iranian family with hereditary diffuse gastric cancer. Eur. J. Surg. Oncol. 2010, 36, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.M.; Chen, C.J.; Chan, D.C.; Wu, H.S.; Liu, Y.C.; Shen, C.Y.; Chang, T.M.; Yu, J.C.; Harn, H.J.; Yu, C.P.; et al. CDH1 polymorphisms and haplotypes in sporadic diffuse and intestinal gastric cancer: A case–control study based on direct sequencing analysis. World J. Surg. Oncol. 2014, 12, 80. [Google Scholar] [CrossRef] [PubMed]
- Medina-Franco, H.; Ramos-De la Medina, A.; Vizcaino, G.; Medina-Franco, J.L. Single nucleotide polymorphisms in the promoter region of the E-cadherin gene in gastric cancer: Case–control study in a young Mexican population. Ann. Surg. Oncol. 2007, 14, 2246–2249. [Google Scholar] [CrossRef] [PubMed]
- Gullo, I.; Devezas, V.; Baptista, M.; Garrido, L.; Castedo, S.; Morais, R.; Wen, X.; Rios, E.; Pinheiro, J.; Pinto-Ribeiro, I.; et al. The phenotypic heterogeneity of hereditary diffuse gastric cancer: The report of one family with early-onset disease. Gastroint. Endos. 2018. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Jang, S.H.; Park, D.M.; Lim, N.J.; Deng, C.; Kim, D.Y.; Green, J.E.; Kim, H.K. Cooperativity of E-cadherin and Smad4 Loss to promote diffuse-type gastric adenocarcinoma and metastasis. Mol. Cancer Res. 2014, 12, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Gall, T.M.H.; Frampton, A.E. Gene of the month: Ecadherin (CDH1). J. Clin. Pathol. 2013, 66, 928–932. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Kim, M.S.; Voon, D.C.; Kim, S.J.; Bae, J.; Mun, D.G.; Ko, S.I.; Kim, H.K.; Lee, S.W.; Kim, D.Y. Multi-omics analysis identifies pathways and genes involved in diffuse-type gastric carcinogenesis induced by E-cadherin, p53, and Smad4 loss in mice. Mol. Carcinog. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ushiku, T.; Ishikawa, S.; Kakiuchi, M.; Tanaka, A.; Katoh, H.; Aburatani, H.; Lauwers, G.Y.; Fukayama, M. RHOA mutation in diffuse-type gastric cancer: A comparative clinicopathology analysis of 87 cases. Gastric Cancer 2016, 19, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Nagamura, Y.; Nakabo, A.; Okabe, A.; Yanagihara, K.; Fukami, K.; Sakai, R.; Yamaguchi, H. Aberrant alternative splicing of RHOA is associated with loss of its expression and activity in diffuse-type gastric carcinoma cells. Biochem. Biophys. Res. Commun. 2018, 495, 1942–1947. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Xu, X.; Chen, M.; Hu, H.; Ge, H.; Wen, S.; Zhou, S.; Pi, R. Advances in the studies of roles of Rho/Rho-kinase in diseases and the development of its inhibitors. Eur. J. Medicinal Chem. 2013, 70, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Thumkeo, D.; Watanabe, S.; Narumiya, S. Physiological roles of Rho and Rho effectors in mammals. Eur. J. Cell Biol. 2013, 92, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.C.; Hait, N.C.; Allegood, J.C.; Price, M.M.; Avni, D.; et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitisassociated cancer. Cancer Cell. 2013, 23, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Anelli, V.; Gault, C.R.; Snider, A.J.; Obeid, L.M. Role of sphingosine kinase-1 in paracrine/transcellular angiogenesis and lymphangiogenesis in vitro. FASEB J. 2010, 24, 2727. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Matsuda, Y.; Moro, K.; Tsuchida, J.; Soma, D.; Hirose, Y.; Kobayashi, T.; Kosugi, S.; Takabe, K.; Komatsu, M.; et al. DNA damage response and sphingolipid signaling in liver diseases. Surg. Today 2016, 46, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Takabe, K.; Spiegel, S. Export of sphingosine-1-phosphate and cancer progression. J. Lipid Res. 2014, 55, 1839. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Takabe, K.; Terracina, K.P.; Soma, D.; Hirose, Y.; Kobayashi, T.; Matsuda, Y.; Wakai, T. Sphingosine-1-phosphate transporters as targets for cancer therapy. Biomed. Res. Int. 2014, 2014, 651727. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 2011, 11, 403–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takabe, K.; Kim, R.H.; Allegood, J.C.; Mitra, P.; Ramchandran, S.; Nagahashi, M.; Harikumar, K.B.; Hait, N.C.; Milstien, S.; Spiegel, S. Estradiol induces export of sphingosine 1-phosphate from breast cancer cells via ABCC1 and ABCG2. J. Biol. Chem. 2010, 285, 10477–10486. [Google Scholar] [CrossRef] [PubMed]
- Takabe, K.; Paugh, S.W.; Milstien, S.; Spiegel, S. “Inside-out” signaling of sphingosine- 1-phosphate: Therapeutic targets. Pharmacol. Rev. 2008, 60, 181. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.M.; Zilz, N.; Beazer-Barclay, Y.; Bryan, T.M.; Hamilton, S.R.; Thibodeau, S.N.; Vogelstein, B.; Kinzler, K.W. APC mutations occur early during colorectal tumorigenesis. Nature 1992, 359, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Nishisho, I.; Nakamura, Y.; Mivoshi, Y.; Miki, Y.; Ando, H.; Horii, A. Mutations of chromosomes 5q21 genes in FAP and cokorctal cancer patients. Science 1991, 253, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Groden, J.; Thliveris, A.; Samovitz, W.S.; Carlson, M.I.; Gilbert, L.; Albertsen, H.; Joslyn, G.; Stevens, J.; Spirio, L.; Robertson, M.; et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991, 66, 589–600. [Google Scholar] [CrossRef]
- Behrens, J.; Von-Kries, J.P.; Kuhl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of β-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.; Goh, L.K.; Wang, H.; Das, K.; Tao, J.; Tan, I.B.; Zhang, S.; Lee, M.; Wu, J.; Lim, K.H.; et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut 2012, 61, 673–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagatsuma, A.K.; Aizawa, M.; Kuwata, T.; Doi, T.; Ohtsu, A.; Fujii, H.; Ochiai, A. Expression profiles of HER2, EGFR, MET and FGFR2 in a large cohort of patients with gastric adenocarcinoma. Gastric Cancer 2015, 18, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Chua, T.C.; Merrett, N.D. Clinicopathologic factors associated with HER2-positive gastric cancer and its impact on survival outcomes-a systematic review. Int. J. Cancer 2012, 130, 2845–2856. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Odagiri, H.; Nakatani, H.; Miyagawa, K.; Naito, K.; Sakamoto, H.; Katoh, O.; Yoshida, T.; Sugimura, T.; Terada, M. K-sam, an amplified gene in stomach cancer, is a member of the heparin binding growth factor receptor genes. Proc. Natl. Acad. Sci. USA 1990, 87, 5983–5987. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi, M.; Otsuki, S.; Fujimori, Y.; Sato, Y.; Nakagawa, M.; Kojima, K. Therapeutic targeting of fibroblast growth factor receptors in gastric cancer. Gastroenterol. Res. Pract. 2015, 2015, 796380. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi, M.; Murase, H.; Otsuki, S.; Kawano, T.; Kojima, K. Different clinical significance of FGFR1–4 expression between diffuse-type and intestinal-type gastric cancer. World J. Surg. Oncol. 2017, 15, 2. [Google Scholar] [CrossRef] [PubMed]
- Ishige, T.; Nishimura, M.; Satoh, M.; Fujimoto, M.; Fukuyo, M.; Semba, T.; Kado, S.; Tsuchida, S.; Sawai, S.; Matsushita, K.; et al. Combined secretomics and transcriptomics revealed cancer-derived GDF15 is involved in diffuse-type gastric cancer progression and fibroblast activation. Sci. Rep. 2016, 6, 21681. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, M.; Hirakawa, K. Cancer-stromal interactions in scirrhous gastric carcinoma. Cancer Microenviron. 2010, 3, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Hata, K.; Yamamoto, Y.; Kiyomatsu, T.; Tanaka, T.; Kazama, S.; Nozawa, H.; Kawai, K.; Tanaka, J.; Nishikawa, T.; Otani, K.; et al. Hereditary gastrointestinal cancer. Surg. Today 2016, 46, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Bellini, M.F.; Cadamuro, A.C.T.; Succi, M.; Proenc, M.A.; Silva, A.E. Alterations of the TP53 gene in gastric and esophageal carcinogenesis. J. Biomed. Biotechnol. 2012, 2012, 891961. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.J.; Kang, H.C.; Park, H.W.; Jang, S.G.; Han, S.Y.; Lim, S.K.; Lee, M.R.; Chang, H.J.; Ku, J.L.; Yang, H.K.; et al. A TP53 -truncating germline mutation (E287X) in a family with characteristics of both hereditary diffuse gastric cancer and Li-Fraumeni syndrome. J. Hum. Genet. 2004, 49, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Kaurah, P.; MacMillan, A.; Boyd, N.; Senz, J.; De Luca, A.; Chun, N.; Suriano, G.; Zaor, S.; Van Manen, L.; Gilpin, C.; et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA 2007, 297, 2360–2372. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Xia, X.; Ding, C.; Zhen, B.; Zhou, Q.; Feng, J.; Yuan, J.; Chen, R.; Li, Y.; Ge, Z.; et al. A proteomic landscape of diffuse-type gastric cancer. Nat. Commun. 2018, 9, 1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donner, I.; Kiviluoto, T.; Ristimaki, A.; Aaltonen, L.A.; Vahteristo, P. Exome sequencing reveals three novel candidate predisposition genes for diffuse gastric cancer. Fam. Cancer 2015, 14, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Xin, Z.C.; Chen, L.; Tian, L.; Yuan, Y.M.; Song, W.D.; Jiang, X.J.; Guo, Y.L. Expression and localization of CKLFSF2 in human spermatogenesis. Asian J. Androl. 2007, 9, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Plate, M.; Li, T.; Wang, Y.; Mo, X.; Zhang, Y.; Ma, D.; Han, W. Identification and characterization of CMTM4, a novel gene with inhibitory effects on HeLa cell growth through Inducing G2/M phase accumulation. Mol. Cells 2010, 29, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Mendoza, M.C.; Pei, X.; Ilter, D.; Mahoney, S.J.; Zhang, Y.; Ma, D.; Blenis, J.; Wang, Y. Down-regulation of CMTM8 induces epithelial to mesenchymal transition-like changes via c-MET/extracellular signal-regulated kinase (ERK) signaling. J. Biol. Chem. 2012, 287, 11850–11858. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, J.; Su, Y.; Fan, Y.; Guo, X.; Li, L.; Su, X.; Rong, R.; Ying, J.; Mo, X.; et al. A novel 3p22.3 gene CMTM7 represses oncogenic EGFR signaling and inhibits cancer cell growth. Oncogene 2014, 33, 3109–3118. [Google Scholar] [CrossRef] [PubMed]
- Schistosomes, liver flukes and Helicobacter pylori. IARC working group on the evaluation of carcinogenic risks to humans. Lyon, 7–14 June 1994. IARC Monogr. Eval. Carcinog. Risks Hum. 1994, 61, 1–241.
- IARC Helicobacter pylori Working Group. Helicobacter pylori Eradication as a Strategy for Preventing Gastric Cancer. Lyon, France 2014: International Agency for Research on Cancer (IARC Working Group Reports, No. 8). Available online: http://www.iarc.fr/en/publications/pdfsonline/wrk/wrk8/index.php. (accessed on 6 December 2013).
- Becker, K.F.; Atkinson, M.J.; Reich, U.; Becker, I.; Nekarda, H.; Siewert, J.R.; Hofler, H. E-cadherin gene mutations provide clues to diffuse type gastric carcinomas. Cancer Res. 1994, 54, 3845–3852. [Google Scholar] [PubMed]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Komoto, K.; Haruma, K.; Kamada, T.; Tanaka, S.; Yoshihara, M.; Sumii, K.; Kajiyama, G.; Talley, N.J. Helicobacter pylori infection and gastric neoplasia: Correlations with histological gastritis and tumor histology. Am. J. Gastroenterol. 1998, 93, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Nishibayashi, H.; Kanayama, S.; Kiyohara, T.; Yamamoto, K.; Miyazaki, Y.; Yasunaga, Y.; Shinomura, Y.; Takeshita, T.; Takeuchi, T.; Morimoto, K.; et al. Helicobacter pylori-induced enlarged-fold gastritis is associated with increased mutagenicity of gastric juice, increased oxidative DNA damage, and an increased risk of gastric carcinoma. J. Gastroenterol. Hepatol. 2003, 18, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Misra, V.; Misra, S.P.; Singh, M.K.; Singh, P.A.; Dwivedi, M. Prevalence of Helicobacter pylori in patients with gastric cancer. Indian J. Pathol. Microbiol. 2007, 50, 702–707. [Google Scholar] [PubMed]
- Awad, H.A.; Hajeer, M.H.; Abulihya, M.W.; Al-Chalabi, M.A.; Al-Khader, A.A. Epidemiologic characteristics of gastric malignancies among Jordan University Hospital patients. Saudi Med. J. 2017, 38, 965–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Kato, J.; Inoue, I.; Yoshimura, N.; Yoshida, T.; Mukoubayashi, C.; Deguchi, H.; Enomoto, S.; Ueda, K.; Maekita, T.; et al. Development of gastric cancer in nonatrophic stomach with highly active inflammation identified by serum levels of pepsinogen and Helicobacter pylori antibody together with endoscopic rugal hyperplastic gastritis. Int. J. Cancer 2012, 131, 2632–2642. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.W.; Choi, I.J.; Cho, S.J.; Lee, J.Y.; Kim, C.G.; Kook, M.C.; Ryu, K.W.; Kim, Y.W. Characteristics of gastric cancer according to Helicobacter pylori infection status. J. Gastroenterol. Hepatol. 2014, 29, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Gong, E.J.; Lee, J.Y.; Bae, S.E.; Park, Y.S.; Choi, K.D.; Song, H.J.; Lee, G.H.; Jung, H.Y.; Jeong, W.J.; Cheon, G.J.; et al. Characteristics of non-cardia gastric cancer with a high serum anti-Helicobacter pylori IgG titer and its association with diffuse-type histology. PLoS ONE 2018, 13, e0195264. [Google Scholar] [CrossRef] [PubMed]
- Jindal, Y.; Singh, A.; Kumar, R.; Varma, K.; Misra, V.; Misra, S.P.; Dwivedi, M. Expression of alpha methylacyl CoA racemase (AMACR) in gastric adenocarcinoma and its correlation with Helicobacter pylori infection. J. Clin. Diag. Res. 2016, 10, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Tatemichi, M.; Sasazuki, S.; Inoue, M.; Tsugane, S. Clinical significance of IgG antibody titer against Helicobacter pylori. Helicobacter 2009, 14, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Du, J.; Liu, F.; Wang, X.; Li, X.; Li, Y. Role of caspase-3/E-cadherin in Helicobacter pylori-induced apoptosis of gastric epithelial cells. Oncotarget 2017, 8, 59204–59216. [Google Scholar] [CrossRef] [PubMed]
- Bagnoli, F.; Buti, L.; Tompkins, L.; Covacci, A.; Amieva, M.R. Helicobacter pylori CagA induces a transition from polarized to invasive phenotypes in MDCK cells. Proc. Natl. Acad. Sci. USA 2005, 102, 16339–16344. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Zhang, P.Y.; Aboul-Soud, M.A. From inflammation to gastric cancer: Role of Helicobacter pylori. Oncol. Lett. 2017, 13, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Ogden, S.R.; Wroblewski, L.E.; Weydig, C.; Romero-Gallo, J.; O’Brien, D.P.; Israel, D.A.; Krishna, U.S.; Fingleton, B.; Reynolds, A.B.; Wessler, S.; et al. p120 and Kaiso regulate Helicobacter pylori-induced expression of matrix metalloproteinase-7. Mol. Biol. Cell 2008, 19, 4110–4121. [Google Scholar] [CrossRef] [PubMed]
- Das, L.; Kokate, S.B.; Dixit, P.; Rath, S.; Rout, N.; Singh, S.P.; Crowe, S.E.; Bhattacharyya, A. Membrane-bound β-catenin degradation is enhanced by ETS2-mediated Siah1 induction in Helicobacter pylori-infected gastric cancer cells. Oncogenesis 2017, 6, e327. [Google Scholar] [CrossRef] [PubMed]
- Murata-Kamiya, N.; Kurashima, Y.; Teishikata, Y.; Yamahashi, Y.; Saito, Y.; Higashi, H.; Aburatani, H.; Akiyama, T.; Peek, R.M. Jr.; Azuma, T.; et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene 2007, 26, 4617–4626. [Google Scholar] [CrossRef] [PubMed]
- Pelengaris, S.; Khan, M. The many faces of c-MYC. Arch. Biochem. Biophys. 2003, 416, 129–136. [Google Scholar] [CrossRef]
- Faria, M.; Patrocínio, R.; Moraes-Filho, M.; Rabenhorst, S. Expressão das proteínas BCL-2 e BAX em tumores astrocíticos humanos. JPBML 2006, 4, 271–278. [Google Scholar] [CrossRef]
- Pelengaris, S.; Khan, M.; Evan, G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Calcagno, D.; Leal, M.; Seabra, A.; Khayat, A.; Chen, E.; Demachki, S.; Assumpção, P.; Faria, M.; Rabenhorst, S.; Ferreira, M.; et al. Interrelationship between chromosome 8 aneuploidy, C-MYC amplification and increased espression in individuals from northern Brazil with gastric adenocarcinoma. World J. Gastroenterol. 2006, 12, 6207–6211. [Google Scholar] [CrossRef] [PubMed]
- De Lima Silva-Fernandes, I.J.; Alves, M.K.S.; Lima, V.P.; Pereira de Lima, M.A.; Barros, M.A.P.; Ferreira, M.V.P.; Rabenhorst, S.H.B. Differential expression of MYC in Helicobacter pylori-related intestinal and diffuse gastric tumors. Virchows Arch. 2011, 458, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Hoy, B.; Brandstetter, H.; Wessler, S. The stability and activity of recombinant Helicobacter pylori HtrA under stress conditions. J. Basic Microbiol. 2013, 53, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Abdi, E.; Latifi-Navid, S.; Zahri, S.; Yazdanbod, A.; Safaralizadeh, R. Helicobacter pylori genotypes determine risk of non-cardia gastric cancer and intestinal- or diffuse-type GC in Ardabil: A very high-risk area in Northwestern Iran. Microb. Pathog. 2017, 107, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Guilford, P.J.; Hopkins, J.B.; Grady, W.M.; Markowitz, S.D.; Willis, J.; Lynch, H.; Rajput, A.; Wiesner, G.L.; Lindor, N.M.; Burgart, L.J.; et al. E-cadherin germline mutations define an inherited cancer syndrome dominated by diffuse gastric cancer. Hum. Mutat. 1999, 14, 249–255. [Google Scholar] [CrossRef]
- Brooks-Wilson, A.R.; Kaurah, P.; Suriano, G.; Leach, S.; Senz, J.; Grehan, N.; Butterfield, Y.S.; Jeyes, J.; Schinas, J.; Bacani, J.; et al. Germline E-cadherin mutations in hereditary diffuse gastric cancer: Assessment of 42 new families and review of genetic screening criteria. J. Med. Genet. 2004, 41, 508–517. [Google Scholar] [CrossRef] [PubMed]
- More, H.; Humar, B.; Weber, W.; Ward, R.; Christian, A.; Lintott, C.; Graziano, F.; Ruzzo, A.M.; Acosta, E.; Boman, B.; et al. Identification of seven novel germline mutations in the human E-cadherin (CDH1) gene. Hum. Mutat. 2007, 28, 203. [Google Scholar] [CrossRef] [PubMed]
- Feroce, I.; Serrano, D.; Biffi, R.; Andreoni, B.; Galimberti, V.; Sonzogni, A.; Bottiglieri, L.; Botteri, E.; Trovato, C.; Marabelli, M.; et al. Hereditary diffuse gastric cancer in two families: A case report. Oncol. Lett. 2017, 14, 1671–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zylberberg, H.M.; Sultan, K.; Rubin, S. Hereditary diffuse gastric cancer: One family’s story. World J. Clin. Cases 2018, 6, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Hakkaart, C.; Ellison-Loschmann, L.; Day, R.; Sporle, A.; Koea, J.; Harawira, P.; Cheng, S.; Gray, M.; Whaanga, T.; Pearce, N.; et al. Germline CDH1 mutations are a significant contributor to the high frequency of early-onset diffuse gastric cancer cases in New Zealand Māori. Fam. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Corso, G.; Marrelli, D.; Pascale, V.; Vindigni, C.; Roviello, F. Frequency of CDH1 germline mutations in gastric carcinoma coming from high- and low-risk areas: Metanalysis and systematic review of the literature. BMC Cancer 2012, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Pinheiro, H.; Figueiredo, J.; Seruca, R.; Carneiro, F. Familial gastric cancer: Genetic susceptibility, pathology, and implications for management. Lancet Oncol. 2015, 16, 60–70. [Google Scholar] [CrossRef]
- Oliveira, C.; Pinheiro, H.; Figueiredo, J.; Seruca, R.; Carneiro, F. E-cadherin alterations in hereditary disorders with emphasis on hereditary diffuse gastric cancer. Prog. Mol. Biol. Transl. Sci. 2013, 116, 337–359. [Google Scholar] [PubMed]
- Guilford, P.; Humar, B.; Blair, V. Hereditary diffuse gastric cancer: Translation of CDH1 germline mutations into clinical practice. Gastric Cancer 2010, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Suriano, G.; Oliveira, C.; Ferreira, P.; Machado, J.C.; Bordin, M.C.; De Wever, O.; Bruyneel, E.A.; Moguilevsky, N.; Grehan, N.; Porter, T.R.; et al. Identification of CDH1 germline missense mutations associated with functional inactivation of the E-cadherin protein in young gastric cancer probands. Hum. Mol. Genet. 2003, 12, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, J.; Seruca, J. Germline missense mutants in hereditary diffuse gastric cancer. Spotlight Fam. Hered. Gastric Cancer 2013, 7, 77–86. [Google Scholar]
- Van der Post, R.S.; Carneiro, F. Emerging concepts in gastric neoplasia heritable gastric cancers and polyposis disorders. Surg. Pathol. 2017, 10, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Kluijt, I.; Siemerink, E.J.; Ausems, M.G.; van Os, T.A.; de Jong, D.; Simoes-Correia, J.; van Krieken, J.H.; Ligtenberg, M.J.; Figueiredo, J.; van Riel, E.; et al. CDH1-related hereditary diffuse gastric cancer syndrome: Clinical variations and implications for counseling. Int. J. Cancer 2012, 131, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.E.; Save, V.; Carneiro, F.; Dwerryhouse, S.; Lao-Sirieix, P.; Hardwick, R.H.; Caldas, C.; Fitzgerald, R.C. Histopathological and molecular analysis of gastrectomy specimens from hereditary diffuse gastric cancer patients has implications for endoscopic surveillance of individuals at risk. J. Pathol. 2008, 216, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Hansford, S.; Kaurah, P.; Li-Chang, H.; Woo, M.; Senz, J.; Pinheiro, H.; Schrader, K.A.; Schaeffer, D.F.; Shumansky, K.; Zogopoulos, G. Hereditary diffuse gastric cancer syndrome: CDH1 mutations and beyond. JAMA Oncol. 2015, 1, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.M.; Laukaitis, C.M. Panel testing reveals nonsense and missense CDH1 mutations in families without hereditary diffuse gastric cancer. Mol. Genet. Genom. Med. 2016, 4, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lajus, T.B.P.; Sales, R.M.D. CDH1 germ-line missense mutation identified by multigene sequencing in a family with no history of diffuse gastric cancer. Gene 2015, 568, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Yurgelun, M.B. Historical Perspective on Familial Gastric Cancer. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, H.; Oliveira, C.; Seruca, R.; Carneiro, F. Hereditary diffuse gastric cancer-pathophysiology and clinical management. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Senz, J.; Kaurah, P.; Pinheiro, H.; Sanges, R.; Haegert, A.; Corso, G.; Schouten, J.; Fitzgerald, R.; Vogelsang, H.; et al. Germline CDH1 deletions in hereditary diffuse gastric cancer families. Hum. Mol. Genet. 2009, 18, 1545–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majewski, I.J.; Kluijt, I.; Cats, A.; Scerri, T.S.; de Jong, D.; Kluin, R.J.; Hansford, S.; Hogervorst, F.B.; Bosma, A.J.; Hofland, I.; et al. An α-E-catenin (CTNNA1) mutation in hereditary diffuse gastric cancer. J. Pathol. 2013, 229, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Gaston, D.; Hansford, S.; Oliveira, C.; Nightingale, M.; Pinheiro, H.; Macgillivray, C.; Kaurah, P.; Rideout, A.L.; Steele, P.; Soares, G.; et al. Germline mutations in MAP3K6 are associated with familial gastric cancer. PLoS Genet. 2014, 10, e1004669. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.E.; Smyrk, T.C.; Zhang, L. Histologic and immunohistochemical differences between hereditary and sporadic diffuse gastric carcinoma. Human Pathol. 2018, 74, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Vogelaar, I.P.; Ligtenberg, M.J.; van der Post, R.S.; de Voer, R.M.; Kets, C.M.; Jansen, T.J.; Jacobs, L.; Schreibelt, G.; de Vries, I.J.; Netea, M.G.; et al. Recurrent candidiasis and early-onset gastric cancer in a patient with a genetically defined partial MYD88 defect. Fam. Cancer 2016, 15, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Factor | Mechanism | Effects | References |
|---|---|---|---|
| Host Factor | |||
| E-cadherin (CDH1) | Mutational alterations | Deregulation of E-cadherin | [28,29] |
| Over expression of transcription repressor | Down regulation of E-cadherin | [30,31,32] | |
| Post-translational modification | Glycosylation modification of E-cadherin | [33] | |
| Promoter hyper-methylation | E-cadherin inactivation | [34] | |
| Promoter polymorphism | Alterations in E-cadherin | [35] | |
| Ras homolog gene family A (RHOA) | Mutational alterations | Loss of E-cadherin activity | [36] |
| Sphingosine-1-phosphate (S1P) | Synthesis | Development of DGC and lymphatic invasion | [37,38] |
| Adenomatous polyposis coli (APC) | Mutations leading to altered expression of APC protein | Accumulation of β-catenin leading to the activation of Wnt-signaling pathway | [39,40] |
| Fibroblast growth factor receptor (FGFR2) | Overexpression | Inhibition in the cellular activities | [41,42] |
| Tumor protein 53 (TP53) | Mutational alteration | Loss of cell regulating mechanism | [43,44,45,46] |
| Helicobacter pylori | |||
| Non-phosphorylated CagA | Binds with E-cadherin | Dissociation of E-cadherin-β-catenin complex | [47] |
| Causes mutational alterations in TP53 | Impairment of E-cadherin synthesis | [48,49] | |
| Causes hyper-methylation of CDH1 | Reduced E-cadherin expression | [50,51] | |
| High temperature requirement A (HtrA) | Causes cleavage of extracellular domain of E-cadherin | Disruption of normal cell junctions | [52,53,54,55,56] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ansari, S.; Gantuya, B.; Tuan, V.P.; Yamaoka, Y. Diffuse Gastric Cancer: A Summary of Analogous Contributing Factors for Its Molecular Pathogenicity. Int. J. Mol. Sci. 2018, 19, 2424. https://doi.org/10.3390/ijms19082424
Ansari S, Gantuya B, Tuan VP, Yamaoka Y. Diffuse Gastric Cancer: A Summary of Analogous Contributing Factors for Its Molecular Pathogenicity. International Journal of Molecular Sciences. 2018; 19(8):2424. https://doi.org/10.3390/ijms19082424
Chicago/Turabian StyleAnsari, Shamshul, Boldbaatar Gantuya, Vo Phuoc Tuan, and Yoshio Yamaoka. 2018. "Diffuse Gastric Cancer: A Summary of Analogous Contributing Factors for Its Molecular Pathogenicity" International Journal of Molecular Sciences 19, no. 8: 2424. https://doi.org/10.3390/ijms19082424
APA StyleAnsari, S., Gantuya, B., Tuan, V. P., & Yamaoka, Y. (2018). Diffuse Gastric Cancer: A Summary of Analogous Contributing Factors for Its Molecular Pathogenicity. International Journal of Molecular Sciences, 19(8), 2424. https://doi.org/10.3390/ijms19082424