GLP-1 Analogue Liraglutide Attenuates Mutant Huntingtin-Induced Neurotoxicity by Restoration of Neuronal Insulin Signaling



Abstract
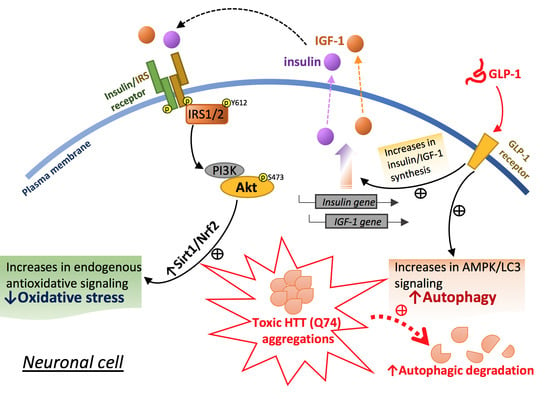
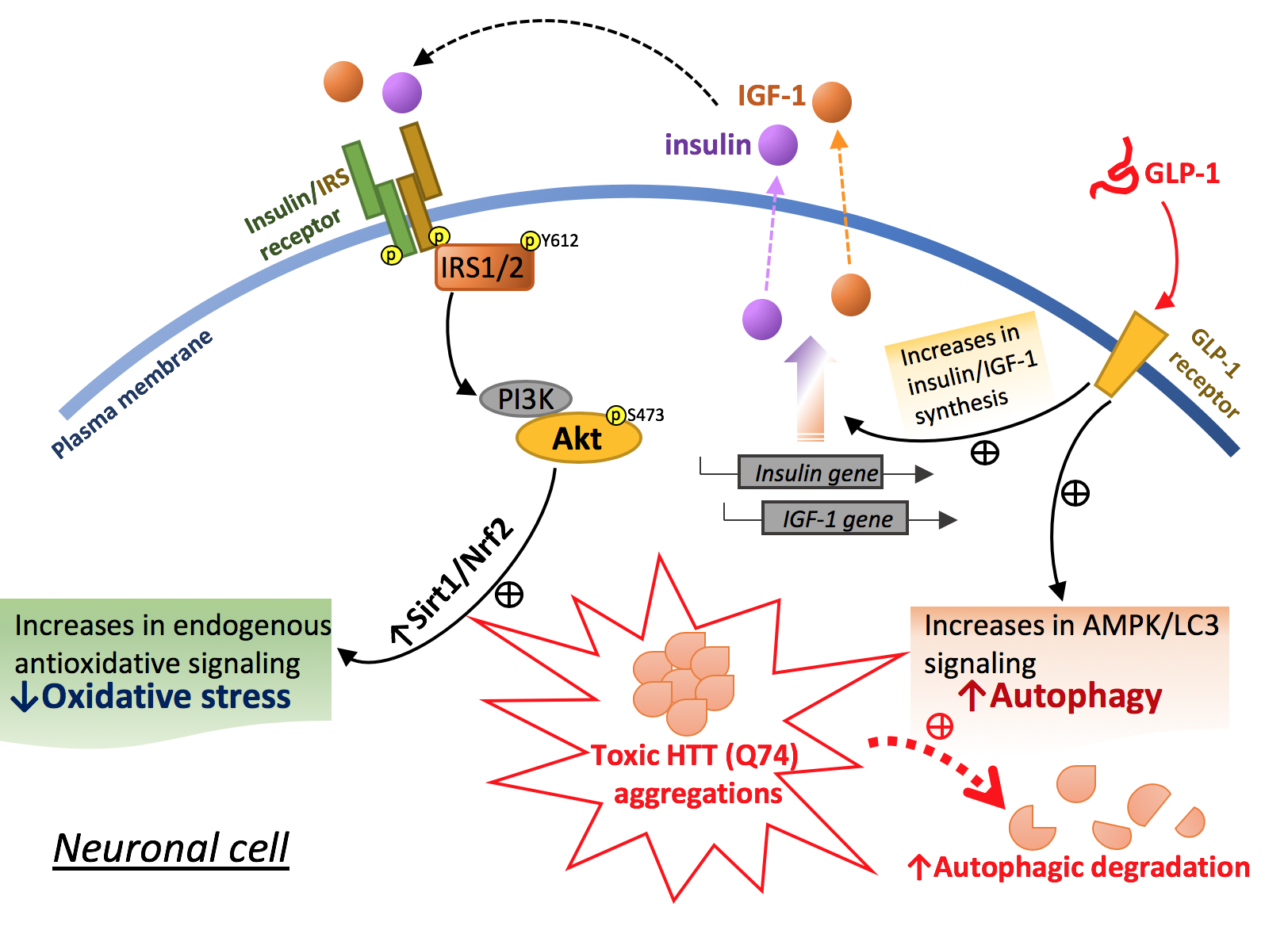
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
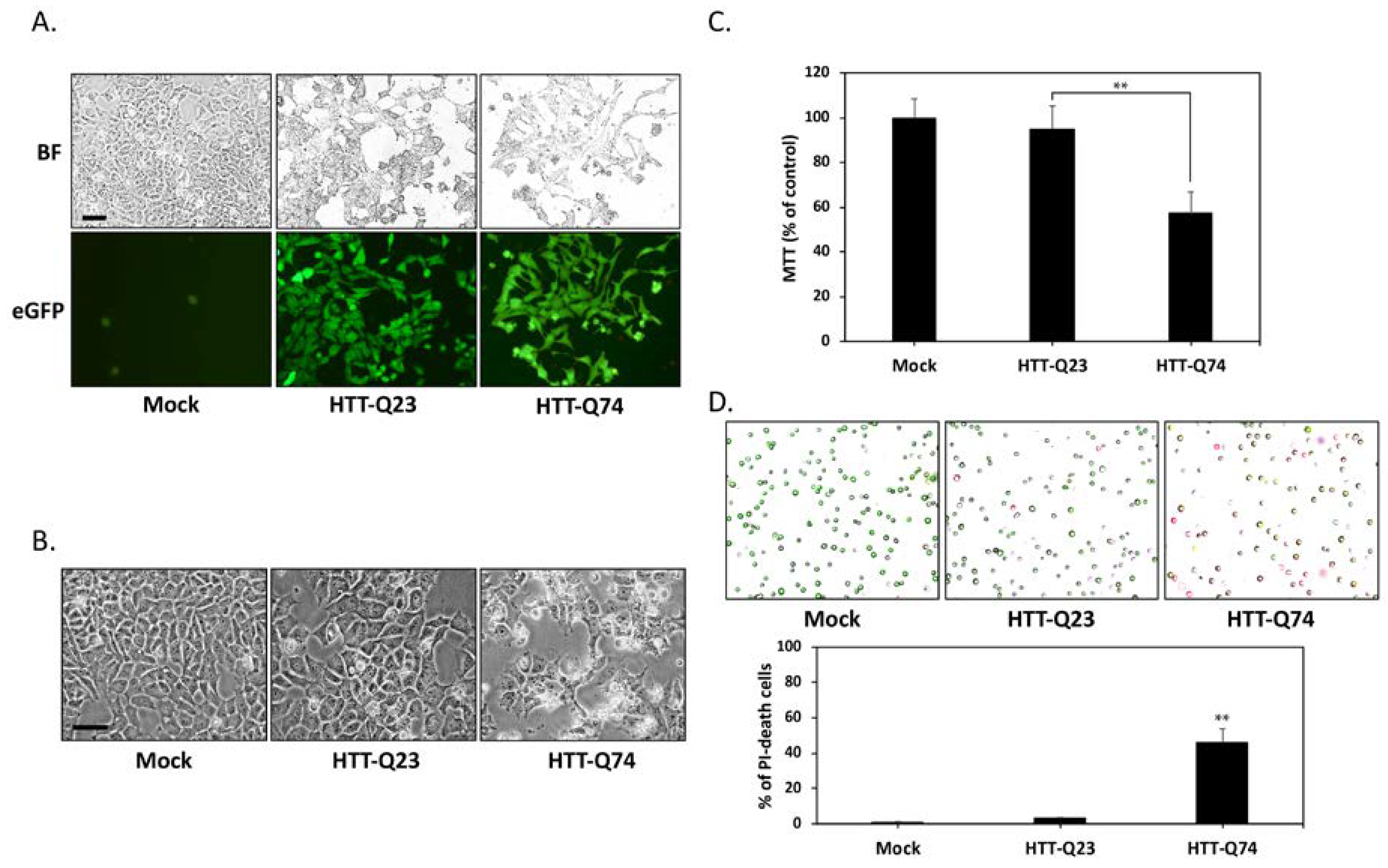
2.1. Q74-mHTT Overexpression Causes Significant Neuronal SK-N-MC Cell Death
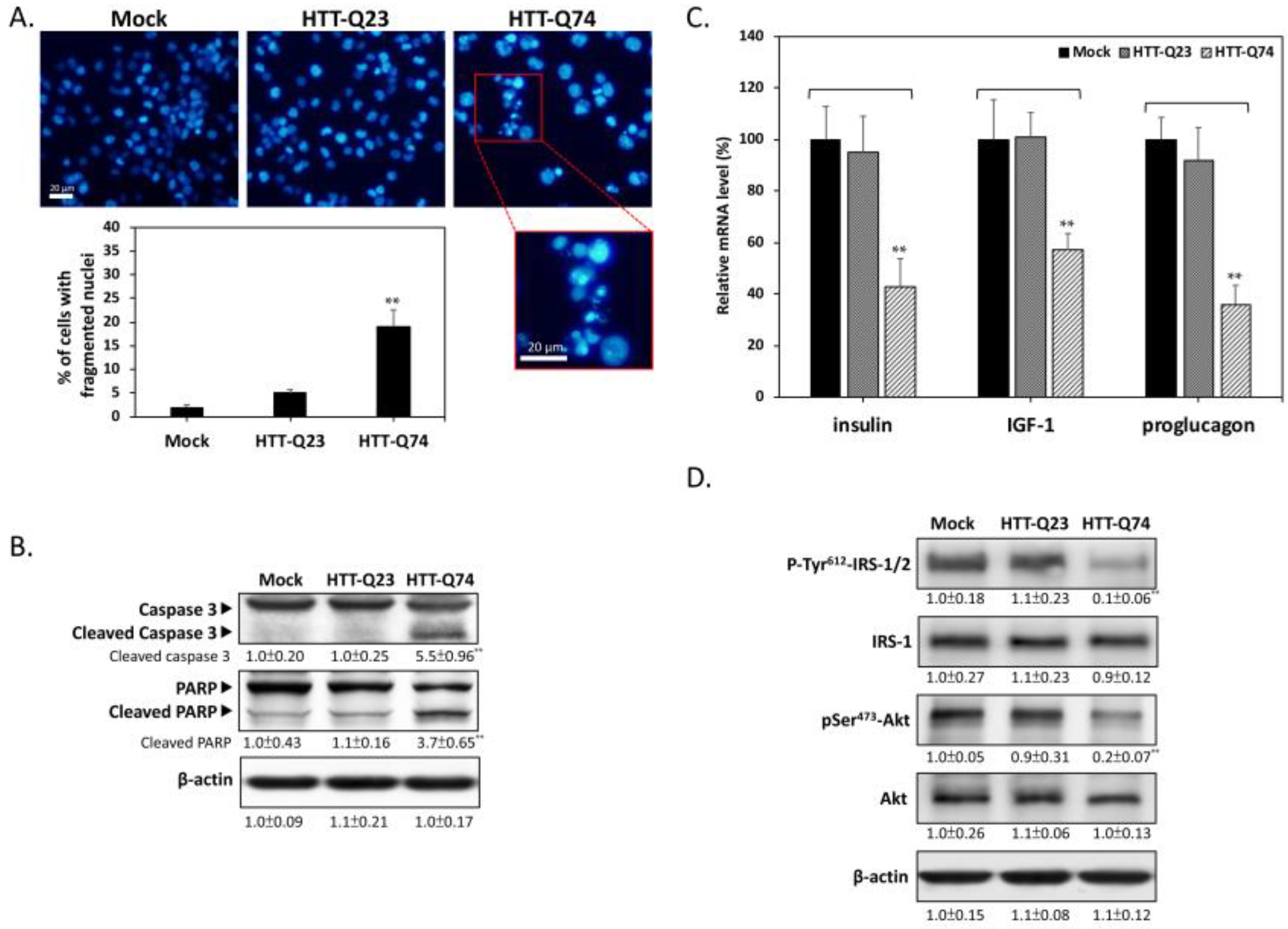
2.2. Q74-mHTT Overexpression Stimulates Apoptosis and Blocks Insulin Signaling
2.3. Liraglutide Alleviates mHTT-Impaired Insulin Downstream Signaling and Oxidative Stress
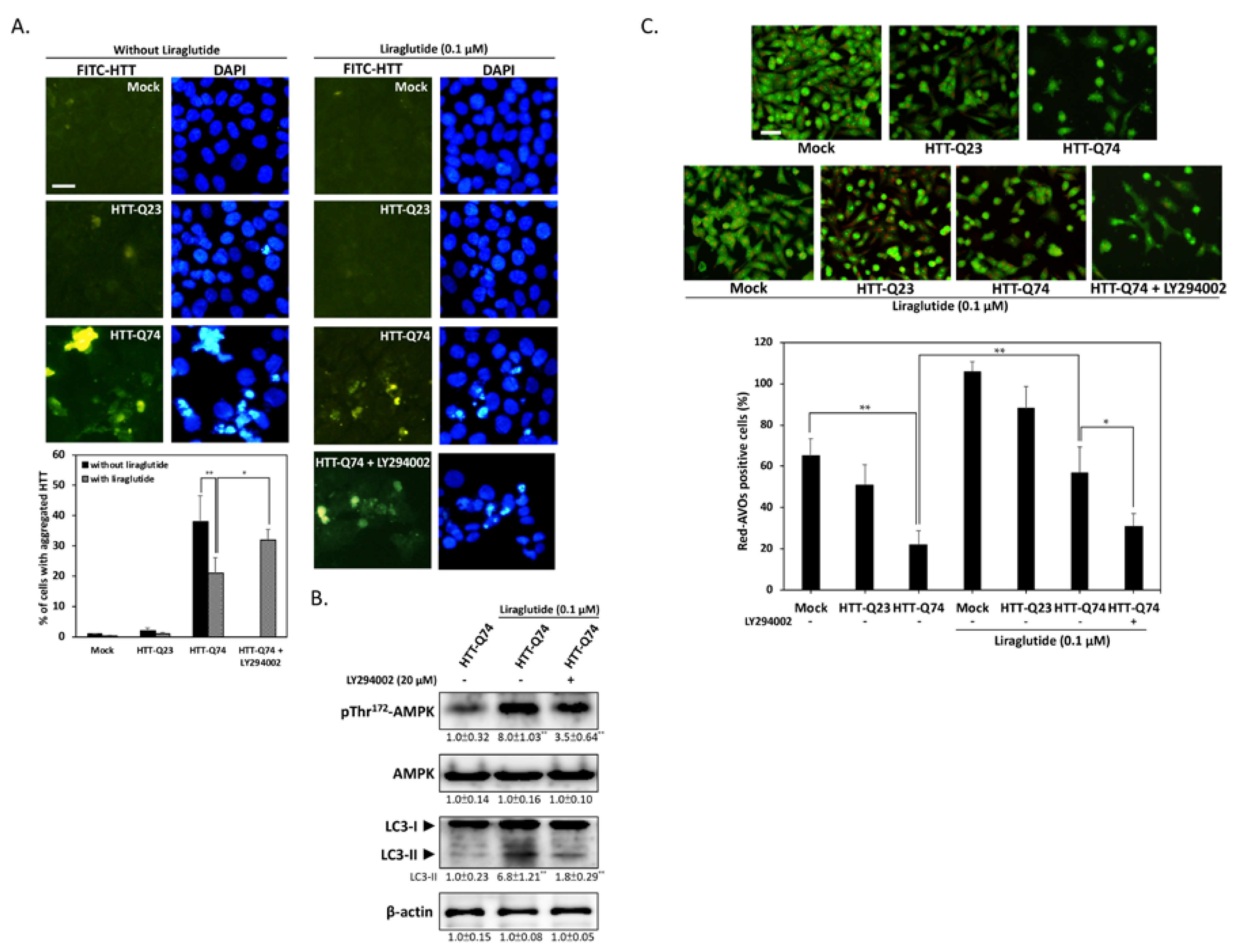
2.4. Liraglutide Increases Autophagy and Alleviates Intracellular HTT Aggregation in HTT-Q74-Overexpressing Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture, Transfection, and MTT Assay
4.3. Microscopic Observation and Acridine Orange–Propidium Iodide Assay
4.4. Assessment of Nuclear Morphology through DAPI Staining
4.5. Western Blot Analysis
4.6. mRNA Expression Analysis through Reverse-Transcription Quantitative PCR
4.7. Measurement of ROS thorough Dihydroethidium Staining
4.8. Immunocytochemistry and AO Staining
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed]
- Warby, S.C.; Montpetit, A.; Hayden, A.R.; Carroll, J.B.; Butland, S.L.; Visscher, H.; Collins, J.A.; Semaka, A.; Hudson, T.J.; Hayden, M.R. Cag Expansion in the Huntington Disease Gene Is Associated with a Specific and Targetable Predisposing Haplogroup. Am. J. Hum. Genet. 2009, 84, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Cummings, D.M.; Alaghband, Y.; Hickey, M.A.; Joshi, P.R.; Hong, S.C.; Zhu, C.; Ando, T.K.; Andre, V.M.; Cepeda, C.; Watson, J.B.; et al. A Critical Window of Cag Repeat-Length Correlates with Phenotype Severity in the R6/2 Mouse Model of Huntington’s Disease. J. Neurophysiol. 2012, 107, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Hong, Y.; Li, X.J.; Li, S.H. Subcellular Clearance and Accumulation of Huntington Disease Protein: A Mini-Review. Front. Mol. Neurosci. 2016, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Elbaum, M. Forms and Phases in Huntingtin Protein Aggregation. Mol. Cell 2018, 70, 567–568. [Google Scholar] [CrossRef] [PubMed]
- Jodeiri Farshbaf, M.; Ghaedi, K. Huntington’s Disease and Mitochondria. Neurotox. Res. 2017, 32, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Vidoni, C.; Castiglioni, A.; Seca, C.; Secomandi, E.; Melone, M.A.; Isidoro, C. Dopamine Exacerbates Mutant Huntingtin Toxicity via Oxidative-Mediated Inhibition of Autophagy in SH-SY5Y Neuroblastoma Cells: Beneficial Effects of Anti-Oxidant Therapeutics. Neurochem. Int. 2016, 101, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Lalic, N.M.; Maric, J.; Svetel, M.; Jotic, A.; Stefanova, E.; Lalic, K.; Dragasevic, N.; Milicic, T.; Lukic, L.; Kostic, V.S. Glucose Homeostasis in Huntington Disease: Abnormalities in Insulin Sensitivity and Early-Phase Insulin Secretion. Arch. Neurol. 2008, 65, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Aziz, N.A.; Pijl, H.; Frolich, M.; Snel, M.; Streefland, T.C.; Roelfsema, F.; Roos, R.A. Systemic Energy Homeostasis in Huntington’s Disease Patients. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1233–1237. [Google Scholar] [CrossRef] [PubMed]
- Montojo, M.T.; Aganzo, M.; Gonzalez, N. Huntington’s Disease and Diabetes: Chronological Sequence of Its Association. J. Huntingt. Dis. 2017, 6, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Stayte, S.; Vissel, B. New Hope for Devastating Neurodegenerative Disease. Brain 2017, 140, 1177–1179. [Google Scholar] [CrossRef] [PubMed]
- Pomytkin, I.; Costa-Nunes, J.P.; Kasatkin, V.; Veniaminova, E.; Demchenko, A.; Lyundup, A.; Lesch, K.P.; Ponomarev, E.D.; Strekalova, T. Insulin Receptor in the Brain: Mechanisms of Activation and the Role in the CNS Pathology and Treatment. CNS Neurosci. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Cremona, M.L.; Rothman, J.E. Autophagy-Mediated Clearance of Huntingtin Aggregates Triggered by the Insulin-Signaling Pathway. J. Cell Biol. 2006, 172, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Naia, L.; Ribeiro, M.; Rodrigues, J.; Duarte, A.I.; Lopes, C.; Rosenstock, T.R.; Hayden, M.R.; Rego, A.C. Insulin and IGF-1 Regularize Energy Metabolites in Neural Cells Expressing Full-Length Mutant Huntingtin. Neuropeptides 2016, 58, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira, C.R.; Rego, A.C. Insulin and IGF-1 Improve Mitochondrial Function in a PI-3K/Akt-Dependent Manner and Reduce Mitochondrial Generation of Reactive Oxygen Species in Huntington’s Disease Knock-in Striatal Cells. Free Radic. Biol. Med. 2014, 74, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Kornelius, E.; Lin, C.L.; Chang, H.H.; Li, H.H.; Huang, W.N.; Yang, Y.S.; Lu, Y.L.; Peng, C.H.; Huang, C.N. DPP-4 Inhibitor Linagliptin Attenuates Abeta-Induced Cytotoxicity through Activation of AMPK in Neuronal Cells. CNS Neurosci. Ther. 2015, 21, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.; Golden, E.; Carlson, O.D.; Pistell, P.; Zhou, J.; Kim, W.; Frank, B.P.; Thomas, S.; Chadwick, W.A.; Greig, N.H.; et al. Exendin-4 Improves Glycemic Control, Ameliorates Brain and Pancreatic Pathologies, and Extends Survival in a Mouse Model of Huntington’s Disease. Diabetes 2009, 58, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Boura-Halfon, S.; Zick, Y. Phosphorylation of IRS Proteins, Insulin Action, and Insulin Resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E581–E591. [Google Scholar] [CrossRef] [PubMed]
- Janssens, J.; Etienne, H.; Idriss, S.; Azmi, A.; Martin, B.; Maudsley, S. Systems-Level G Protein-Coupled Receptor Therapy across a Neurodegenerative Continuum by the GLP-1 Receptor System. Front. Endocrinol. (Lausanne) 2014, 5, 142. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Wang, S.E.; Hsu, C.H.; Sheu, S.J.; Wu, C.H. Oral Treatment with Herbal Formula B307 Alleviates Cardiac Failure in Aging R6/2 Mice with Huntington’s Disease Via Suppressing Oxidative Stress, Inflammation, and Apoptosis. Clin. Interv. Aging 2015, 10, 1173–1187. [Google Scholar] [PubMed]
- Ehrnhoefer, D.E.; Martin, D.D.O.; Schmidt, M.E.; Qiu, X.; Ladha, S.; Caron, N.S.; Skotte, N.H.; Nguyen, Y.T.N.; Vaid, K.; Southwell, A.L.; et al. Preventing Mutant Huntingtin Proteolysis and Intermittent Fasting Promote Autophagy in Models of Huntington Disease. Acta Neuropathol. Commun. 2018, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Degradation of Misfolded Proteins in Neurodegenerative Diseases: Therapeutic Targets and Strategies. Exp. Mol. Med. 2015, 47, E147. [Google Scholar] [CrossRef] [PubMed]
- Scotter, E.L.; Vance, C.; Nishimura, A.L.; Lee, Y.B.; Chen, H.J.; Urwin, H.; Sardone, V.; Mitchell, J.C.; Rogelj, B.; Rubinsztein, D.C.; et al. Differential Roles of the Ubiquitin Proteasome System and Autophagy in the Clearance of Soluble and Aggregated TDP-43 Species. J. Cell Sci. 2014, 127, 1263–1278. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.H.; Wang, Y.; Kegel, K.B.; Kazantsev, A.; Apostol, B.L.; Thompson, L.M.; Yoder, J.; Aronin, N.; DiFiglia, M. Autophagy Regulates the Processing of Amino Terminal Huntingtin Fragments. Hum. Mol. Genet. 2003, 12, 3231–3244. [Google Scholar] [CrossRef] [PubMed]
- Kvam, E.; Nannenga, B.L.; Wang, M.S.; Jia, Z.; Sierks, M.R.; Messer, A. Conformational Targeting of Fibrillar Polyglutamine Proteins in Live Cells Escalates Aggregation and Cytotoxicity. PLoS ONE 2009, 4, E5727. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A. Diabetes Mellitus in Huntington Disease. Clin. Genet. 1985, 27, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Nah, J.; Yuan, J.; Jung, Y.K. Autophagy in Neurodegenerative Diseases: From Mechanism to Therapeutic Approach. Mol. Cells 2015, 38, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korolenko, T.A.; Pupyshev, A.B.; Pospelova, T.I.; Gritsyk, O.B.; Panov, A.V.; Vavilin, V.A.; Shintyapina, A.B.; Tikhonova, M.A. Autophagy and Neurodegeneration; Nova Science Publishers: Hauppauge, NY, USA, 2017; pp. 1–34. ISBN 978-1-53612-260-2. [Google Scholar]
- Abd-Elrahman, K.S.; Hamilton, A.; Hutchinson, S.R.; Liu, F.; Russell, R.C.; Ferguson, S.S.G. mGluR5 Antagonism Increases Autophagy and Prevents Disease Progression in the zQ175 Mouse Model of Huntington’s Disease. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.S. The Role of Mammalian Target of Rapamycin (mTOR) in Insulin Signaling. Nutrients 2017, 9, 1176. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A Pharmacologic Target for Autophagy Regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Moloudizargari, M.; Asghari, M.H.; Ghobadi, E.; Fallah, M.; Rasouli, S.; Abdollahi, M. Autophagy, Its Mechanisms and Regulation: Implications in Neurodegenerative Diseases. Ageing Res. Rev. 2017, 40, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Panagaki, T.; Michael, M.; Holscher, C. Liraglutide Restores Chronic ER Stress, Autophagy Impairments and Apoptotic Signalling in SH-SY5Y Cells. Sci. Rep. 2017, 7, 16158. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.I.; Sjogren, M.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I.; Bjorkqvist, M. Dual Therapy with Liraglutide and Ghrelin Promotes Brain and Peripheral Energy Metabolism in the R6/2 Mouse Model of Huntington’s Disease. Sci. Rep. 2018, 8, 8961. [Google Scholar] [CrossRef] [PubMed]
- Weids, A.J.; Ibstedt, S.; Tamas, M.J.; Grant, C.M. Distinct Stress Conditions Result in Aggregation of Proteins with Similar Properties. Sci. Rep. 2016, 6, 24554. [Google Scholar] [CrossRef] [PubMed]
- Chartoumpekis, D.V.; Yagishita, Y.; Fazzari, M.; Palliyaguru, D.L.; Rao, U.N.; Zaravinos, A.; Khoo, N.K.; Schopfer, F.J.; Weiss, K.R.; Michalopoulos, G.K.; et al. Nrf2 Prevents Notch-Induced Insulin Resistance and Tumorigenesis in Mice. JCI Insight 2018, 3, 97735. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhao, Z.; Ke, L.; Li, Z.; Li, W.; Zhang, Z.; Zhou, Y.; Feng, X.; Zhu, W. Resveratrol Improves Glucose Uptake in Insulin-Resistant Adipocytes via Sirt1. J. Nutr. Biochem. 2018, 55, 209–218. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.-C.; Lin, T.-C.; Ho, H.-L.; Kuo, C.-Y.; Li, H.-H.; Korolenko, T.A.; Chen, W.-J.; Lai, T.-J.; Ho, Y.-J.; Lin, C.-L. GLP-1 Analogue Liraglutide Attenuates Mutant Huntingtin-Induced Neurotoxicity by Restoration of Neuronal Insulin Signaling. Int. J. Mol. Sci. 2018, 19, 2505. https://doi.org/10.3390/ijms19092505
Chang C-C, Lin T-C, Ho H-L, Kuo C-Y, Li H-H, Korolenko TA, Chen W-J, Lai T-J, Ho Y-J, Lin C-L. GLP-1 Analogue Liraglutide Attenuates Mutant Huntingtin-Induced Neurotoxicity by Restoration of Neuronal Insulin Signaling. International Journal of Molecular Sciences. 2018; 19(9):2505. https://doi.org/10.3390/ijms19092505
Chicago/Turabian StyleChang, Ching-Chi, Tzu-Chin Lin, Hsiao-Li Ho, Chien-Yin Kuo, Hsin-Hua Li, Tatiana A. Korolenko, Wei-Jen Chen, Te-Jen Lai, Ying-Jui Ho, and Chih-Li Lin. 2018. "GLP-1 Analogue Liraglutide Attenuates Mutant Huntingtin-Induced Neurotoxicity by Restoration of Neuronal Insulin Signaling" International Journal of Molecular Sciences 19, no. 9: 2505. https://doi.org/10.3390/ijms19092505
APA StyleChang, C. -C., Lin, T. -C., Ho, H. -L., Kuo, C. -Y., Li, H. -H., Korolenko, T. A., Chen, W. -J., Lai, T. -J., Ho, Y. -J., & Lin, C. -L. (2018). GLP-1 Analogue Liraglutide Attenuates Mutant Huntingtin-Induced Neurotoxicity by Restoration of Neuronal Insulin Signaling. International Journal of Molecular Sciences, 19(9), 2505. https://doi.org/10.3390/ijms19092505