Repression of the Glucocorticoid Receptor Aggravates Acute Ischemic Brain Injuries in Adult Mice


 and
and 
Abstract
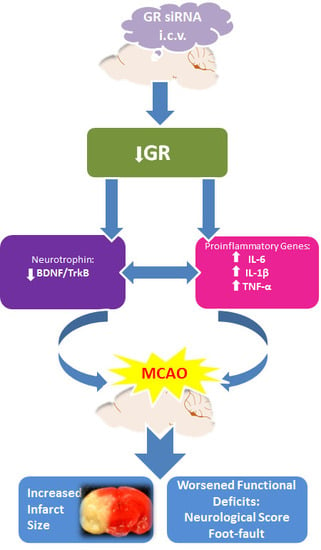
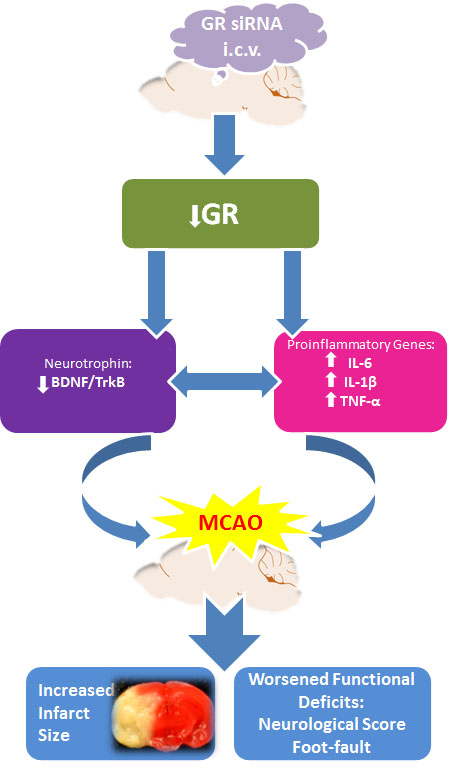
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
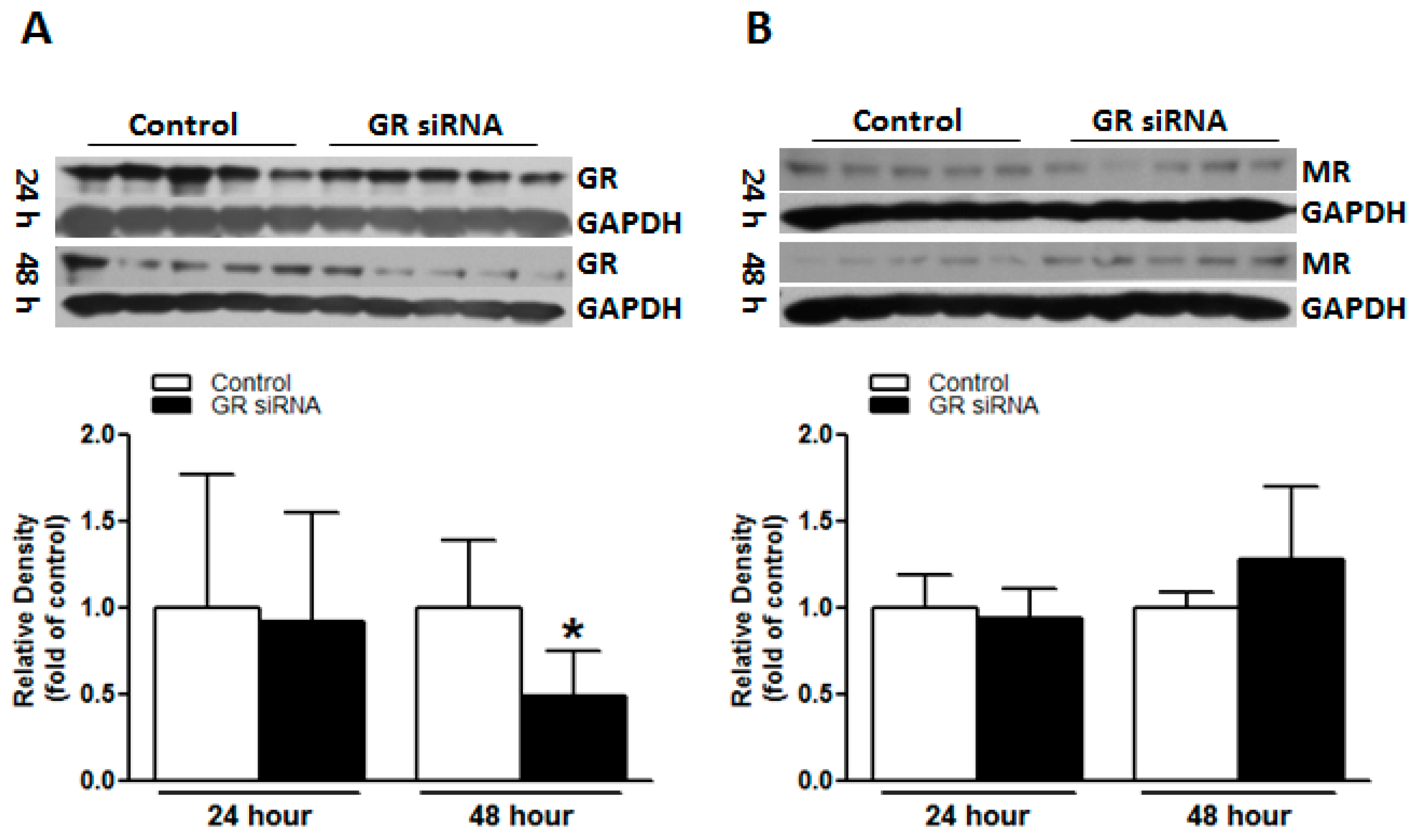
2.1. GR siRNA Treatment Repressed GR Expression in the Brain
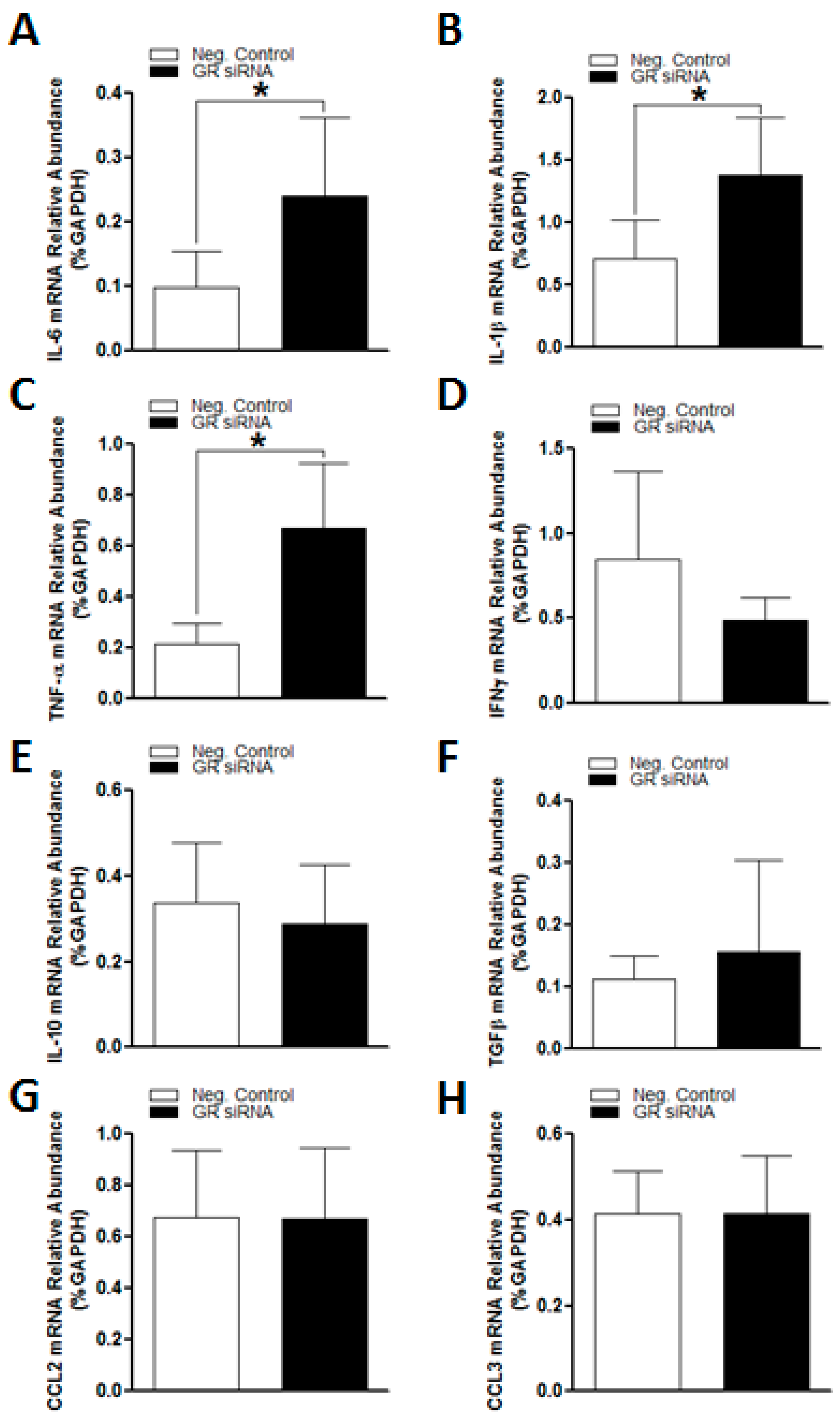
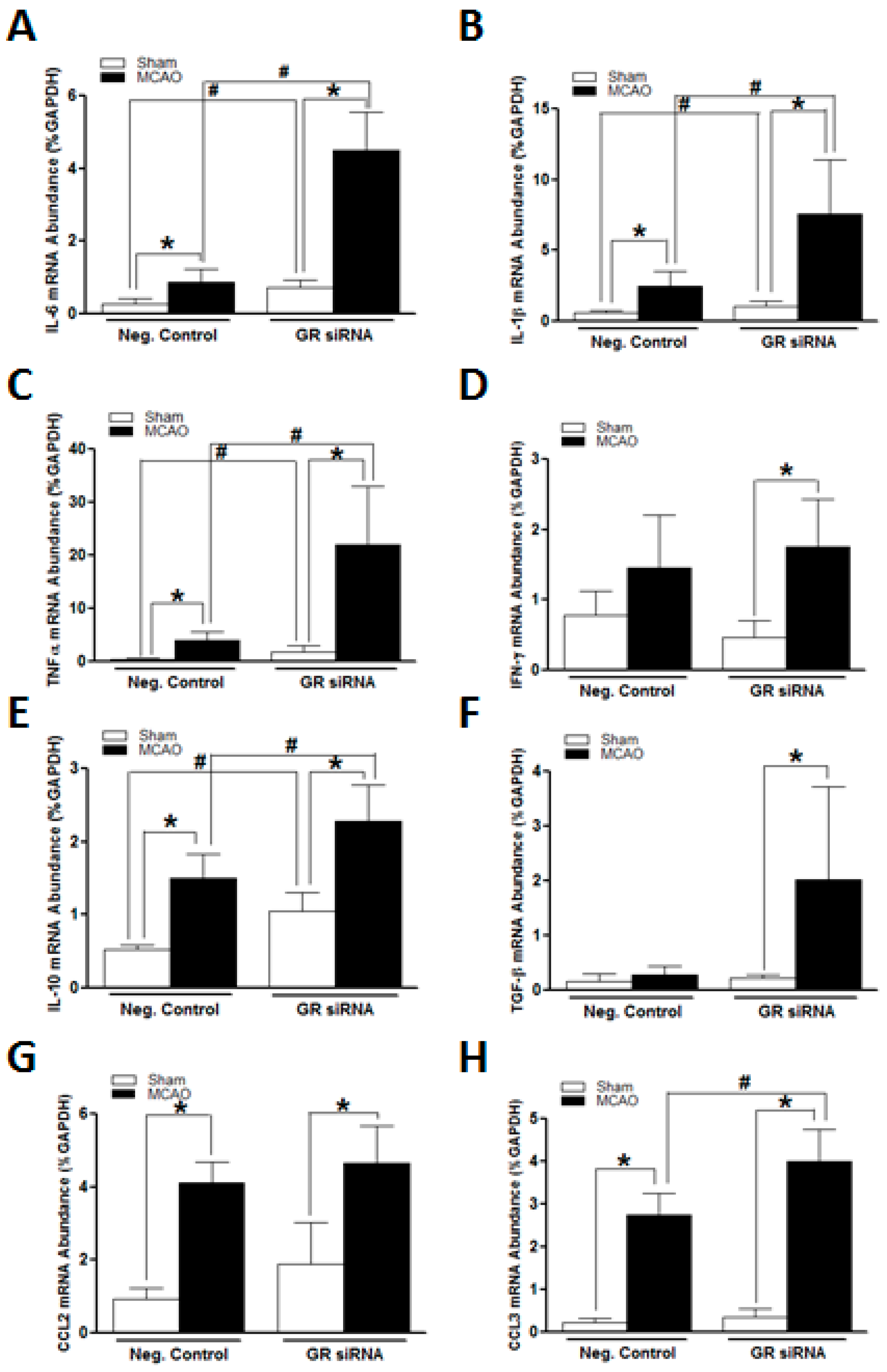
2.2. GR Repression Induced Heightened Inflammatory Responses in the Mouse Brain
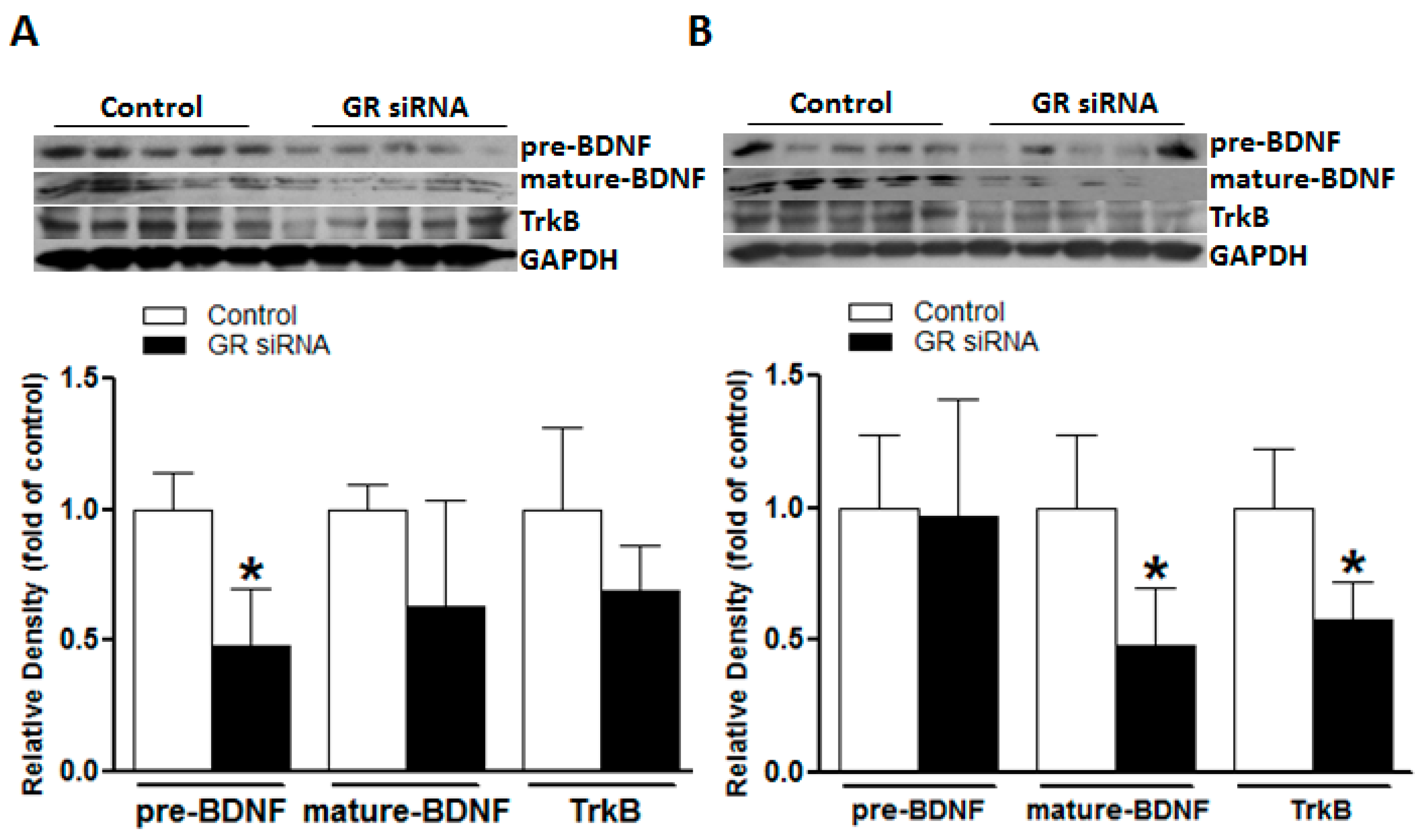
2.3. GR Repression Suppressed BDNF/TrkB Signaling in Mice Brains
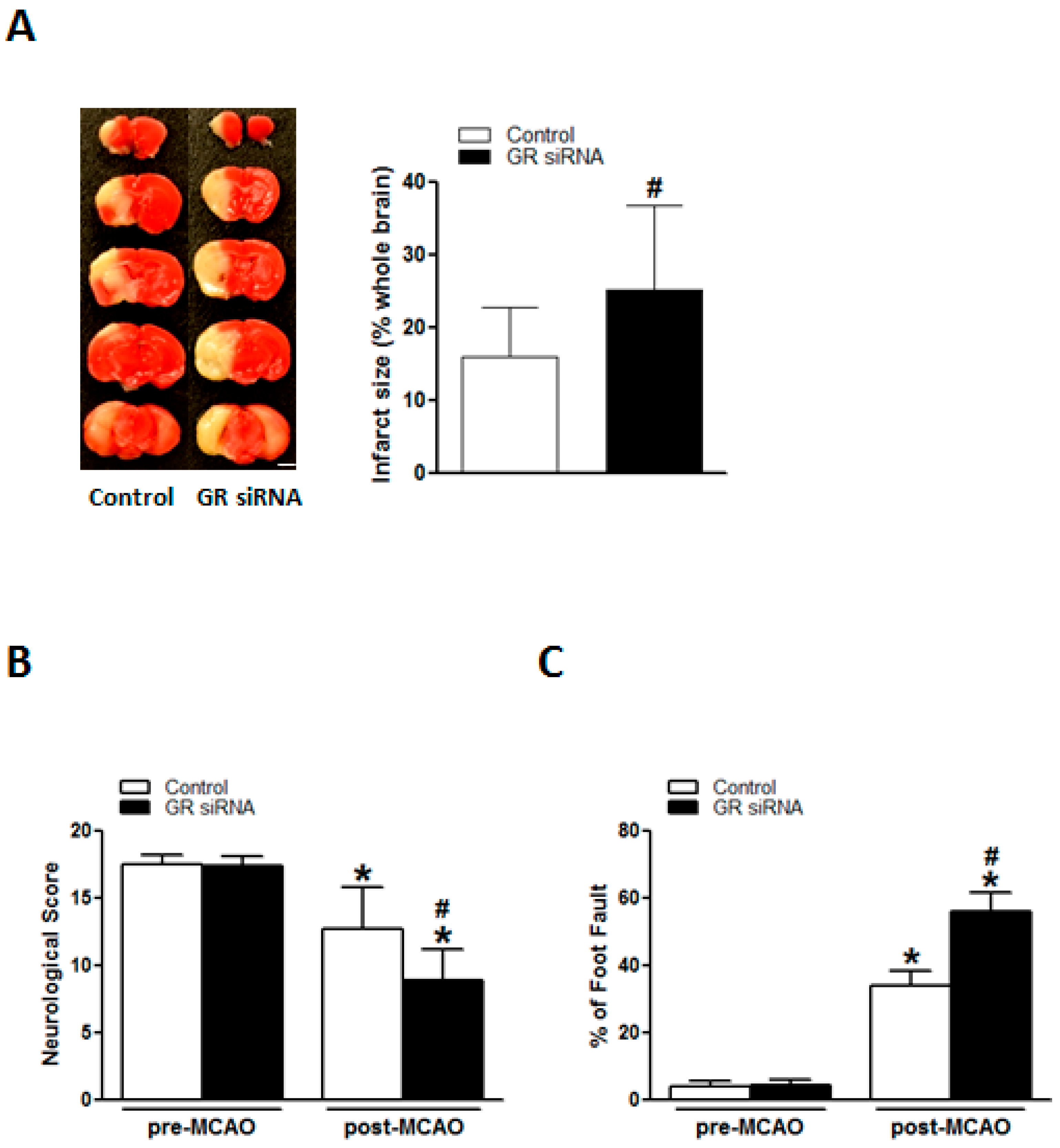
2.4. GR Repression Increased Infarction Size and Worsened Neurobehavioral Deficits in MCAO Insult
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Middle Cerebral Artery Occlusion (MCAO)
4.3. Intracerebroventricular Injection (i.c.v)
4.4. Measurement of Infarction Size
4.5. Neurobehavioral Tests
4.6. Western Blotting
4.7. Real-time RT-PCR
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart disease and stroke statistics-2017 update: A report from the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Ma, Q.; Li, Y.; Li, B.; Zhang, L. Inhibition of microRNA-210 suppresses pro-inflammatory response and reduces acute brain injury of ischemic stroke in mice. Exp. Neurol. 2018, 300, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Howard, G.; Goff, D.C. Population shifts and the future of stroke: Forecasts of the future burden of stroke. Ann. N. Y. Acad. Sci. 2012, 1268, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gonzalez, P.; Zhang, L. Fetal stress and programming of hypoxic/ischemic-sensitive phenotype in the neonatal brain: Mechanisms and possible interventions. Prog. Neurobiol. 2012, 98, 145–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Rodriguez, P.J.; Li, Y.; Martinez, F.; Zhang, L. Dexamethasone protects neonatal hypoxic-ischemic brain injury via L-PGDS-dependent PGD2-DP1-pERK signaling pathway. PLoS ONE 2014, 9, e114470. [Google Scholar] [CrossRef] [PubMed]
- Harding, B.; Conception, K.; Li, Y.; Zhang, L. Glucocorticoids Protect Neonatal Rat Brain in Model of Hypoxic-Ischemic Encephalopathy (HIE). Int. J. Mol. Sci. 2016, 18, 17. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, P.J.; Xiong, F.; Li, Y.; Zhou, J.; Zhang, L. Fetal hypoxia increases vulnerability of hypoxic-ischemic brain injury in neonatal rats: Role of glucocorticoid receptors. Neurobiol. Dis. 2014, 65, 172–179. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; Joels, M. Functional implications of brain corticosteroid receptor diversity. Cell Mol. Neurobiol. 1993, 13, 433–455. [Google Scholar] [CrossRef] [PubMed]
- de Kloet, E.R.; Sutanto, W.; van den Berg, D.T.; Carey, M.P.; van Haarst, A.D.; Hornsby, C.D.; Meijer, O.C.; Rots, N.Y.; Oitzl, M.S. Brain mineralocorticoid receptor diversity: Functional implications. J. Steroid. Biochem. Mol. Biol. 1993, 47, 183–190. [Google Scholar] [CrossRef]
- De Kloet, E.R.; Vreugdenhil, E.; Oitzl, M.S.; Joels, M. Brain corticosteroid receptor balance in health and disease. Endocr. Rev. 1998, 19, 269–301. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Ozawa, H.; Matsuda, K.; Nishi, M.; Kawata, M. Colocalization of mineralocorticoid receptor and glucocorticoid receptor in the hippocampus and hypothalamus. Neurosci. Res. 2005, 51, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Zhe, D.; Fang, H.; Yuxiu, S. Expressions of hippocampal mineralocorticoid receptor (MR) and glucocorticoid receptor (GR) in the single-prolonged stress-rats. Acta Histochem. Cytochem. 2008, 41, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Manji, H.; Lu, B. New insights into BDNF function in depression and anxiety. Nat. Neurosci. 2007, 10, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Jeanneteau, F.; Garabedian, M.J.; Chao, M.V. Activation of Trk neurotrophin receptors by glucocorticoids provides a neuroprotective effect. Proc. Natl. Acad. Sci. USA 2008, 105, 4862–4867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, I.M.; Harkany, T.; Horvath, K.M.; Luiten, P.G. Action of glucocorticoids on survival of nerve cells: Promoting neurodegeneration or neuroprotection? J. Neuroendocrinol. 2001, 13, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Flavin, M.P. Influence of dexamethasone on neurotoxicity caused by oxygen and glucose deprivation in vitro. Exp. Neurol. 2004, 139, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.S.; Seidler, F.J.; Slotkin, T.A. Prenatal dexamethasone exposure causes loss of neonatal hypoxia tolerance: Cellular mechanisms. Pediatr. Res. 1994, 35, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Tombaugh, G.C.; Yang, S.H.; Swanson, R.A.; Sapolsky, R.M. Glucocorticoids exacerbate hypoxic and hypoglycemic hippocampal injury in vitro: Biochemical correlates and a role for astrocytes. J. Neurochem. 1992, 59, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Tuor, U.I. Dexamethasone and the prevention of neonatal hypoxic-ischemic brain damage. Ann. N. Y. Acad. Sci. 1995, 765, 179–195. [Google Scholar] [CrossRef] [PubMed]
- Tuor, U.I. Glucocorticoids and the prevention of hypoxic-ischemic brain damage. Neurosci. Biobehav. Rev. 1997, 21, 175–179. [Google Scholar] [CrossRef]
- Whitelaw, A.; Thoresen, M. Antenatal steroids and the developing brain. Arch. Dis. Child Fetal. Neonatal. Ed. 2000, 83, F154–F157. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.P.; Patel, P.D.; Akil, H.; Watson, S.J. Localization and regulation of glucocorticoid and mineralocorticoid receptor messenger RNAs in the hippocampal formation of the rat. Mol. Endocrinol. 1989, 3, 1886–1894. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, K.R.; Zhang, L. Corticosteroids and perinatal hypoxic-ischemic brain injury. Drug. Discov. Today 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Oyamada, N.; Sone, M.; Miyashita, K.; Park, K.; Taura, D.; Inuzuka, M.; Sonoyama, T.; Tsujimoto, H.; Fukunaga, Y.; Tamura, N.; et al. The role of mineralocorticoid receptor expression in brain remodeling after cerebral ischemia. Endocrinology 2008, 149, 3764–3777. [Google Scholar] [CrossRef] [PubMed]
- Pizza, V.; Agresta, A.; D’Acunto, C.W.; Festa, M.; Capasso, A. Neuroinflamm-aging and neurodegenerative diseases: An overview. CNS Neurol. Disord. Drug Targets 2011, 10, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Tobin, M.K.; Bonds, J.A.; Minshall, R.D.; Pelligrino, D.A.; Testai, F.D.; Lazarov, O. Neurogenesis and inflammation after ischemic stroke: What is known and where we go from here. J. Cereb. Blood. Flow. MeTable 2014, 34, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Vandewalle, J.; Luypaert, A.; De Bosscher, K.; Libert, C. Therapeutic Mechanisms of Glucocorticoids. Trends Endocrinol. MeTable 2018, 29, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Vandevyver, S.; Dejager, L.; Tuckermann, J.; Libert, C. New insights into the anti-inflammatory mechanisms of glucocorticoids: An emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology 2013, 154, 993–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gururajan, A.; Hill, R.A.; van den Buuse, M. Brain-derived neurotrophic factor heterozygous mutant rats show selective cognitive changes and vulnerability to chronic corticosterone treatment. Neuroscience 2015, 284, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T. Possible protective action of neurotrophic factors and natural compounds against common neurodegenerative diseases. Neural. Regen. Res. 2014, 9, 1506–1508. [Google Scholar] [CrossRef] [PubMed]
- Makar, T.K.; Nimmagadda, V.K.; Singh, I.S.; Lam, K.; Mubariz, F.; Judge, S.I.; Trisler, D.; Bever, C.T. TrkB agonist, 7,8-dihydroxyflavone, reduces the clinical and pathological severity of a murine model of multiple sclerosis. J. Neuroimmunol. 2016, 292, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Makar, T.K.; Trisler, D.; Sura, K.T.; Sultana, S.; Patel, N.; Bever, C.T. Brain derived neurotrophic factor treatment reduces inflammation and apoptosis in experimental allergic encephalomyelitis. J. Neurol. Sci. 2008, 270, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Liu, X.; Zhang, N.; Zhu, X.; Liang, H.; Sun, L.; Cheng, Y. Neuroprotective Effects of Brain-Derived Neurotrophic Factor and Noggin-Modified Bone Mesenchymal Stem Cells in Focal Cerebral Ischemia in Rats. J. Stroke Cerebrovasc. Dis. 2016, 25, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Alboni, S.; Tascedda, F.; Corsini, D.; Benatti, C.; Caggia, F.; Capone, G.; Barden, N.; Blom, J.M.; Brunello, N. Stress induces altered CRE/CREB pathway activity and BDNF expression in the hippocampus of glucocorticoid receptor-impaired mice. Neuropharmacology 2011, 60, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Pandya, C.; Kutiyanawalla, A.; Turecki, G.; Pillai, A. Glucocorticoid regulates TrkB protein levels via c-Cbl dependent ubiquitination: A decrease in c-Cbl mRNA in the prefrontal cortex of suicide subjects. Psychoneuroendocrinology 2014, 45, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.Y.; Bambah-Mukku, D.; Pollonini, G.; Alberini, C.M. Glucocorticoid receptors recruit the CaMKIIα-BDNF-CREB pathways to mediate memory consolidation. Nat. Neurosci. 2012, 15, 1707–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Ke, J.; Li, Y.; Ma, Q.; Dasgupta, C.; Huang, X.; Zhang, L.; Xiao, D. Inhibition of miRNA-210 reverses nicotine-induced brain hypoxic-ischemic injury in neonatal rats. Int. J. Biol. Sci. 2017, 13, 76–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q.; Dasgupta, C.; Li, Y.; Bajwa, N.M.; Xiong, F.; Harding, B.; Hartman, R.; Zhang, L. Inhibition of microRNA-210 provides neuroprotection in hypoxic-ischemic brain injury in neonatal rats. Neurobiol. Dis. 2016, 89, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wong, S.; Snyder, E.Y.; Hamblin, M.H.; Lee, J.P. Human neural stem cells rapidly ameliorate symptomatic inflammation in early-stage ischemic-reperfusion cerebral injury. Stem Cell Res. Ther. 2014, 5, 129. [Google Scholar] [CrossRef] [PubMed]
- Bederson, J.B.; Pitts, L.H.; Germano, S.M.; Nishimura, M.C.; Davis, R.L.; Bartkowski, H.M. Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke 1986, 17, 1304–1308. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.H.; Wagner, S.; Liu, K.F.; Hu, X.J. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke 1995, 26, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Liu, Y.; Lu, J.; Cerqueira, B.; Misra, V.; Duong, T.Q. Intraarterial transplantation of human umbilical cord blood mononuclear cells in hyperacute stroke improves vascular function. Stem Cell Res. Ther. 2017, 8, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Huang, L.; Ma, Q.; Concepcion, K.R.; Song, M.A.; Zhang, P.; Fu, Y.; Xiao, D.; Zhang, L. Repression of the Glucocorticoid Receptor Aggravates Acute Ischemic Brain Injuries in Adult Mice. Int. J. Mol. Sci. 2018, 19, 2428. https://doi.org/10.3390/ijms19082428
Li Y, Huang L, Ma Q, Concepcion KR, Song MA, Zhang P, Fu Y, Xiao D, Zhang L. Repression of the Glucocorticoid Receptor Aggravates Acute Ischemic Brain Injuries in Adult Mice. International Journal of Molecular Sciences. 2018; 19(8):2428. https://doi.org/10.3390/ijms19082428
Chicago/Turabian StyleLi, Yong, Lei Huang, Qingyi Ma, Katherine R. Concepcion, Minwoo A. Song, Peng Zhang, Yingjie Fu, Daliao Xiao, and Lubo Zhang. 2018. "Repression of the Glucocorticoid Receptor Aggravates Acute Ischemic Brain Injuries in Adult Mice" International Journal of Molecular Sciences 19, no. 8: 2428. https://doi.org/10.3390/ijms19082428
APA StyleLi, Y., Huang, L., Ma, Q., Concepcion, K. R., Song, M. A., Zhang, P., Fu, Y., Xiao, D., & Zhang, L. (2018). Repression of the Glucocorticoid Receptor Aggravates Acute Ischemic Brain Injuries in Adult Mice. International Journal of Molecular Sciences, 19(8), 2428. https://doi.org/10.3390/ijms19082428