Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus acutissima


Abstract
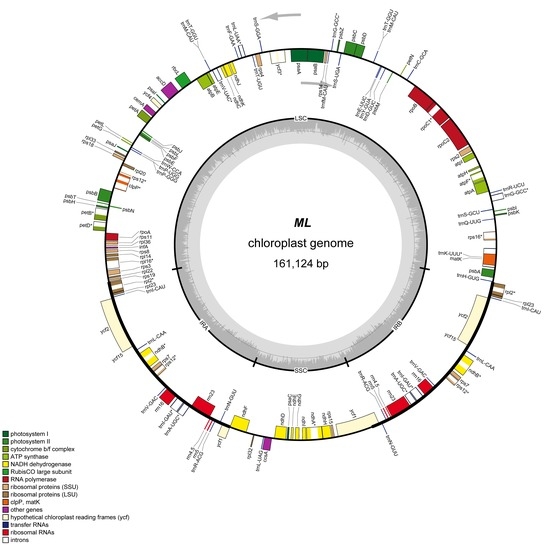
:
1. Introduction
2. Results and Discussion
2.1. Features of Q. Acutissima cpDNA
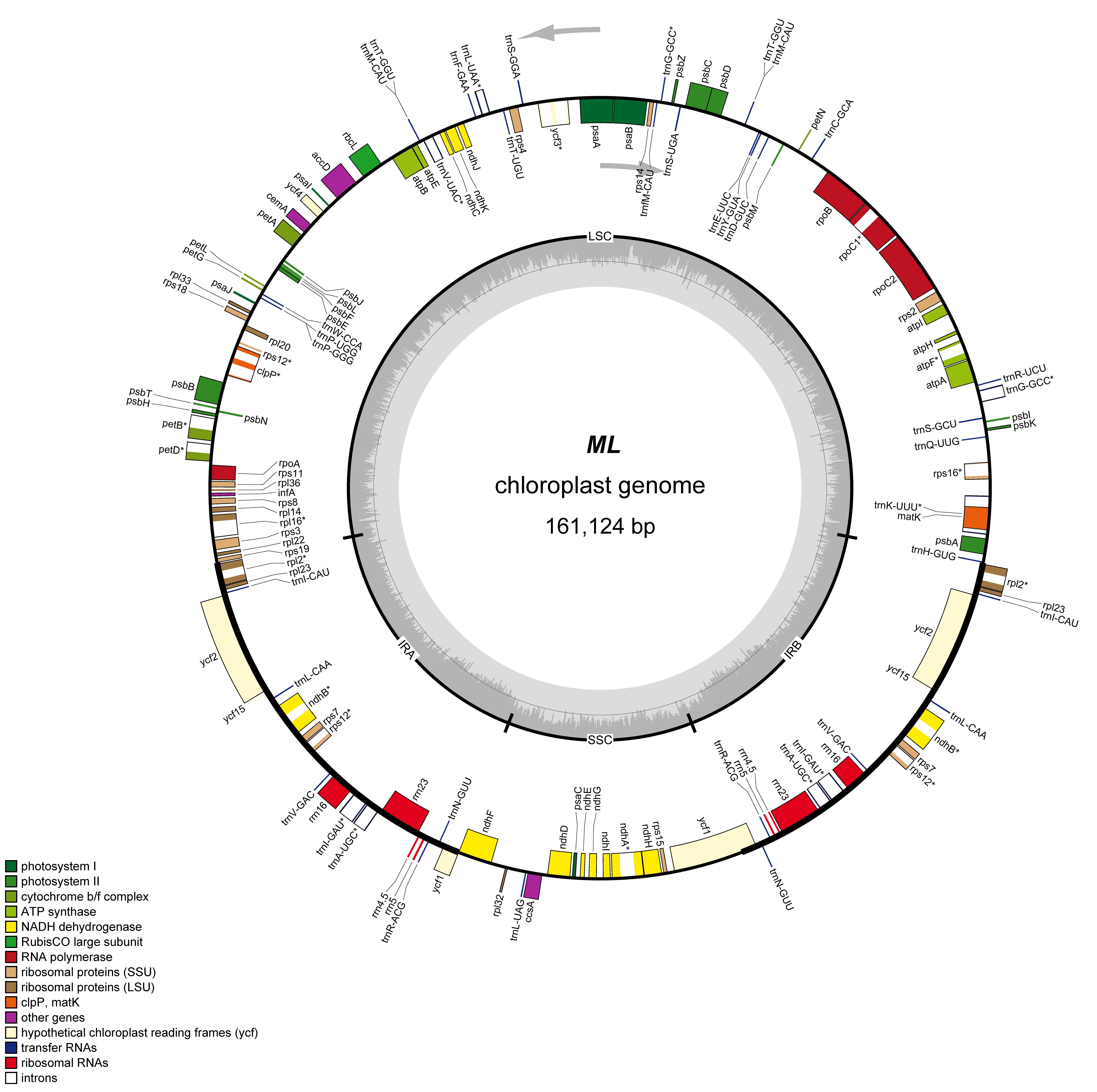
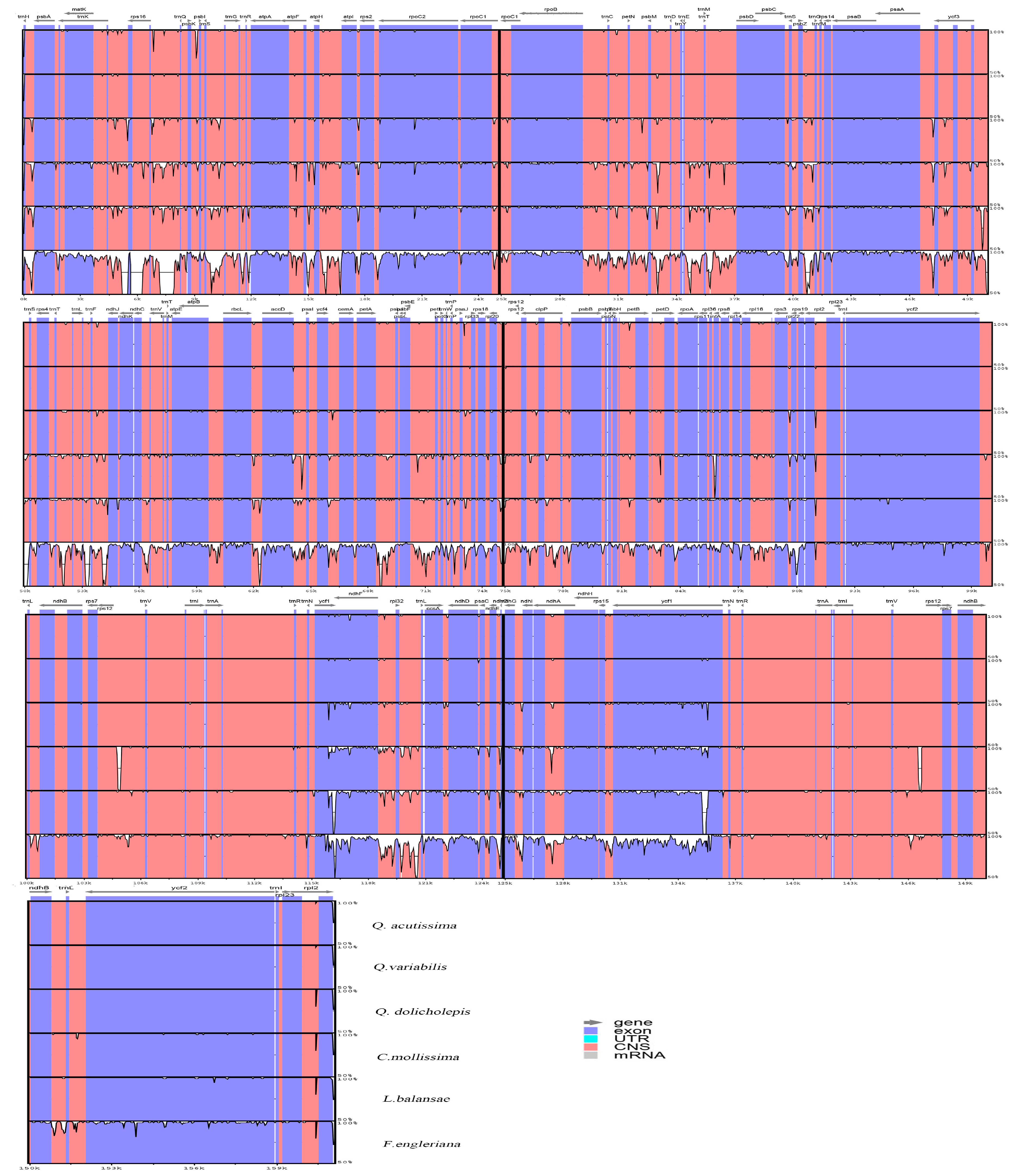
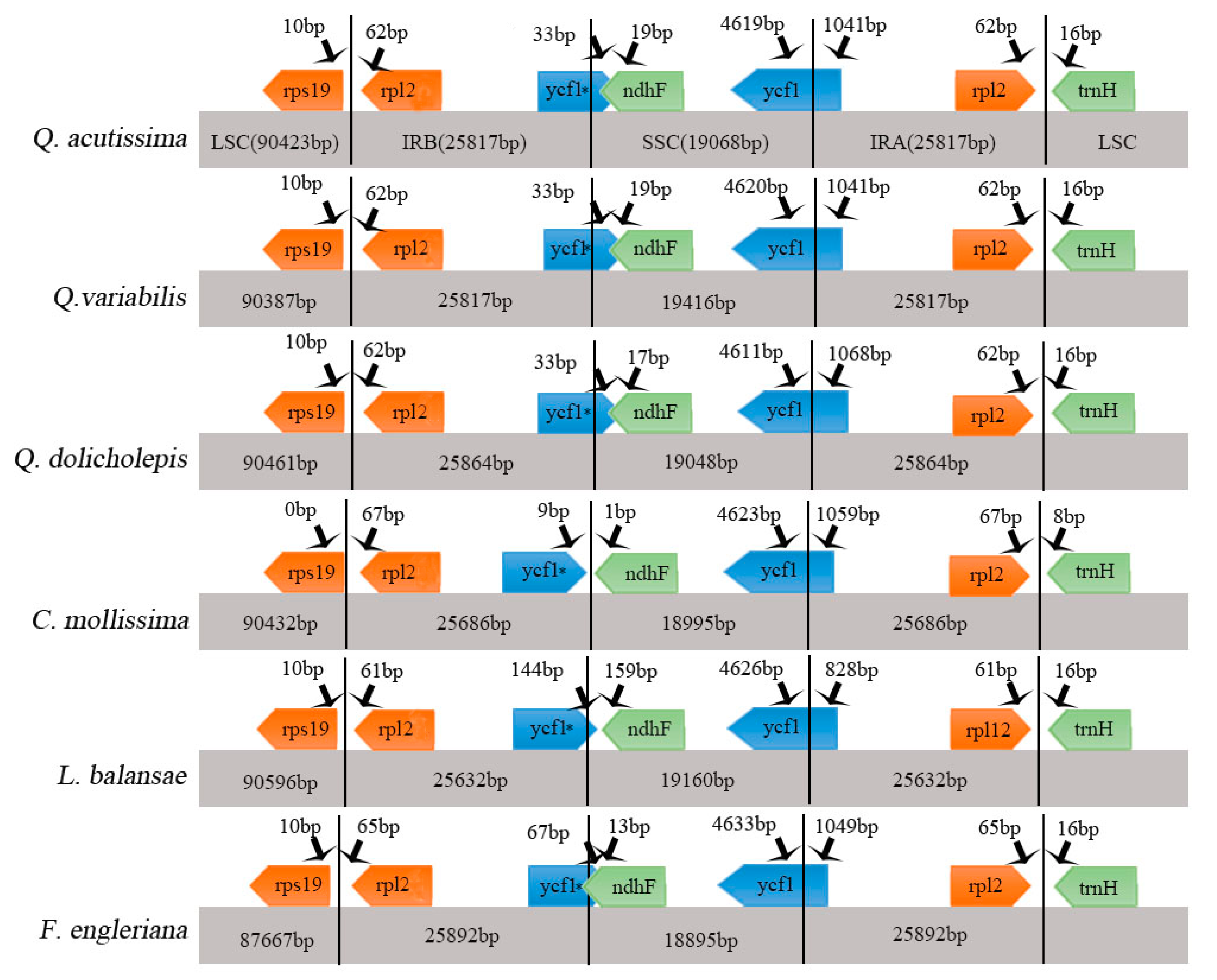
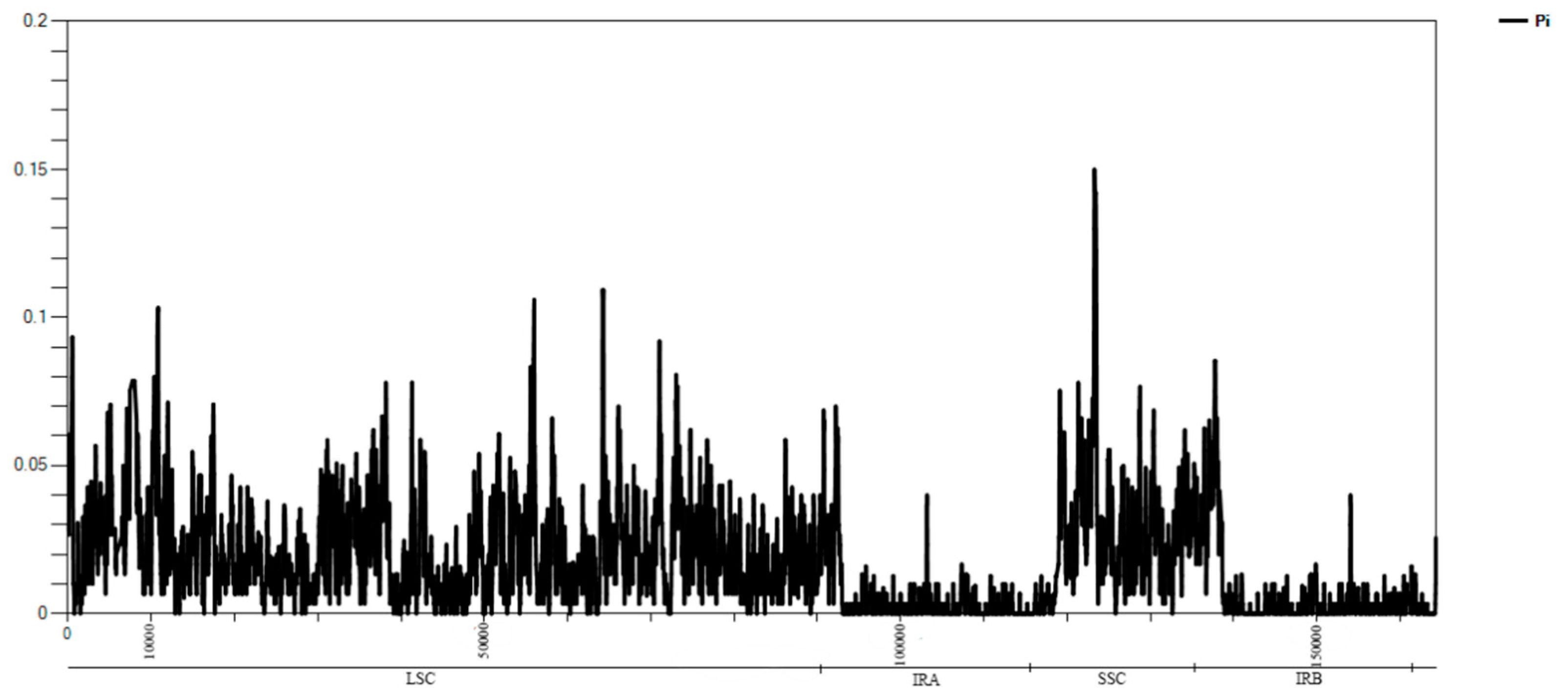
2.2. Comparative Analysis of Genomic Structure
2.3. Long-Repeat and SSR Analysis
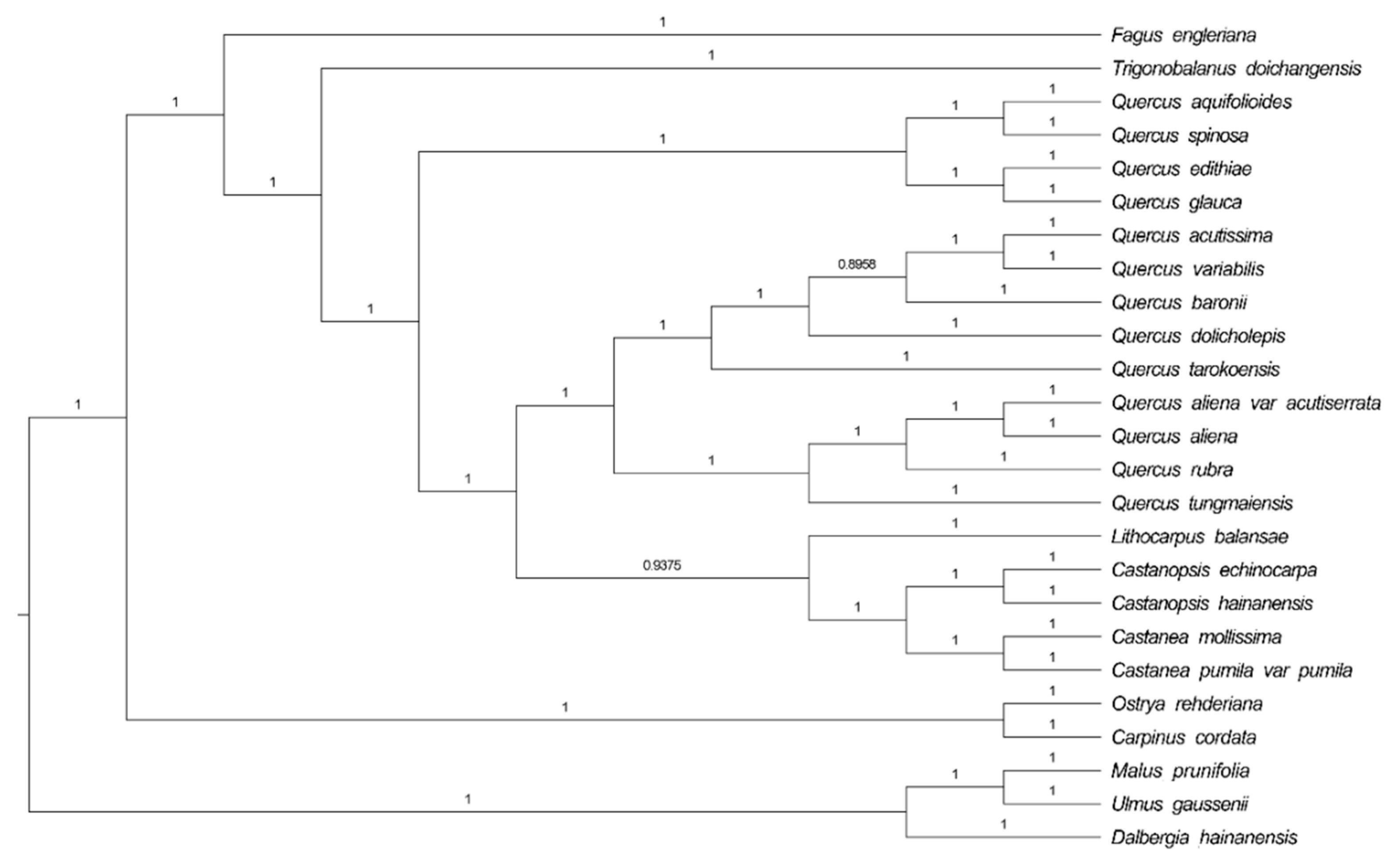
2.4. Phylogenetic Analysis
3. Materials and Methods
3.1. Sampling, DNA Extraction, Sequencing, and Assembly
3.2. Annotation and Analysis of the cpDNA Sequences
3.3. Genome Comparison
3.4. Phylogenetic Analysis
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| LSC | Large single copy |
| SSC | Small single copy |
| IR | Inverted repeat |
| Cp | Chloroplast |
| BI | Bayesian inference |
| A | Adenine |
| T | Thymine |
| G | Guanine |
| C | Cytosine |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Number of CDS | Number of tRNA | Number of rRNA | Total |
|---|---|---|---|---|
| LSC region | 62 | 25 | 0 | 87 |
| SSC region | 13 | 1 | 0 | 14 |
| IRA region | 6 | 7 | 4 | 17 |
| IRB region | 7 | 7 | 4 | 18 |
| Amino Acid | Codon | No. | RSCU | tRNA | Amino Acid | Codon | No. | RSCU | tRNA |
|---|---|---|---|---|---|---|---|---|---|
| Ala | GCG | 164 | 0.47 | Pro | CCA | 313 | 1.13 | trnP-TGG | |
| Ala | GCC | 224 | 0.64 | Pro | CCC | 226 | 0.82 | ||
| Ala | GCU | 630 | 1.79 | Pro | CCU | 409 | 1.48 | ||
| Ala | GCA | 388 | 1.1 | Pro | CCG | 161 | 0.58 | ||
| Cys | UGU | 221 | 1.44 | Gln | CAG | 215 | 0.45 | ||
| Cys | UGC | 86 | 0.56 | trnC-GCA | Gln | CAA | 731 | 1.55 | trnQ-TTG |
| Asp | GAC | 209 | 0.39 | trnD-GTC | Arg | CGU | 337 | 1.26 | trnR-ACG |
| Asp | GAU | 870 | 1.61 | Arg | AGA | 500 | 1.87 | trnR-TCT | |
| Glu | GAA | 1064 | 1.5 | trnE-TTC | Arg | CGA | 358 | 1.34 | |
| Glu | GAG | 357 | 0.5 | Arg | AGG | 183 | 0.68 | ||
| Phe | UUU | 983 | 1.3 | Arg | CGG | 118 | 0.44 | ||
| Phe | UUC | 535 | 0.7 | trnF-GAA | Arg | CGC | 109 | 0.41 | |
| Gly | GGU | 580 | 1.27 | Ser | AGC | 125 | 0.37 | trnS-GCT | |
| Gly | GGG | 330 | 0.72 | Ser | UCU | 557 | 1.66 | ||
| Gly | GGA | 706 | 1.55 | Ser | UCA | 397 | 1.18 | trnS-TGA | |
| Gly | GGC | 206 | 0.45 | trnG-GCC | Ser | UCC | 349 | 1.04 | trnS-GGA |
| His | CAU | 486 | 1.54 | Ser | AGU | 391 | 1.17 | ||
| His | CAC | 145 | 0.46 | trnH-GTG | Ser | UCG | 193 | 0.58 | |
| Ile | AUC | 458 | 0.58 | Thr | ACU | 538 | 1.6 | ||
| Ile | AUA | 758 | 0.97 | Thr | ACG | 160 | 0.48 | ||
| Ile | AUU | 1139 | 1.45 | Thr | ACC | 247 | 0.73 | trnT-GGT | |
| Lys | AAG | 379 | 0.5 | Thr | ACA | 402 | 1.19 | trnT-TGT | |
| Lys | AAA | 1062 | 1.4 | Val | GUU | 508 | 1.41 | ||
| Leu | UUG | 572 | 1.22 | trnL-CAA | Val | GUC | 181 | 0.5 | trnV-GAC |
| Leu | UUA | 894 | 1.9 | Val | GUA | 547 | 1.52 | ||
| Leu | CUU | 583 | 1.24 | Val | GUG | 207 | 0.57 | ||
| Leu | CUA | 373 | 0.79 | trnL-TAG | Trp | UGG | 462 | 1 | trnW-CCA |
| Leu | CUC | 204 | 0.43 | Tyr | UAC | 212 | 0.42 | trnY-GTA | |
| Leu | CUG | 198 | 0.42 | Tyr | UAU | 792 | 1.58 | ||
| Met | AUG | 620 | 1 | trnI-CAT | Stop | UAA | 47 | 1.6 | |
| Asn | AAU | 1004 | 1.5 | Stop | UAG | 22 | 0.75 | ||
| Asn | AAC | 304 | 0.46 | Stop | UGA | 19 | 0.65 |


References
- Aldrich, P.R.; Cavender-Bares, J. Quercus. Wild Crop Relat. Genom. Breed. Resour. 2011, 89–129. [Google Scholar] [CrossRef]
- Manos, P.S.; Cannon, C.H.; Oh, S.H. Phylogenetic relationships and taxonomic status of the paleoendemic Fagaceae of western North America: Recognition of a new genus, Notholithocarpus. Madroño 2008, 55, 181–190. [Google Scholar] [CrossRef]
- Oh, S.H.; Manos, P.S. Molecular phylogenetics and cupule evolution in Fagaceae as inferred from nuclear crabs claw sequences. Taxon 2008, 57, 434–451. [Google Scholar]
- Pelser, P.B.; Kennedy, A.H.; Tepe, E.J.; Shidler, J.B.; Nordenstam, B.; Kadereit, J.W.; Watson, L.E. Patterns and causes of incongruence between plastid and nuclear Senecioneae (Asteraceae) phylogenies. Am. J. Bot. 2010, 97, 856–873. [Google Scholar] [CrossRef] [PubMed]
- Pérezescobar, O.A.; Balbuena, J.A.; Gottschling, M. Rumbling orchids: How to assess divergent evolution between chloroplast endosymbionts and the nuclear host. Syst. Biol. 2016, 65, 51. [Google Scholar] [CrossRef] [PubMed]
- Curtu, A.L.; Gailing, O.; Leinemann, L.; Finkeldey, R. Genetic variation and differentiation within a natural community of five oak species (Quercus spp.). Plant Biol. 2006, 9, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmit, J.; Kleinschmit, J.G.R.; Vukelic, J.; Anic, I. Quercus robur-Quercus petraea: A critical review of the species concept. Glasnik Za Šumske Pokuse 2000, 37, 441–452. [Google Scholar]
- Denk, T.; Grimm, G.W. The oaks of western Eurasia: Traditional classifications and evidence from two nuclear markers. Taxon 2010, 59, 351–366. [Google Scholar]
- Kremer, A.; Abbott, A.G.; Carlson, J.E.; Manos, P.S.; Plomion, C.; Sisco, P.; Staton, M.E.; Ueno, S.; Vendramin, G.G. Genomics of Fagaceae. Tree Genet. Genomes 2012, 8, 583–610. [Google Scholar] [CrossRef] [Green Version]
- Simeone, M.C.; Piredda, R.; Papini, A.; Vessella, F.; Schirone, B. Application of plastid and nuclear markers to DNA barcoding of Euro-Mediterranean oaks (Quercus, Fagaceae): Problems, prospects and phylogenetic implications. Bot. J. Linn. Soc. 2013, 172, 478–499. [Google Scholar] [CrossRef]
- Hipp, A.L. Should hybridization make us skeptical of the oak phylogeny? Int. Oaks 2015, 26, 9–17. [Google Scholar]
- Denk, T.; Grimm, G.W.; Manos, P.S.; Deng, M.; Hipp, A.L. An updated infrageneric classification of the oaks: Review of previous taxonomic schemes and synthesis of evolutionary patterns. In Oaks Physiological Ecology. Exploring the Functional Diversity of Genus Quercus L.; Springer: Cham, Switzerland, 2017; pp. 13–38. [Google Scholar]
- Zhou, Z.; Wilkinson, H.; Wu, Z. Taxonomical and evolutionary implications of the leaf anatomy and architecture of Quercus L. Subgenus Quercus from China. Cathaya 1995, 7, 1–34. [Google Scholar]
- Pu, C.; Zhou, Z.; Luo, Y. A cladistic analysis of Quercus (Fagaceae) in China based on leaf epidermic and architecture. Acta Bot. Yunnanica 2002, 24, 689–698. [Google Scholar]
- Peng, Y.S.; Chen, L.; Li, J.Q. Study on Numerical Taxonomy of Quercus L. (Fagaceae) in China. J. Plant Sci. 2007, 25, 149–157. [Google Scholar]
- Zhang, X.; Yao, L.I.; Fang, Y. Geographical distribution and prediction of potential ranges of Quercus acutissima in China. Acta Bot. Boreali-Occident. Sin. 2014, 34, 1685–1692. [Google Scholar]
- Zhang, X.; Li, Y.; Liu, C.; Xia, T.; Zhang, Q.; Fang, Y. Phylogeography of the temperate tree species Quercus acutissima in China: Inferences from chloroplast DNA variations. Biochem. Syst. Ecol. 2015, 63, 190–197. [Google Scholar] [CrossRef]
- Hui, L.; Xie, H.; Jiang, Z.; Li, C.; Zhang, G. Photosynthetic response of potted Quercus acutissima Carruth seedlings under different soil moisture conditions. Sci. Soil Water Conserv. 2013, 11, 93–97. [Google Scholar]
- Fang, S.; Liu, Z.; Cao, Y.; Liu, D.; Yu, M.; Tang, L. Sprout development, biomass accumulation and fuelwood characteristics from coppiced plantations of Quercus acutissima. Biomass Bioenergy 2011, 35, 3104–3114. [Google Scholar] [CrossRef]
- Wu, T.; Wang, G.G.; Wu, Q.; Cheng, X.; Yu, M.; Wang, W.; Yu, X. Patterns of leaf nitrogen and phosphorus stoichiometry among Quercus acutissima provenances across China. Ecol Complex. 2014, 17, 32–39. [Google Scholar] [CrossRef]
- Choi, H.S.; Kim, Y.Y.; Hong, K.N.; Hong, Y.P.; Hyun, J.O. Genetic structure of a population of Quercus acutissima in Korea revealed by microsatellite markers. Korean J. Genet. 2005, 27, 267–271. [Google Scholar]
- Huang, L.; Xiao, L.I.; Yan, J. Studies on Introduction of North American Oaks. China Forestry Science and Technology. 2005. Available online: http://xueshu.baidu.com/s?wd=paperuri%3A%2866d7b49f4975cf2de13aa699e48387b1%29&filter=sc_long_sign&tn=SE_xueshusource_2kduw22v&sc_vurl=http%3A%2F%2Fen.cnki.com.cn%2FArticle_en%2FCJFDTOTAL-LKKF200501009.htm&ie=utf-8&sc_us=11198188077522908127 (accessed on 16 August 2018).
- Shen, X.; Wu, M.; Liao, B.; Liu, Z.; Bai, R.; Xiao, S.; Li, X.; Zhang, B.; Xu, J.; Chen, S. Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant Artemisia annua. Molecules 2017, 22, 1330. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Guo, L.; Zhao, W.; Xu, J.; Li, Y.; Zhang, X.; Shen, X.; Wu, M.; Hou, X. Complete chloroplast genome sequence and phylogenetic analysis of Paeonia ostii. Molecules 2018, 23, 246. [Google Scholar] [CrossRef] [PubMed]
- Lobry, J.R. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. Evol. 1996, 13, 660–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Necsulea, A.; Lobry, J. A new method for assessing the effect of replication on DNA base composition asymmetry. Mol. Biol. Evol. 2007, 24, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Tillier, E.R.; Collins, R.A. The contributions of replication orientation, gene direction, and signal sequences to base-composition asymmetries in bacterial genomes. J. Mol. Evol. 2000, 50, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; Depamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J. Methods for obtaining and analyzing whole chloroplast genome sequences. Method Enzymol. 2005, 395, 348. [Google Scholar]
- Shetty, S.M.; Md Shah, M.U.; Makale, K.; Mohd-Yusuf, Y.; Khalid, N.; Othman, R.Y. Complete chloroplast genome sequence of Musa balbisiana corroborates structural heterogeneity of inverted repeats in wild progenitors of cultivated bananas and plantains. Plant Genome 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, E.; Takahashi, Y.; Lemieux, C.; Turmel, M.; Rochaix, J.D. The chloroplast ycf3 and ycf4 open reading frames of Chlamydomonas reinhardtii are required for the accumulation of the photosystem I complex. Embo J. 1997, 16, 6095–6104. [Google Scholar] [CrossRef] [PubMed]
- Cavender Bares, J.; González Rodríguez, A.; Eaton, D.A.R.; Hipp, A.A.L.; Beulke, A.; Manos, P.S. Phylogeny and biogeography of the American live oaks (Quercus subsection Virentes): A genomic and population genetics approach. Mol Ecol. 2015, 24, 3668–3687. [Google Scholar] [CrossRef] [PubMed]
- Kode, V.; Mudd, E.A.; Iamtham, S.; Day, A. The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 2005, 44, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Tang, P.; Li, Z.; Li, D.; Liu, Y.; Huang, H. The first complete chloroplast genome sequences in Actinidiaceae: Genome structure and comparative analysis. PLoS ONE 2015, 10, e129347. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. Ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Bédard, J.; Hirano, M.; Hirabayashi, Y.; Oishi, M.; Imai, M.; Takase, M.; Ide, T.; Nakai, M. Uncovering the protein translocon at the chloroplast inner envelope membrane. Science 2013, 339, 571. [Google Scholar] [CrossRef] [PubMed]
- Provan, J. Novel chloroplast microsatellites reveal cytoplasmic variation in Arabidopsis thaliana. Mol. Ecol. 2000, 9, 2183–2185. [Google Scholar] [CrossRef] [PubMed]
- Flannery, M.L.; Mitchell, F.J.; Coyne, S.; Kavanagh, T.A.; Burke, J.I.; Salamin, N.; Dowding, P.; Hodkinson, T.R. Plastid genome characterisation in Brassica and Brassicaceae using a new set of nine SSRs. Theor. Appl. Genet. 2006, 113, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Tsuda, Y.; Uchiyama, K.; Saito, Y.; Tsuda, Y.; Uchiyama, K.; Fukuda, T.; Seto, Y.; Kim, P.G.; Shen, H.L.; et al. Genetic Variation in Quercus acutissima Carruth., in Traditional Japanese Rural Forests and Agricultural Landscapes, Revealed by Chloroplast Microsatellite Markers. Forests 2017, 8, 451. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, J.; Feng, L.; Zhou, T.; Bai, G.; Yang, J.; Zhao, G. Plastid genome comparative and phylogenetic analyses of the key genera in Fagaceae: Highlighting the effect of codon composition bias in phylogenetic inference. Front. Plant Sci. 2018, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wuyun, T.N.; Du, H.; Wang, D.; Cao, D. Complete chloroplast genome sequences of Eucommia ulmoides: Genome structure and evolution. Tree Genet. Genomes 2016, 12, 12. [Google Scholar] [CrossRef]
- Doyle, J.J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. Novoplasty: De novo assembly of organelle genomes from whole genome DNA. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [PubMed]
- Chang, L.; Shi, L.; Zhu, Y.; Chen, H.; Zhang, J.; Lin, X.; Guan, X. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genom. 2012, 13, 715. [Google Scholar]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. Organellar Genome DRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Mudunuri, S.B.; Nagarajaram, H.A. IMEx: Imperfect Microsatellite Extractor. Bioinformatics 2007, 23, 1181–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Lam-Tung, N.; Schmidt, H.A.; Arndt, V.H.; Quang, M.B. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]




| Genome Features | Q. acutissima | Q. variabilis | Q. dolicholepis | C. mollissima | L. balansae | F. engleriana |
|---|---|---|---|---|---|---|
| Genome size (bp) | 161,124 | 161,077 | 161,237 | 160,799 | 161,020 | 158,346 |
| LSC length (bp) | 90,423 | 90,387 | 90,461 | 90,432 | 90,596 | 87,667 |
| SSC length (bp) | 19,068 | 19,056 | 19,048 | 18,995 | 19,160 | 18,895 |
| IR length (bp) | 51,632 | 51,634 | 51,728 | 51,372 | 51,264 | 51,784 |
| Number of genes | 136 | 134 | 134 | 130 | 134 | 131 |
| Number of protein–coding genes | 88 | 86 | 86 | 83 | 87 | 83 |
| Number of tRNA genes | 40 | 40 | 40 | 37 | 39 | 40 |
| Number of rRNA genes | 8 | 8 | 8 | 8 | 8 | 8 |
| Region | A (%) | T (U) (%) | C (%) | G (%) | A + T (%) | G + C (%) |
|---|---|---|---|---|---|---|
| LSC | 31.99 | 33.4 | 17.74 | 16.88 | 65.39 | 34.62 |
| SSC | 34.46 | 34.71 | 16.24 | 14.6 | 69.17 | 30.84 |
| IR | 28.61 | 28.61 | 21.39 | 21.39 | 57.22 | 42.78 |
| Total | 31.69 | 32.24 | 18.46 | 17.62 | 63.93 | 36.08 |
| Function | Genes |
|---|---|
| RNAs, transfer | trnH-GUG, trnK-UUU, trnQ-UUG, trnS-GCU, trnG-GCC, trnR-UCU, trnC-GCA, trnD-GUC, trnY-GUA, trnE-UUC, trnT-GGU, trnM-CAU, trnS-UGA, trnG-GCC, trnfM-CAU, trnS-GGA, trnT-UGU, trnL-UAA, trnF-GAA, trnV-UAC, trnM-CAU, trnT-GGU, trnW-CCA, trnP-UGG, trnP-GGG, trnI *-CAU, trnL-CAA *, trnV-GAC, trnI-GAU *, trnA-UGC, trnR-ACG, trnN-GUU, trnL-UAG, trnN-GUU, trnR-ACG, trnA-UGC, trnV-GAC |
| RNAs, ribosomal | rrn23 *, rrn16 *, rrn5 *, rrn4.5 * |
| Transcription and splicing | rpoC1 *, rpoC2, rpoA, rpoB |
| Translation, ribosomal proteins | |
| Small subunit | rps2, rps3, rps4, rps7, rps8, rps11, rps12 **, rps14, rps15, rps16 *, rps18, rps19 |
| Large subunit | rpl2 *, rpl14, rpl16 *, rpl20, rpl22, rpl23, rpl32, rpl33, rpl36 |
| Photosynthesis | |
| ATP synthase | atpE, atpB, atpA, atpF *, atpH, atpI |
| Photosystem I | psaI, psaB, psaA, psaC, psaJ, ycf3 *, ycf4 |
| Photosystem II | psbD, psbC, psbZ, psbT, psbH, psbK, psbI, psbJ, psbF, psbE, psbM, psbN, psbL, psbA, psbB |
| Calvin cycle | rbcL |
| Cytochrome complex | petN, petA, petL, petG, petB *, petD * |
| NADH dehydrogenase | ndhB *, ndhI, ndhK, ndhC, ndhF, ndhD, ndhG, ndhE, ndhA, ndhH, ndhJ |
| Others | inFA, ycf15 *, ycf1 *, ycf2 *, accD, cemA, ccsA, clpP ** |
| Gene | Location | Exon I (bp) | Intron I (bp) | Exon II (bp) | Intron II (bp) | Exon III (bp) |
|---|---|---|---|---|---|---|
| rps16 | LSC | 42 | 898 | 195 | ||
| atpF | LSC | 144 | 780 | 411 | ||
| rpoC1 | LSC | 432 | 827 | 1626 | ||
| ycf3 | LSC | 127 | 718 | 228 | 778 | 155 |
| clpP | LSC | 69 | 844 | 294 | 649 | 228 |
| petB | LSC | 6 | 841 | 642 | ||
| petD | LSC | 9 | 640 | 474 | ||
| rpl16 | LSC | 9 | 1102 | 399 | ||
| rpl2 | RepeatA | 390 | 628 | 471 | ||
| ndhB | RepeatA | 777 | 680 | 756 | ||
| rps12 | RepeatA | 10 | 537 | 231 | ||
| ndhA | SSC | 551 | 1040 | 541 | ||
| rps12 | RepeatB | 232 | 536 | 26 | ||
| ndhB | RepeatB | 777 | 680 | 756 | ||
| rpl2 | RepeatB | 390 | 628 | 471 | ||
| trnG-GCC | LSC | 23 | 734 | 37 | ||
| trnK-UUU | LSC | 37 | 2505 | 35 | ||
| trnL-UAA | LSC | 35 | 483 | 50 | ||
| trnV-UAC | LSC | 36 | 630 | 37 | ||
| trnI-GAU | RepeatA | 42 | 950 | 35 | ||
| trnA-UGC | RepeatA | 38 | 800 | 35 | ||
| TRNA-UGC | RepeatB | 38 | 800 | 35 | ||
| trnI-GAU | RepeatB | 42 | 950 | 35 |
| ID | Repeat Start I | Type | Size (bp) | Repeat Start 2 | Mismatch (bp) | E-Value | Gene | Region |
|---|---|---|---|---|---|---|---|---|
| 1 | 6831 | F | 46 | 6853 | 0 | 1.47 × 10−18 | IGS | LSC |
| 2 | 11,847 | R | 31 | 11,847 | 0 | 1.58 × 10−9 | IGS | LSC |
| 3 | 6818 | R | 26 | 6818 | 0 | 1.62 × 10−6 | rps16 | LSC |
| 4 | 47,242 | F | 25 | 47,264 | 0 | 6.49 × 10−6 | IGS | LSC |
| 5 | 6831 | F | 24 | 6875 | 0 | 2.59 × 10−5 | IGS | LSC |
| 6 | 115,801 | F | 24 | 135,722 | 0 | 2.59 × 10−5 | ycf1 | IRA; IRB |
| 7 | 113,545 | F | 23 | 113,576 | 0 | 1.04 × 10−4 | IGS | IRA |
| 8 | 118,844 | R | 23 | 118,844 | 0 | 1.04 × 10−4 | IGS | IRA |
| 9 | 137,948 | F | 23 | 137,979 | 0 | 1.04 × 10−4 | IGS | IRB |
| 10 | 11,371 | F | 22 | 41,193 | 0 | 4.15 × 10−4 | trnG-GCC (exon), trnG-GCC | LSC |
| 11 | 9536 | F | 21 | 39,849 | 0 | 1.66 × 10−3 | trnS-UGA, trnS-GCU | LSC |
| 12 | 10,319 | F | 21 | 18,682 | 0 | 1.66 × 10−3 | IGS | LSC |
| 13 | 117,049 | R | 21 | 117,049 | 0 | 1.66 × 10−3 | ndhF | SSC |
| 14 | 36,478 | F | 20 | 53,719 | 0 | 6.64 × 10−3 | IGS | LSC |
| 15 | 53,720 | F | 20 | 130,481 | 0 | 6.64 × 10−3 | IGS | LSC; SSC |
| 16 | 55,907 | R | 20 | 55,907 | 0 | 6.64 × 10−3 | atpB | LSC |
| 17 | 57,271 | F | 20 | 142,064 | 0 | 6.64 × 10−3 | trnV-UAC, trnA-UGC | LSC; IRB |
| 18 | 105,331 | F | 20 | 105,349 | 0 | 6.64 × 10−3 | IGS | IRA |
| 19 | 146,178 | F | 20 | 146,196 | 0 | 6.64 × 10−3 | IGS | IRB |
| 20 | 4930 | F | 19 | 36,476 | 0 | 2.66 × 10−2 | IGS | LSC |
| 21 | 8915 | R | 19 | 8915 | 0 | 2.66 × 10−2 | IGS | LSC |
| 22 | 13,541 | R | 19 | 76,642 | 0 | 2.66 × 10−2 | atpA | LSC |
| 23 | 18,685 | R | 19 | 118,842 | 0 | 2.66 × 10−2 | clpP | LSC; SSC |
| 24 | 21,297 | R | 19 | 54,183 | 0 | 2.66 × 10−2 | rpoC2 | LSC |
| 25 | 36,479 | F | 19 | 130,481 | 0 | 2.66 × 10−2 | IGS | LSC; SSC |
| 26 | 39,957 | R | 19 | 39,957 | 0 | 2.66 × 10−2 | IGS | LSC |
| 27 | 62,040 | R | 19 | 62,040 | 0 | 2.66 × 10−2 | IGS | LSC |
| 28 | 64,751 | R | 19 | 64,751 | 0 | 2.66 × 10−2 | IGS | LSC |
| 29 | 69,026 | R | 19 | 69,026 | 0 | 2.66 × 10−2 | IGS | LSC |
| 30 | 71,277 | R | 19 | 71,277 | 0 | 2.66 × 10−2 | IGS | LSC |
| 31 | 72,561 | R | 19 | 72,561 | 0 | 2.66 × 10−2 | IGS | LSC |
| 32 | 4430 | R | 18 | 4430 | 0 | 1.06 × 10−1 | IGS | LSC |
| 33 | 4437 | F | 18 | 24,828 | 0 | 1.06 × 10−1 | rpoC1 (intron) | SSC |
| 34 | 4935 | F | 18 | 52,105 | 0 | 1.06 × 10−1 | IGS | LSC |
| 35 | 4938 | F | 18 | 118,695 | 0 | 1.06 × 10−1 | IGS | LSC |
| 36 | 6813 | F | 18 | 6847 | 0 | 1.06 × 10−1 | IGS | LSC |
| 37 | 6813 | F | 18 | 6869 | 0 | 1.06 × 10−1 | IGS | LSC |
| 38 | 6817 | F | 18 | 127,945 | 0 | 1.06 × 10−1 | ndhA (intron) | LSC |
| 39 | 7369 | F | 18 | 7387 | 0 | 1.06 × 10−1 | IGS | LSC; SSC |
| 40 | 7465 | R | 18 | 7465 | 0 | 1.06 × 10−1 | IGS | LSC; SSC |
| 41 | 8589 | R | 18 | 34,768 | 0 | 1.06 × 10−1 | IGS | LSC; SSC |
| 42 | 9996 | R | 18 | 9996 | 0 | 1.06 × 10−1 | IGS | LSC |
| 43 | 10,283 | F | 18 | 31,730 | 0 | 1.06 × 10−1 | IGS | LSC |
| 44 | 10,322 | R | 18 | 118,843 | 0 | 1.06 × 10−1 | IGS | LSC; IRA |
| 45 | 10,548 | F | 18 | 133,365 | 0 | 1.06 × 10−1 | ycf1 | LSC |
| 46 | 31,728 | F | 18 | 125,951 | 0 | 1.06 × 10−1 | IGS | LSC |
| 47 | 39,812 | F | 18 | 40,698 | 0 | 1.06 × 10−1 | trnS-UGA | LSC; SSC |
| 48 | 40,022 | R | 18 | 69,093 | 0 | 1.06 × 10−1 | IGS | LSC |
| 49 | 40,700 | F | 18 | 123,827 | 0 | 1.06 × 10−1 | IGS | LSC |
| 50 | 43,446 | F | 18 | 45,670 | 0 | 1.06 × 10−1 | psaB | SSC |
| 51 | 40,022 | R | 18 | 69,093 | 0 | 1.06 × 10−1 | IGS | LSC |
| 52 | 40,700 | F | 18 | 123,827 | 0 | 1.06 × 10−1 | IGS | LSC |
| 53 | 43,446 | F | 18 | 45,670 | 0 | 1.06 × 10−1 | psaB, psaA | LSC |
| ID | Repeat Motif | Length (bp) | Start | End | Region | Gene | ID | Repeat Motif | Length (bp) | Start | End | Region | Gene |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | (A)10 | 9 | 1809 | 1818 | LSC | 34 | (T)10 | 9 | 55,713 | 55,722 | LSC | ||
| 2 | (C)14 | 13 | 4433 | 4446 | LSC | 35 | (T)10 | 9 | 59,591 | 59,600 | LSC | ||
| 3 | (T)11 | 10 | 4697 | 4707 | LSC | 36 | (T)10 | 9 | 60,063 | 60,072 | LSC | ||
| 4 | (A)10 | 9 | 4939 | 4948 | LSC | trnK-UUU | 37 | (T)10 | 9 | 64,092 | 64,101 | LSC | accD |
| 5 | (T)11 | 10 | 7001 | 7011 | LSC | 38 | (A)11 | 10 | 64,266 | 64,276 | LSC | ||
| 6 | (T)10 | 9 | 7746 | 7755 | LSC | 39 | (AT)7 | 13 | 64,570 | 64,583 | LSC | ||
| 7 | (A)10 | 9 | 8174 | 8183 | LSC | 40 | (T)14 | 13 | 64,945 | 64,958 | LSC | ||
| 8 | (A)12 | 11 | 8590 | 8601 | LSC | psbK | 41 | (T)13 | 12 | 66,170 | 66,182 | LSC | |
| 9 | (A)11 | 10 | 8920 | 8930 | LSC | 42 | (T)11 | 10 | 68,616 | 68,626 | LSC | petA | |
| 10 | (A)10 | 9 | 9465 | 9474 | LSC | 43 | (T)11 | 10 | 70,730 | 70,740 | LSC | ||
| 11 | (A)10 | 9 | 10,161 | 10,170 | LSC | 44 | (T)11 | 10 | 71,398 | 71,408 | LSC | ||
| 12 | (A)11 | 10 | 13,547 | 13,557 | LSC | 45 | (T)11 | 10 | 73,389 | 73,399 | LSC | ||
| 13 | (T)12 | 11 | 15,345 | 15,356 | LSC | 46 | (AT)6 | 11 | 77,274 | 77,285 | LSC | clpP | |
| 14 | (T)10 | 9 | 16,160 | 16,169 | LSC | 47 | (TA)7 | 13 | 82,928 | 82,941 | LSC | petD | |
| 15 | (A)12 | 11 | 18,692 | 18,703 | LSC | rpoC2 | 48 | (A)11 | 10 | 85,781 | 85,791 | LSC | |
| 16 | (T)12 | 11 | 21,295 | 21,306 | LSC | rpoC2 | 49 | (T)10 | 9 | 86,100 | 86,109 | LSC | |
| 17 | (T)14 | 13 | 25,299 | 25,312 | LSC | 50 | (T)10 | 9 | 88,820 | 88,829 | LSC | ||
| 18 | (T)10 | 9 | 28,563 | 28,572 | LSC | 51 | (T)11 | 10 | 114,070 | 114,080 | IRA | ||
| 19 | (T)10 | 9 | 29,651 | 29,660 | LSC | 52 | (T)12 | 11 | 118,582 | 118,593 | SSC | ||
| 20 | (T)11 | 10 | 30,275 | 30,285 | LSC | 53 | (A)11 | 10 | 118,695 | 118,705 | SSC | ||
| 21 | (C)14 | 13 | 30,428 | 30,441 | LSC | 54 | (T)11 | 10 | 119,000 | 119,010 | SSC | ||
| 22 | (T)11 | 10 | 31,731 | 31,741 | LSC | 55 | (A)10 | 9 | 119,794 | 119,803 | SSC | ||
| 23 | (A)10 | 9 | 32,094 | 32,103 | LSC | 56 | (T)11 | 10 | 122,199 | 122,209 | SSC | ndhD | |
| 24 | (A)10 | 9 | 33,986 | 33,995 | LSC | 57 | (A)10 | 9 | 122,546 | 122,555 | SSC | ||
| 25 | (A)13 | 12 | 34,775 | 34,787 | LSC | 58 | (AT)8 | 15 | 123,832 | 123,847 | SSC | ||
| 26 | (A)10 | 9 | 34,955 | 34,964 | LSC | 59 | (T)11 | 10 | 125,812 | 125,822 | SSC | ||
| 27 | (A)10 | 9 | 36,485 | 36,494 | LSC | 60 | (T)11 | 10 | 125,954 | 125,964 | SSC | ||
| 28 | (AT)6 | 11 | 39,819 | 39,830 | LSC | 61 | (T)11 | 10 | 130,262 | 130,272 | SSC | ||
| 29 | (T)10 | 9 | 41,238 | 41,247 | LSC | trnfM-CAU | 62 | (A)10 | 9 | 130,487 | 130,496 | SSC | |
| 30 | (T)11 | 10 | 53,217 | 53,227 | LSC | 63 | (T)10 | 9 | 133,465 | 133,474 | SSC | ycf1 | |
| 31 | (A)10 | 9 | 53,726 | 53,735 | LSC | 64 | (T)13 | 12 | 134,042 | 134,054 | SSC | ycf1 | |
| 32 | (T)15 | 14 | 54,110 | 54,124 | LSC | 65 | (A)11 | 10 | 137,468 | 137,478 | SSC | ||
| 33 | (A)11 | 10 | 54,990 | 55,000 | LSC |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Li, Y.; Zang, M.; Li, M.; Fang, Y. Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus acutissima. Int. J. Mol. Sci. 2018, 19, 2443. https://doi.org/10.3390/ijms19082443
Li X, Li Y, Zang M, Li M, Fang Y. Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus acutissima. International Journal of Molecular Sciences. 2018; 19(8):2443. https://doi.org/10.3390/ijms19082443
Chicago/Turabian StyleLi, Xuan, Yongfu Li, Mingyue Zang, Mingzhi Li, and Yanming Fang. 2018. "Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus acutissima" International Journal of Molecular Sciences 19, no. 8: 2443. https://doi.org/10.3390/ijms19082443
APA StyleLi, X., Li, Y., Zang, M., Li, M., & Fang, Y. (2018). Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus acutissima. International Journal of Molecular Sciences, 19(8), 2443. https://doi.org/10.3390/ijms19082443