Role of Nitric Oxide in the Cardiovascular and Renal Systems

 ,
,
Abstract
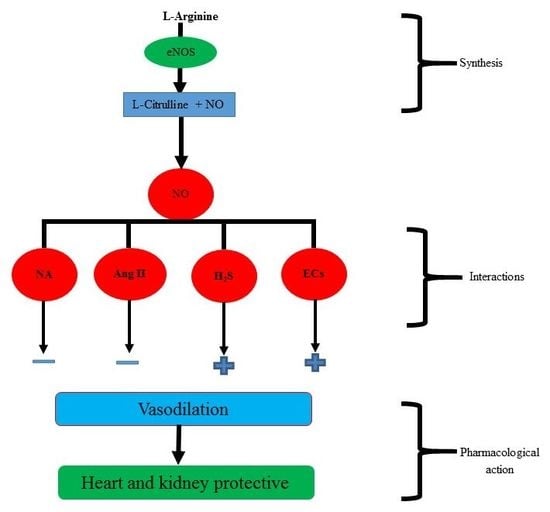
:
{kind=link}
{kind=link}
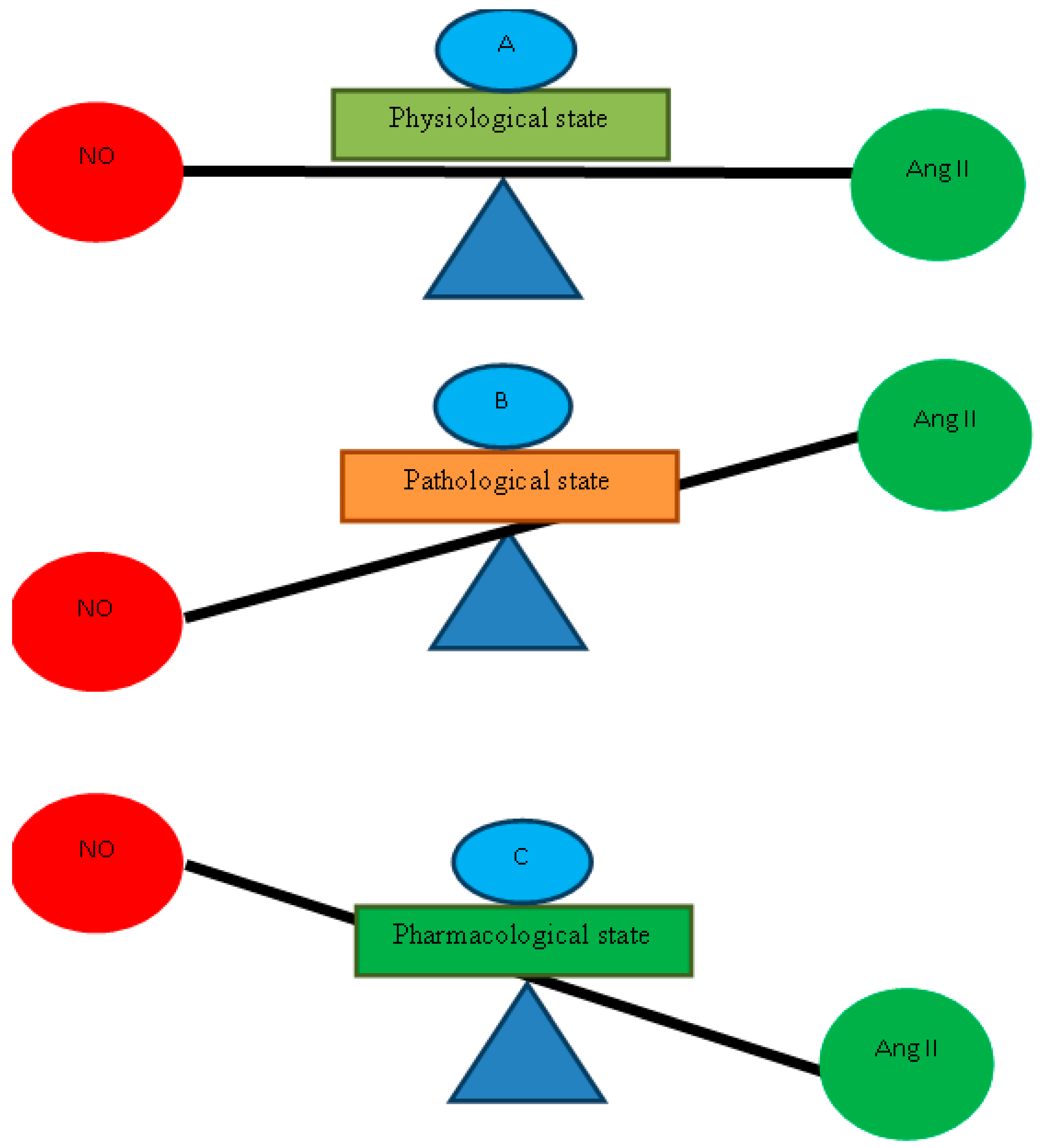{kind=link}
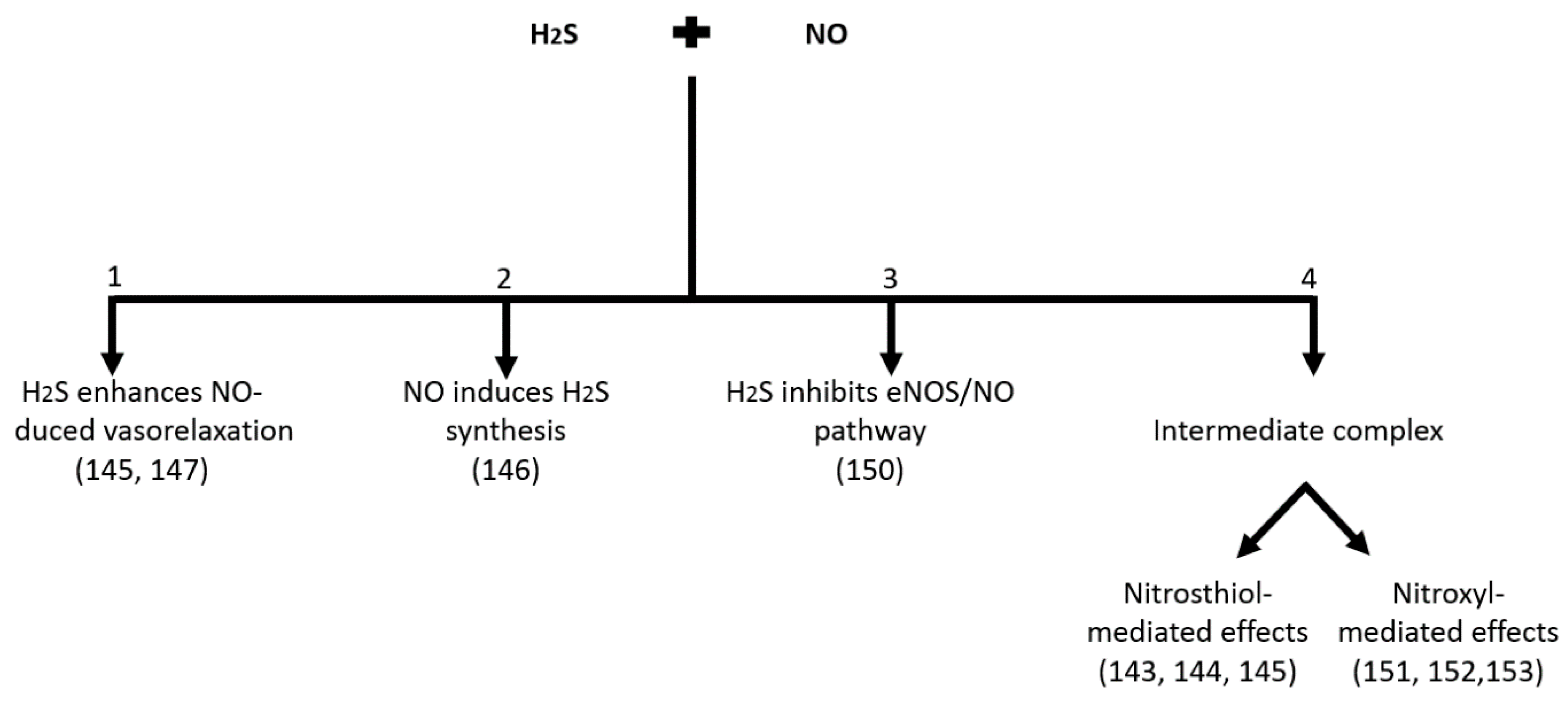{kind=link}
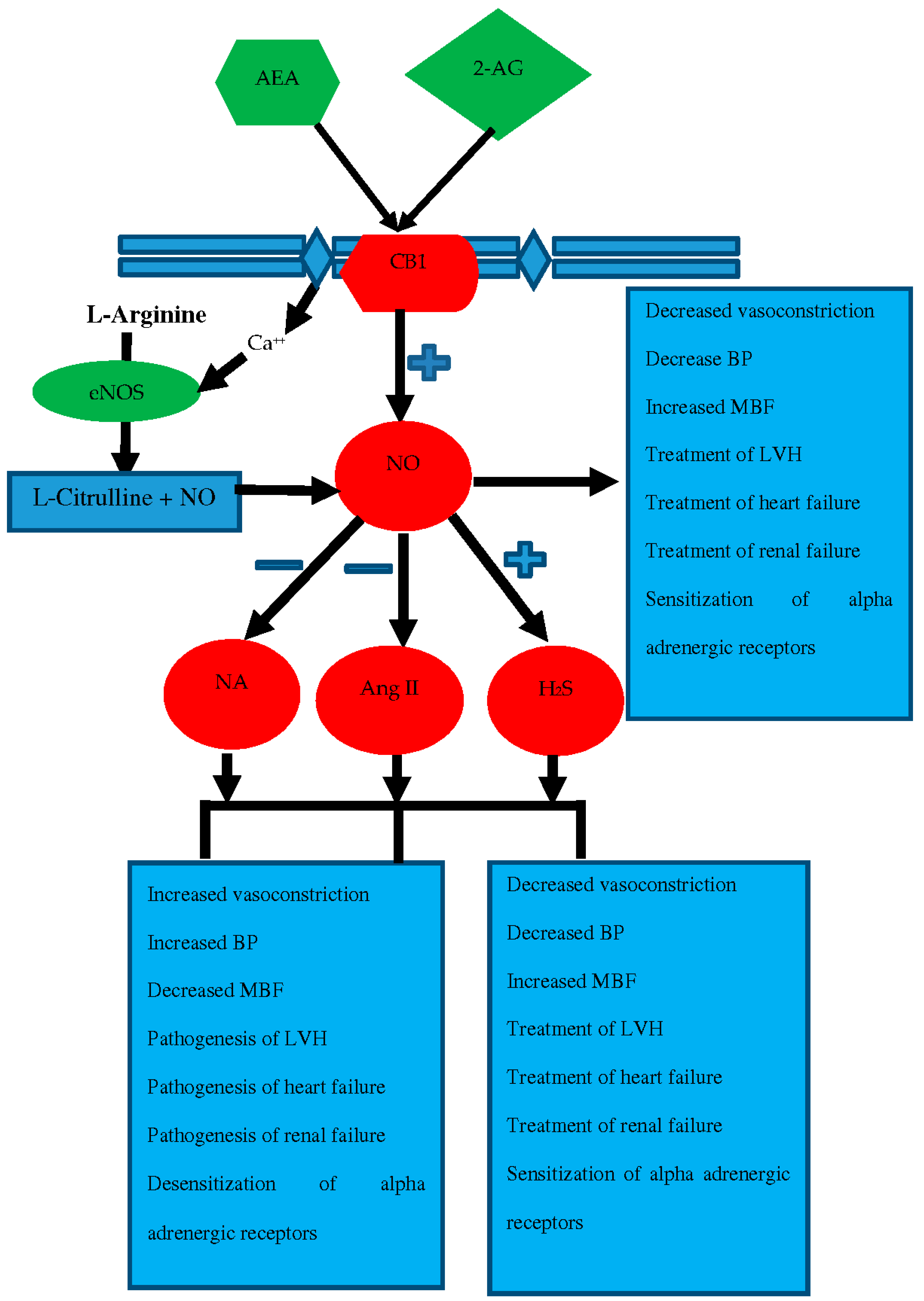{kind=link}
1. Nitric Oxide Background and Production
2. Role of Nitric Oxide in Hypertension
3. Role of Nitric Oxide in Left Ventricular Hypertrophy (LVH)
Alteration of Expression of Endothelial Nitric Oxide Synthase (eNOS) in LVH
4. Roles of Nitric Oxide in the Kidney
5. Interaction with Alpha Adrenergic Receptors of the Kidney
Renal α1-Adrenergic Receptors Subtypes
6. Interaction of Nitric Oxide with Other Systems
6.1. Nitric Oxide with Angiotensin II
6.2. Nitric Oxide with Gaseous Transmitters Like Hydrogen Sulfide
6.3. Nitric Oxide with Noradrenaline
6.4. Nitric Oxide with Endocannabinoids
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NO | Nitric oxide |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| EDRF | Endothelial derived vasorelaxant factor |
| ECs | Endocannabinoid system |
| eNOS | Endothelial nitric oxide synthase |
| nNOS | Neuronal nitric oxide synthase |
| iNOS | Inducible nitric oxide synthase |
| cGMP | Cyclic 3′,5′-guanosine monophosphate |
| GC | Guanylate cyclase |
| l-NMMA | NG-monomethyl-l-arginine |
| l-NAME | Nw-nitro-l-arginine methyl ester |
| sGC | Soluble guanylyl cyclase |
| PDE | phosphodiesterase |
| SHR | Spontaneously hypertensive rats |
| LVH | Left ventricular hypertrophy |
| TAC | Transverse aortic constriction |
| ADMA | NG,NG-dimethylarginine |
| PCT | Proximal convoluted tubules |
| LPS | Lipopolysaccharide |
| H2S | Hydrogen sulfide |
| BH4 | Tetrahydrobiopterin |
| NE | Norepinephrine |
| NA | Noradrenaline |
| SNP | Sodium nitroprusside |
| AEA | Arachidonoyl ethanolamine (Anandamide) |
| 2-AG | 2-arachidonoylglycerol |
| FAAH | Fatty acid amide hydrolase |
| MAGL | Monoacylglycerol lipase |
| CB1 | Cannabinoid receptor type 1 |
| CB2 | Cannabinoid receptor type 2 |
| DAG | Diacylglycerol lipase |
| THC | Tetrahydrocannabinol |
References
- Llorens, S.; Jordan, J.; Nava, E. The nitric oxide pathway in the cardiovascular system. J. Physiol. Biochem. 2002, 58, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.R.; Emanuel, K.; Sears, C.E.; Zhang, Y.H.; Casadei, B. Are myocardial eNOS and nNOS involved in the beta-adrenergic and muscarinic regulation of inotropy? A systematic investigation. Cardiovasc. Res. 2006, 70, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Sears, C.E.; Ashley, E.A.; Casadei, B. Nitric oxide control of cardiac function: Is neuronal nitric oxide synthase a key component? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 1021–1044. [Google Scholar] [CrossRef] [PubMed]
- Massion, P.B.; Feron, O.; Dessy, C.; Balligand, J.L. Nitric oxide and cardiac function: Ten years after, and continuing. Circ. Res. 2003, 93, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Casadei, B.; Sears, C.E. Nitric-oxide-mediated regulation of cardiac contractility and stretch responses. Prog. Biophys. Mol. Biol. 2003, 82, 67–80. [Google Scholar] [CrossRef]
- Haynes, W.G.; Noon, J.P.; Walker, B.R.; Webb, D.J. Inhibition of nitric oxide synthesis increases blood pressure in healthy humans. J. Hypertens. 1993, 11, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Lepori, M.; Sartori, C.; Trueb, L.; Owlya, R.; Nicod, P.; Scherrer, U. Haemodynamic and sympathetic effects of inhibition of nitric oxide synthase by systemic infusion of NG-monomethyl-L-arginine into humans are dose dependent. J. Hypertens. 1998, 16, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Vallance, P.; Collier, J.; Moncada, S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet 1989, 334, 997–1000. [Google Scholar] [CrossRef]
- Pucci, M.; Lin, L.; Nasjletti, A. Pressor and renal vasoconstrictor effects of NG-nitro-L-arginine as affected by blockade of pressor mechanisms mediated by the sympathetic nervous system, angiotensin, prostanoids and vasopressin. J. Pharmacol. Exp. Ther. 1992, 261, 240–245. [Google Scholar] [PubMed]
- Ahmad, A.; Sattar, M.A.; Azam, M.; Khan, S.A.; Bhatt, O.; Johns, E.J. Interaction between nitric oxide and renal α(1)-adrenoreceptors mediated vasoconstriction in rats with left ventricular hypertrophyin Wistar Kyoto rats. PLoS ONE 2018, 13, e0189386. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Alkaitis, M.S.; Crabtree, M.J. Recoupling the cardiac nitric oxide synthases: Tetrahydrobiopterin synthesis and recycling. Curr. Heart Fail. Rep. 2012, 9, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Wang-Rosenke, Y.; Neumayer, H.H.; Peters, H. NO signaling through cGMP in renal tissue fibrosis and beyond: Key pathway and novel therapeutic target. Curr. Med. Chem. 2008, 15, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, W.K.; Beavo, J.A. Cyclic GMP and regulation of cyclic nucleotide hydrolysis. Adv. Pharmacol. 1994, 26, 87–114. [Google Scholar] [PubMed]
- Santillo, M.F.; Mapa, M.S.T. Phosphodiesterase (PDE5) inhibition assay for rapid detection of erectile dysfunction drugs and analogs in sexual enhancement products. Drug Test. Anal. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ko, F.N.; Wu, C.C.; Kuo, S.C.; Lee, F.Y.; Teng, C.M. YC-1, a novel activator of platelet guanylate cyclase. Blood 1994, 84, 4226–4233. [Google Scholar] [PubMed]
- Friebe, A.; Koesling, D. Mechanism of YC-1-induced activation of soluble guanylyl cyclase. Mol. Pharmacol. 1998, 53, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Stasch, J.P.; Becker, E.M.; Alonso-Alija, C.; Apeler, H.; Dembowsky, K.; Feurer, A.; Gerzer, R.; Minuth, T.; Perzborn, E.; Pleiss, U.; et al. NO-independent regulatory site on soluble guanylate cyclase. Nature 2001, 410, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Stasch, J.P.; Schmidt, P.; Alonso-Alija, C.; Apeler, H.; Dembowsky, K.; Haerter, M.; Heil, M.; Minuth, T.; Perzborn, E.; Pleiss, U.; et al. NO- and haem-independent activation of soluble guanylyl cyclase: Molecular basis and cardiovascular implications of a new pharmacological principle. Br. J. Pharmacol. 2002, 136, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, C.; Boudier, A.; Dahboul, F.; Parent, M.; Leroy, P. S-nitrosation/denitrosation in cardiovascular pathologies: Facts and concepts for the rational design of S-nitrosothiols. Curr. Pharm. Des. 2013, 19, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Wynia-Smith, S.L.; Smith, B.C. Nitrosothiol formation and S-nitrosation signaling through nitric oxide synthases. Nitric Oxide 2017, 63, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Gokce, N. L-arginine and hypertension. J. Nutr. 2004, 134, 2807S–2811S. [Google Scholar] [CrossRef] [PubMed]
- Grandvuillemin, I.; Buffat, C.; Boubred, F.; Lamy, E.; Fromonot, J.; Charpiot, P.; Simoncini, S.; Sabatier, F.; Dignat-George, F.; Peyter, A.C.; et al. Arginase up-regulation and eNOS uncoupling contribute to impaired endothelium-dependent vasodilation in a rat model of intrauterine growth restriction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Node, K.; Kitakaze, M.; Yoshikawa, H.; Kosaka, H.; Hori, M. Reduced plasma concentrations of nitrogen oxide in individuals with essential hypertension. Hypertension 1997, 30, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Panza, J.A.; Quyyumi, A.A.; Brushm, J.E., Jr.; Epstein, S.E. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N. Engl. J. Med. 1990, 323, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Sasaki, S.; Kurisu, S.; Yoshimizu, A.; Sasaki, N.; Matsuura, H.; Kajiyama, G.; Oshima, T. Regular aerobic exercise augments endothelium-dependent vascular relaxation in normotensive as well as hypertensive subjects role of endothelium-derived nitric oxide. Circulation 1999, 100, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Podjarny, E.; Hasdan, G.; Bernheim, J.; Rashid, G.; Green, J.; Korzets, Z.; Bernheim, J. Effect of chronic tetrahydrobiopterin supplementation on blood pressure and proteinuria in 5/6 nephrectomized rats. Nephrol. Dial. Transplant. 2004, 19, 2223–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinozaki, K.; Nishio, Y.; Okamura, T.; Yoshida, Y.; Maegawa, H.; Kojima, H.; Masada, M.; Toda, N.; Kikkawa, R.; Kashiwagi, A. Oral administration of tetrahydrobiopterin prevents endothelial dysfunction and vascular oxidative stress in the aortas of insulin-resistant rats. Circ. Res. 2000, 87, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L.; Huang, Z.; Mashimo, H.; Bloch, K.D.; Moskowitz, M.A.; Bevan, J.A.; Fishman, M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995, 377, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Calver, A.; Collier, J.; Moncada, S.; Vallance, P. Effect of local intra-arterial NG-monomethyl-L-arginine in patients with hypertension: The nitric oxide dilator mechanism appears abnormal. J. Hypertens. 1992, 10, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Lahera, V.; Salom, M.G.; Miranda-Guardiola, F.; Moncada, S.; Romero, J.C. Effects of NG-nitro-L-arginine methyl ester on renal function and blood pressure. Am. J. Physiol. 1991, 261, F1033–F1037. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Virdis, A.; Mattei, P.; Ghiadoni, L.; Sudano, I.; Salvetti, A. Defective L-arginine–nitric oxide pathway in offspring of essential hypertensive patients. Circulation 1996, 94, 1298–1303. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Sanders, P.W. L-arginine abrogates salt-sensitive hypertension in Dahl/Rapp rats. J. Clin. Investig. 1991, 88, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Palloshi, A.; Fragasso, G.; Piatti, P.; Monti, L.D.; Setola, E.; Valsecchi, G.; Galluccio, E.; Chierchia, S.L.; Margonato, A. Effect of oral l-arginine on blood pressure and symptoms and endothelial function in patients with systemic hypertension, positive exercise tests, and normal coronary arteries. Am. J. Cardiol. 2004, 93, 933–935. [Google Scholar] [CrossRef] [PubMed]
- Gokce, N.; Holbrook, M.; Duffy, S.J.; Demissie, S.; Cupples, L.A.; Biegelsen, E.; Keaney, J.F.; Loscalzo, J.; Vita, J.A. Effects of race and hypertension on flow-mediated and nitroglycerin-mediated dilation of the brachial artery. Hypertension 2001, 38, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.L. The effects of sustained-release-L-arginine formulation on blood pressure and vascular compliance in 29 healthy individuals. Altern. Med. Rev. 2006, 11, 23–29. [Google Scholar] [PubMed]
- Higashi, Y.; Oshima, T.; Ozono, R.; Watanabe, M.; Matsuura, H.; Kajiyama, G. Effects of L-arginine infusion on renal hemodynamics in patients with mild essential hypertension. Hypertension 1995, 25, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Oshima, T.; Ono, N.; Hiraga, H.; Yoshimura, M.; Watanabe, M.; Matsuura, H.; Kambe, M.; Kajiyama, G. Intravenous administration of L-arginine inhibits angiotensin-converting enzyme in humans. J. Clin. Endocrinol. Metab. 1995, 80, 2198–2202. [Google Scholar] [PubMed]
- Zanfolin, M.; Faro, R.; Araujo, E.G.; Guaraldo, A.M.; Antunes, E.; De Nucci, G. Protective effects of BAY 41-2272 (sGC stimulator) on hypertension, heart, and cardiomyocyte hypertrophy induced by chronic L-NAME treatment in rats. J. Cardiovasc. Pharmacol. 2006, 47, 391–395. [Google Scholar] [PubMed]
- Hayakawa, H.; Raij, L. The Link Among Nitric Oxide Synthase Activity, Endothelial Function, and Aortic and Ventricular Hypertrophy in Hypertension. Hypertension 1997, 29, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzoni, D.; Muiesan, M.L.; Porteri, E.; Castellano, M.; Zulli, R.; Bettoni, G.; Salvetti, M.; Monteduro, C.; Agabiti-Rosei, E. Effects of long-term antihypertensive treatment with lisinopril on resistance arteries in hypertensive patients with left ventricular hypertrophy. J. Hypertens. 1997, 15, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Bernatova, I.; Pechanova, O.; Simko, F. Captopril prevents NO-deficient hypertension and left ventricular hypertrophy without affecting nitric oxide synthase activity in rats. Physiol. Res. Acad. Sci. Bohemoslov. 1995, 45, 311–316. [Google Scholar]
- Simko, F.; Simko, J. The potential role of nitric oxide in the hypertrophic growth of the left ventricle. Physiol. Res. 2000, 49, 37–46. [Google Scholar] [PubMed]
- Paulis, L.; Matuskova, J.; Adamcova, M.; Pelouch, V.; Simko, J.; Krajcirovicova, K.; Potacova, A.; Hulin, I.; Janega, P.; Pechanova, O. Regression of left ventricular hypertrophy and aortic remodelling in NO-deficient hypertensive rats: Effect of l-arginine and spironolactone. Acta Physiol. 2008, 194, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Crabos, M.; Coste, P.; Paccalin, M.; Tariosse, L.; Daret, D.; Besse, P.; Bonoron-Adèle, S. Reduced basal NO-mediated dilation and decreased endothelial NO-synthase expression in coronary vessels of spontaneously hypertensive rats. J. Mol. Cell. Cardiol. 1997, 29, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, H.; Nakata, M.; Kohno, K.; Koga, Y.; Nomura, G.; Toshima, H.; Imaizumi, T. Chronic L-Arginine Administration Attenuates Cardiac Hypertrophy in Spontaneously Hypertensive Rats. Hypertension 1996, 27, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Kristek, F. Long-term administration of L-arginine did not influence blood pressure, heart rate, cardiac hypertrophy or arterial wall thickness of spontaneously hypertensive rats. Exp. Physiol. 1998, 83, 595–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stier, C.T., Jr.; Sim, G.J.; Levine, S. Dietary arginine fails to protect against cerebrovascular damage in stroke-prone hypertensive rats. Brain Res. 1991, 549, 354–356. [Google Scholar] [CrossRef]
- Siani, A.; Pagano, E.; Iacone, R.; Iacoviello, L.; Scopacasa, F.; Strazzullo, P. Blood pressure and metabolic changes during dietary L-arginine supplementation in humans. Am. J. Hypertens. 2000, 13, 547–551. [Google Scholar] [CrossRef] [Green Version]
- Miyata, N.; Cowley, A.W. Renal intramedullary infusion of L-arginine prevents reduction of medullary blood flow and hypertension in Dahl salt-sensitive rats. Hypertension 1999, 33, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Granger, J.P.; Kirchner, K.A. L-arginine improves transmission of perfusion pressure to the renal interstitium in Dahl salt-sensitive rats. Am. J. Physiol. 1994, 266, R1730–R1735. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, M.; Kawashima, S.; Yamashita, T.; Hirase, T.; Ohashi, Y.; Inoue, N.; Hirata, K.-I.; Yokoyama, M. Overexpression of endothelial nitric oxide synthase attenuates cardiac hypertrophy induced by chronic isoproterenol infusion. Circ. J. 2002, 66, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Barouch, L.A.; Cappola, T.P.; Harrison, R.W.; Crone, J.K.; Rodriguez, E.R.; Burnett, A.L.; Hare, J.M. Combined loss of neuronal and endothelial nitric oxide synthase causes premature mortality and age-related hypertrophic cardiac remodeling in mice. J. Mol. Cell. Cardiol. 2003, 35, 637–644. [Google Scholar] [CrossRef]
- Raij, L. Nitric Oxide in Hypertension: Relationship With Renal Injury and Left Ventricular Hypertrophy. Hypertension 1998, 31, 189–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, L.; Li, C.; Xia, D.; Qu, P.; Li, Z.; Zhang, W.; Feng, X. Relationship between hypertensive left ventricular hypertrophy and levels of endothelin and nitric oxide. Hypertens. Res. 2000, 23, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Sattar, M.A.; Rathore, H.A.; Abdulla, M.H.; Khan, S.A.; Abdullah, N.A.; Johns, E.J. Enhanced expression of endothelial nitric oxide synthase in the myocardium ameliorates the progression of left ventricular hypertrophy in L-arginine treated Wistar-Kyoto rats. J. Physiol. Pharmacol. 2016, 67, 31–44. [Google Scholar] [PubMed]
- Barouch, L.A.; Harrison, R.W.; Skaf, M.W.; Rosas, G.O.; Cappola, T.P.; Kobeissi, Z.A.; Hobai, I.A.; Lemmon, C.A.; Burnett, A.L.; O’Rourke, B. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature 2002, 416, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Kazakov, A.; Müller, P.; Jagoda, P.; Semenov, A.; Böhm, M.; Laufs, U. Endothelial nitric oxide synthase of the bone marrow regulates myocardial hypertrophy, fibrosis, and angiogenesis. Cardiovasc. Res. 2012, 93, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Buys, E.S.; Raher, M.J.; Blake, S.L.; Neilan, T.G.; Graveline, A.R.; Passeri, J.J.; Llano, M.; Perez-Sanz, T.M.; Ichinose, F.; Janssens, S.; et al. Cardiomyocyte-restricted restoration of nitric oxide synthase 3 attenuates left ventricular remodeling after chronic pressure overload. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H620–H627. [Google Scholar] [CrossRef] [PubMed]
- Scherrer-Crosbie, M.; Ullrich, R.; Bloch, K.D.; Nakajima, H.; Nasseri, B.; Aretz, H.T.; Lindsey, M.L.; Vançon, A.C.; Huang, P.L.; Lee, R.T.; et al. Endothelial Nitric Oxide Synthase Limits Left Ventricular Remodeling After Myocardial Infarction in Mice. Circulation 2001, 104, 1286–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichinose, F.; Bloch, K.D.; Wu, J.C.; Hataishi, R.; Aretz, H.T.; Picard, M.H.; Scherrer-Crosbie, M. Pressure overload-induced LV hypertrophy and dysfunction in mice are exacerbated by congenital NOS3 deficiency. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1070–H1075. [Google Scholar] [CrossRef] [PubMed]
- Ruetten, H.; Dimmeler, S.; Gehring, D.; Ihling, C.; Zeiher, A.M. Concentric left ventricular remodeling in endothelial nitric oxide synthase knockout mice by chronic pressure overload. Cardiovasc. Res. 2005, 66, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takimoto, E.; Champion, H.C.; Li, M.; Ren, S.; Rodriguez, E.R.; Tavazzi, B.; Lazzarino, G.; Paolocci, N.; Gabrielson, K.L.; Wang, Y. Oxidant stress from nitric oxide synthase–3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J. Clin. Investig. 2005, 115, 1221–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.Y.; Wauquier, F.; Eid, A.A.; Roman, L.J.; Ghosh-Choudhury, G.; Khazim, K.; Block, K.; Gorin, Y. Nox4 NADPH oxidase mediates peroxynitrite-dependent uncoupling of endothelial nitric-oxide synthase and fibronectin expression in response to angiotensin II: Role of mitochondrial reactive oxygen species. J. Biol. Chem. 2013, 288, 28668–28686. [Google Scholar] [CrossRef] [PubMed]
- Galougahi, K.K.; Liu, C.C.; Gentile, C.; Kok, C.; Nunez, A.; Garcia, A.; Fry, N.A.; Davies, M.J.; Hawkins, C.L.; Rasmussen, H.H.; et al. Glutathionylation mediates angiotensin II-induced eNOS uncoupling, amplifying NADPH oxidase-dependent endothelial dysfunction. J. Am. Heart Assoc. 2014, 3, e000731. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Chen, J.; Seshan, S.V.; Khodadadian, J.J.; Gallagher, R.; El Chaar, M.; Vaughan, E.D., Jr.; Poppas, D.P.; Felsen, D. Dietary arginine supplementation attenuates renal damage after relief of unilateral ureteral obstruction in rats. Kidney Int. 2005, 68, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Reis, F.; Rocha-Pereira, P.; Teixeira de Lemos, E.; Parada, B.; Baptista, S.; Figueiredo, A.; Santos-Silva, A.; Costa-Almeida, C.; Mota, A.; Teixeira, F. Oxidative stress in cyclosporine-induced hypertension: Evidence of beneficial effects or tolerance development with nitrate therapy. Transplant. Proc. 2007, 39, 2494–2500. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Easton, L.K.; Booth, L.C.; Schlaich, M.P.; Head, G.A.; Moritz, K.M.; Denton, K.M. Renal Nitric Oxide Deficiency and Chronic Kidney Disease in Young Sheep Born with a Solitary Functioning Kidney. Sci. Rep. 2016, 6, 26777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lankadeva, Y.R.; Singh, R.R.; Moritz, K.M.; Parkington, H.C.; Denton, K.M.; Tare, M. Renal dysfunction is associated with a reduced contribution of nitric oxide and enhanced vasoconstriction after a congenital renal mass reduction in sheep. Circulation 2015, 131, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Abd El Motteleb, D.M.; Elshazly, S.M. Renoprotective effect of sitagliptin against hypertensive nephropathy induced by chronic administration of L-NAME in rats: Role of GLP-1 and GLP-1 receptor. Eur. J. Pharmacol. 2013, 720, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Leone, A.; Moncada, S.; Vallance, P.; Calver, A.; Collier, J. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992, 339, 572–575. [Google Scholar] [CrossRef]
- Reyes, A.A.; Purkerson, M.L.; Karl, I.; Klahr, S. Dietary Supplementation With l-Arginine Ameliorates the Progression of Renal Disease in Rats With Subtotal Nephrectomy. Am. J. Kidney Dis. 1992, 20, 168–176. [Google Scholar] [CrossRef]
- Sakuma, I.; Togashi, H.; Yoshioka, M.; Saito, H.; Yanagida, M.; Tamura, M.; Kobayashi, T.; Yasuda, H.; Gross, S.S.; Levi, R. NG-methyl-L-arginine, an inhibitor of L-arginine-derived nitric oxide synthesis, stimulates renal sympathetic nerve activity in vivo. A role for nitric oxide in the central regulation of sympathetic tone? Circ. Res. 1992, 70, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Ashab, I.; Peer, G.; Blum, M.; Wollman, Y.; Chernihovsky, T.; Hassner, A.; Schwartz, D.; Cabili, S.; Silverberg, D.; Iaina, A. Oral administration of L-arginine and captopril in rats prevents chronic renal failure by nitric oxide production. Kidney Int. 1995, 47, 1515–1521. [Google Scholar] [CrossRef] [PubMed]
- Chintala, M.; Chin, P.S.; Vemulapalli, S.; Watkins, R.; Sybertz, E. Inhibition of endothelial derived relaxing factor (EDRF) aggravates ischemic acute renal failure in anesthetized rats. Naunyn-Schmiedebergs Arch. Pharmacol. 1993, 348, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Goor, Y.; Peer, G.; Iaina, A.; Blum, M.; Wollman, Y.; Chernihovsky, T.; Silverberg, D.; Cabili, S. Nitric oxide in ischaemic acute renal failure of streptozotocin diabetic rats. Diabetologia 1996, 39, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Knox, F.G. Production and functional roles of nitric oxide in the proximal tubule. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, R1117–R1124. [Google Scholar] [CrossRef] [PubMed]
- Hye Khan, M.A.; Sattar, M.A.; Abdullah, N.A.; Johns, E.J. Influence of combined hypertension and renal failure on functional α(1)-adrenoceptor subtypes in the rat kidney. Br. J. Pharmacol. 2008, 153, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Abdulla, M.H.; Sattar, M.A.; Johns, E.J.; Abdullah, N.A.; Hye Khan, M.A.; Rathore, H.A. High-fructose feeding impacts on the adrenergic control of renal haemodynamics in the rat. Br. J. Nutr. 2012, 107, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Sattar, M.A.; Abdullah, N.A.; Abdulla, M.H.; Salman, I.M.; Kazi, R.N.; Swarup, K.R.L.A.; Rathore, H.A.; Basri, F.; Hussain, N.M.; et al. Functional Subtypes of Renal α-Adrenoceptor in Spontaneously Hypertensive Rats with Streptozotocin-Induced Experimental Diabetic Nephropathy. Kidney Blood Press. Res. 2009, 32, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.; Pettinger, W.A.; Abel, P.W.; Jeffries, W.B. Regional distribution of alpha 1-adrenoceptor subtypes in rat kidney. J. Pharmacol. Exp. Ther. 1991, 258, 263–268. [Google Scholar] [PubMed]
- Sattar, M.A.; Johns, E.J. Evidence for an alpha 1-adrenoceptor subtype mediating adrenergic vasoconstriction in Wistar normotensive and stroke-prone spontaneously hypertensive rat kidney. J. Cardiovasc. Pharmacol. 1994, 23, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Villalobos-Molina, R.; López-Guerrero, J.J.; Ibarra, M. α1D- and α1A-adrenoceptorsmediate contraction in rat renal artery. Eur. J. Pharmacol. 1997, 322, 225–227. [Google Scholar] [CrossRef]
- Salomonsson, M.; Brannstrom, K.; Arendshorst, W.J. α1-adrenoceptor subtypes in rat renal resistance vessels: In vivo and in vitro studies. Am. J. Physiol. 2000, 278, F138–F147. [Google Scholar] [CrossRef] [PubMed]
- Sattar, M.A.; Johns, E.J. α-1 adrenoceptor subtypes involved in mediating adrenergically induced antinatriuresis and antidiuresis in two kidney, one clip Goldblatt and deoxycorticosterone acetate-salt hypertensive rats. J. Pharmacol. Exp. Ther. 1996, 277, 245–252. [Google Scholar] [PubMed]
- Sattar, M.A.; Abdullah, N.A.; Khan, M.A.; Johns, E.J. α1A- and α1D-adrenoceptors are the major functional subtypes of renal α1-adrenoceptors in streptozotocin-induced diabetic and normal Sprague-Dawley rats. Auton. Autacoid Pharmacol. 2008, 28, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Abdulla, M.; Sattar, M.; Johns, E.; Abdullah, N.; Khan, M.A. Evidence for the role of α1A-adrenoceptor subtype in the control of renal haemodynamics in fructose-fed Sprague–Dawley rat. Eur. J. Nutr. 2011, 50, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Kazi, R.N.; Munavvar, A.S.; Abdullah, N.A.; Khan, A.H.; Johns, E.J. Influence of high dietary sodium intake on the functional subtypes of α1-adrenoceptors in the renal cortical vasculature of Wistar–Kyoto rats. Auton. Autacoid Pharmacol. 2009, 29, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.A.; Munavvar, A.S.; Abdullah, N.A.; Johns, E.J. Role of α1 Adrenoceptor Subtypes in Renal Haemodynamics in Heart Failure And Diabetic Sd Rats. Can. J. Pure Appl. Sci. 2007, 1, 21–34. [Google Scholar]
- Ahmad, A.; Sattar, M.A.; Rathore, H.A.; Abdulla, M.H.; Khan, S.A.; Abdullah, N.A.; Kaur, G.; Johns, E.J. Functional contribution of α1D-adrenoceptors in the renal vasculature of left ventricular hypertrophy induced with isoprenaline and caffeine in Wistar–Kyoto rats. Can. J. Physiol. Pharmacol. 2014, 92, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Theilig, F.; Bostanjoglo, M.; Pavenstadt, H.; Grupp, C.; Holland, G.; Slosarek, I.; Gressner, A.M.; Russwurm, M.; Koesling, D.; Bachmann, S. Cellular distribution and function of soluble guanylyl cyclase in rat kidney and liver. J. Am. Soc. Nephrol. 2001, 12, 2209–2220. [Google Scholar] [PubMed]
- Pfeilschifter, J. Signalling pathways of nitric oxide. Kidney Blood Press. Res. 2000, 23, 159–161. [Google Scholar] [PubMed]
- Flanagan, E.T.; Buckley, M.M.; Aherne, C.M.; Lainis, F.; Sattar, M.; Johns, E.J. Impact of cardiac hypertrophy on arterial and cardiopulmonary baroreflex control of renal sympathetic nerve activity in anaesthetized rats. Exp. Physiol. 2008, 93, 1058–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, H.; Hess, D.T.; Zhang, R.; Sugi, K.; Gao, H.; Tan, B.L.; Bowles, D.E.; Milano, C.A.; Jain, M.K.; Koch, W.J.; et al. S-Nitrosylation of beta-Arrestins Biases Receptor Signaling and Confers Ligand Independence. Mol. Cell 2018, 70, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Nelson, C.D.; Garrison, T.R.; Miller, W.E.; Lefkowitz, R.J. Desensitization, internalization, and signaling functions of beta-arrestins demonstrated by RNA interference. Proc. Natl. Acad. Sci. USA 2003, 100, 1740–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.S.; Tian, X.; Benovic, J.L. β-Arrestins and G protein-coupled receptor trafficking. Methods Enzymol. 2013, 521, 91–108. [Google Scholar] [PubMed]
- Ozawa, K.; Whalen, E.J.; Nelson, C.D.; Mu, Y.; Hess, D.T.; Lefkowitz, R.J.; Stamler, J.S. S-nitrosylation of beta-arrestin regulates beta-adrenergic receptor trafficking. Mol. Cell 2008, 31, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.F.; Torres, M.T.; Silva, L.S.; Alves, F.L.; de Sa Pinheiro, A.A.; Miranda, A.; Capurro, M.L.; de la Fuente-Nunez, C.; Oliveira, V.X., Jr. Angiotensin II-derived constrained peptides with antiplasmodial activity and suppressed vasoconstriction. Sci. Rep. 2017, 7, 14326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waghe, P.; Sarath, T.S.; Gupta, P.; Kandasamy, K.; Choudhury, S.; Kutty, H.S.; Mishra, S.K.; Sarkar, S.N. Arsenic causes aortic dysfunction and systemic hypertension in rats: Augmentation of angiotensin II signaling. Chem. Biol. Interact. 2015, 237, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, J.; Dong, H.; Khan, S.A.; Chu, M.L.; Tsuda, T. Fibulin-2 deficiency attenuates angiotensin II-induced cardiac hypertrophy by reducing transforming growth factor-β signalling. Clin. Sci. 2014, 126, 275–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikusic, N.L.R.; Kouyoumdzian, N.M.; Uceda, A.; Del Mauro, J.S.; Pandolfo, M.; Gironacci, M.M.; Puyo, A.M.; Toblli, J.E.; Fernandez, B.E.; Choi, M.R. Losartan prevents the imbalance between renal dopaminergic and renin angiotensin systems induced by fructose overload. L-dopa/dopamine index as new potential biomarker of renal dysfunction. Metabolism 2018. [Google Scholar] [CrossRef]
- Zou, A.P.; Wu, F.; Cowley, A.W., Jr. Protective effect of angiotensin II-induced increase in nitric oxide in the renal medullary circulation. Hypertension 1998, 31, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Chin, S.Y.; Wang, C.T.; Majid, D.S.; Navar, L.G. Renoprotective effects of nitric oxide in angiotensin II-induced hypertension in the rat. Am. J. Physiol. 1998, 274, F876–F882. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Sattar, M.A.; Rathore, H.A.; Abdulla, M.H.; Khan, S.A.; Azam, M.; Abdullah, N.A.; Johns, E.J. Up Regulation of cystathione gamma lyase and Hydrogen Sulphide in the Myocardium Inhibits the Progression of Isoproterenol-Caffeine Induced Left Ventricular Hypertrophy in Wistar Kyoto Rats. PLoS ONE 2016, 11, e0150137. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Sattar, M.; Khan, S.A.; Abdullah, N.A.; Johns, E.J.; Afzal, S. Increased Oxidative Stress and Downregulation of Endothelial Nitric Oxide Synthase (ENOS) in the Kidney Atten- Uate the Responsiveness of (XLB Adrenergic Receptors in the Kidney of Rats with Left Ventricular Hypertrophy. Acta Pol. Pharm. 2017, 74, 413–423. [Google Scholar] [PubMed]
- Mimran, A.; Ribstein, J.; DuCailar, G. Contrasting effect of antihypertensive treatment on the renal response to L-arginine. Hypertension 1995, 26, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.O.; Antunes, E.; de Nucci, G.; Lovisolo, S.M.; Zatz, R. Chronic inhibition of nitric oxide synthesis. A new model of arterial hypertension. Hypertension 1992, 20, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Engels, K.; Baylis, C. Angiotensin II and α 1-adrenergic tone in chronic nitric oxide blockade-induced hypertension. Am. J. Physiol. 1994, 266, R1470–R1476. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mendoza, A.; Hong, E.; Escalante, B. The role of nitric oxide in angiotensin II-induced renal vasoconstriction in renovascular hypertension. J. Hypertens. 1998, 16, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Symons, J.D.; Stebbins, C.L.; Musch, T.I. Interactions between angiotensin II and nitric oxide during exercise in normal and heart failure rats. J. Appl. Physiol. 1999, 87, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Toth, A.D.; Gyombolai, P.; Szalai, B.; Varnai, P.; Turu, G.; Hunyady, L. Angiotensin type 1A receptor regulates beta-arrestin binding of the β2-adrenergic receptor via heterodimerization. Mol. Cell Endocrinol. 2017, 442, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Kashihara, T.; Nakada, T.; Kojima, K.; Takeshita, T.; Yamada, M. Angiotensin II activates CaV 1.2 Ca2+ channels through beta-arrestin2 and casein kinase 2 in mouse immature cardiomyocytes. J. Physiol. 2017, 595, 4207–4225. [Google Scholar] [CrossRef] [PubMed]
- Ichiki, T.; Usui, M.; Kato, M.; Funakoshi, Y.; Ito, K.; Egashira, K.; Takeshita, A. Downregulation of angiotensin II type 1 receptor gene transcription by nitric oxide. Hypertension 1998, 31, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Wang, R. Hydrogen sulfide-based therapeutics: Exploiting a unique but ubiquitous gasotransmitter. Nat. Rev. Drug Discov. 2015, 14, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Hu, Q.; Ma, F.; Zhu, Y.Z. Vasorelaxant Effect of a New Hydrogen Sulfide-Nitric Oxide Conjugated Donor in Isolated Rat Aortic Rings through cGMP Pathway. Oxid. Med. Cell. Longev. 2016, 2016, 7075682. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, A.; Habara, Y. Hydrogen Sulfide Regulates Ca2+ Homeostasis Mediated by Concomitantly Produced Nitric Oxide via a Novel Synergistic Pathway in Exocrine Pancreas. Antioxid. Redox Signal. 2014, 20, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Sattar, M.A.; Rathore, H.A.; Khan, S.A.; Lazhari, M.I.; Afzal, S.; Hashmi, F.; Abdullah, N.A.; Johns, E.J. A critical review of pharmacological significance of Hydrogen Sulfide in hypertension. Indian J. Pharmacol. 2015, 47, 243–247. [Google Scholar] [PubMed]
- Vacek, T.P.; Rehman, S.; Neamtu, D.; Yu, S.; Givimani, S.; Tyagi, S.C. Matrix metalloproteinases in atherosclerosis: Role of nitric oxide, hydrogen sulfide, homocysteine, and polymorphisms. Vasc. Health Risk Manag. 2015, 11, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, R. H(2)S-induced vasorelaxation and underlying cellular and molecular mechanisms. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H474–H480. [Google Scholar] [CrossRef] [PubMed]
- Dhaese, I.; Lefebvre, R.A. Myosin light chain phosphatase activation is involved in the hydrogen sulfide-induced relaxation in mouse gastric fundus. Eur. J. Pharmacol. 2009, 606, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Gainey, L.F., Jr.; Greenberg, M.J. Hydrogen sulfide is synthesized in the gills of the clam Mercenaria mercenaria and acts seasonally to modulate branchial muscle contraction. Biol. Bull. 2005, 209, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Bibli, S.I.; Yang, G.; Zhou, Z.; Wang, R.; Topouzis, S.; Papapetropoulos, A. Role of cGMP in hydrogen sulfide signaling. Nitric Oxide 2015, 46, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Ping, C.; Mok, Y.Y.; Ling, L.; Whiteman, M.; Bhatia, M.; Moore, P. Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulphide? Br. J. Pharmacol. 2006, 149, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteman, M.; Li, L.; Kostetski, I.; Chu, S.H.; Siau, J.L.; Bhatia, M.; Moore, P.K. Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochem. Biophys. Res. Commun. 2006, 343, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Hosoki, R.; Matsuki, N.; Kimura, H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Ndisang, J.F.; Wang, R. Modulation of endogenous production of H2S in rat tissues. Can. J. Physiol. Pharmacol. 2003, 81, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Grossi, L. Hydrogen sulfide induces nitric oxide release from nitrite. Bioorg. Med. Chem. Lett. 2009, 19, 6092–6094. [Google Scholar] [CrossRef] [PubMed]
- Grossi, L.; Montevecchi, P.C. A Kinetic Study of S-Nitrosothiol Decomposition. Chem. A Eur. J. 2002, 8, 380–387. [Google Scholar] [CrossRef]
- Grossi, L.; D’Angelo, S. Sodium nitroprusside: Mechanism of NO release mediated by sulfhydryl-containing molecules. J. Med. Chem. 2005, 48, 2622–2626. [Google Scholar] [CrossRef] [PubMed]
- Kubo, S.; Doe, I.; Kurokawa, Y.; Nishikawa, H.; Kawabata, A. Direct inhibition of endothelial nitric oxide synthase by hydrogen sulfide: Contribution to dual modulation of vascular tension. Toxicology 2007, 232, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Yong, Q.-C.; Hu, L.-F.; Wang, S.; Huang, D.; Bian, J.-S. Hydrogen sulfide interacts with nitric oxide in the heart: Possible involvement of nitroxyl. Cardiovasc. Res. 2010, 88, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Irvine, J.C.; Ritchie, R.H.; Favaloro, J.L.; Andrews, K.L.; Widdop, R.E.; Kemp-Harper, B.K. Nitroxyl (HNO): The Cinderella of the nitric oxide story. Trends Pharmacol. Sci. 2008, 29, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Paolocci, N.; Katori, T.; Champion, H.C.; John, M.E.S.; Miranda, K.M.; Fukuto, J.M.; Wink, D.A.; Kass, D.A. Positive inotropic and lusitropic effects of HNO/NO− in failing hearts: Independence from β-adrenergic signaling. Proc. Natl. Acad. Sci. USA 2003, 100, 5537–5542. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.K.; Xue, B.; Shi, H. Activation of the sympathetic nervous system suppresses mouse white adipose tissue hyperplasia through the β1 adrenergic receptor. Physiol. Rep. 2018, 6, e13645. [Google Scholar] [CrossRef] [PubMed]
- Ali Elnabtity, A.M.; Selim, M.F. Norepinephrine versus Ephedrine to Maintain Arterial Blood Pressure during Spinal Anesthesia for Cesarean Delivery: A Prospective Double-blinded Trial. Anesth. Essays Res. 2018, 12, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Kolo, L.L.; Westfall, T.C.; Macarthur, H. Nitric oxide decreases the biological activity of norepinephrine resulting in altered vascular tone in the rat mesenteric arterial bed. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H296–H303. [Google Scholar] [CrossRef] [PubMed]
- Esler, M.; Jennings, G.; Biviano, B.; Lambert, G.; Hasking, G. Mechanism of elevated plasma noradrenaline in the course of essential hypertension. J. Cardiovasc. Pharmacol. 1986, 8 (Suppl. 5), S39–S43. [Google Scholar] [CrossRef] [PubMed]
- Schlaich, M.P.; Kaye, D.M.; Lambert, E.; Sommerville, M.; Socratous, F.; Esler, M.D. Relation between cardiac sympathetic activity and hypertensive left ventricular. Circulation 2003, 108, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Klahr, S. The role of nitric oxide in hypertension and renal disease progression. Nephrol. Dial. Transplant. 2001, 16 (Suppl. 1), 60–62. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Zhou, Z.; Wang, L.; Yang, C.; Wang, J.; Wu, T.; Song, L. Mutual modulation between norepinephrine and nitric oxide in hemocytes during the mollusc immune response. Sci. Rep. 2014, 4, 6963. [Google Scholar] [CrossRef] [PubMed]
- Kvetnansky, R.; Pacak, K.; Tokarev, D.; Jelokova, J.; Jezova, D.; Rusnak, M. Chronic blockade of nitric oxide synthesis elevates plasma levels of catecholamines and their metabolites at rest and during stress in rats. Neurochem. Res. 1997, 22, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Thatikunta, P.; Chakder, S.; Rattan, S. Nitric oxide synthase inhibitor inhibits catecholamines release caused by hypogastric sympathetic nerve stimulation. J. Pharmacol. Exp. Ther. 1993, 267, 1363–1368. [Google Scholar] [PubMed]
- Yamamoto, R.; Wada, A.; Asada, Y.; Yuhi, T.; Yanagita, T.; Niina, H.; Sumiyoshi, A. Functional relation between nitric oxide and noradrenaline for the modulation of vascular tone in rat mesenteric vasculature. Naunyn Schmiedebergs Arch. Pharmacol. 1994, 349, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Tyce, G.M.; Chritton, S.L.; Barnes, R.D.; Ward, L.E.; Hunter, L.W.; Rorie, D.K. The adrenal gland as a source of dihydroxyphenylalanine and catecholamine metabolites. Adv. Pharmacol. 1998, 42, 370–373. [Google Scholar] [PubMed]
- Schwarz, P.; Diem, R.; Dun, N.J.; Forstermann, U. Endogenous and exogenous nitric oxide inhibits norepinephrine release from rat heart sympathetic nerves. Circ. Res. 1995, 77, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, D.G.; Goligorsky, M.S.; Schmid, P.C.; Krebsbach, R.J.; Schmid, H.H.; Das, S.K.; Dey, S.K.; Arreaza, G.; Thorup, C.; Stefano, G.; et al. Production and physiological actions of anandamide in the vasculature of the rat kidney. J. Clin. Investig. 1997, 100, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Fowler, C.J. Transport of endocannabinoids across the plasma membrane and within the cell. FEBS J. 2013, 280, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Di Marzo, V. An introduction to the endocannabinoid system: From the early to the latest concepts. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ford, W.R.; Honan, S.A.; White, R.; Hiley, C.R. Evidence of a novel site mediating anandamide-induced negative inotropic and coronary vasodilatator responses in rat isolated hearts. Br. J. Pharmacol. 2002, 135, 1191–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, E.A.; Fuller, S.A.; Edgemond, W.S.; Campbell, W.B. Physiological and behavioural effects of the endogenous cannabinoid, arachidonylethanolamide (anandamide), in the rat. Br. J. Pharmacol. 1996, 119, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lake, K.D.; Compton, D.R.; Varga, K.; Martin, B.R.; Kunos, G. Cannabinoid-induced hypotension and bradycardia in rats mediated by CB1-like cannabinoid receptors. J. Pharmacol. Exp. Ther. 1997, 281, 1030–1037. [Google Scholar] [PubMed]
- Lake, K.D.; Martin, B.R.; Kunos, G.; Varga, K. Cardiovascular effects of anandamide in anesthetized and conscious normotensive and hypertensive rats. Hypertension 1997, 29, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Varga, K.; Lake, K.D.; Huangfu, D.; Guyenet, P.G.; Kunos, G. Mechanism of the hypotensive action of anandamide in anesthetized rats. Hypertension 1996, 28, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Luce, V.; Fernandez Solari, J.; Rettori, V.; De Laurentiis, A. The inhibitory effect of anandamide on oxytocin and vasopressin secretion from neurohypophysis is mediated by nitric oxide. Regul. Pept. 2014, 188, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Niederhoffer, N.; Schmid, K.; Szabo, B. The peripheral sympathetic nervous system is the major target of cannabinoids in eliciting cardiovascular depression. Naunyn Schmiedebergs Arch. Pharmacol. 2003, 367, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Pfitzer, T.; Niederhoffer, N.; Szabo, B. Search for an endogenous cannabinoid-mediated effect in the sympathetic nervous system. Naunyn Schmiedebergs Arch. Pharmacol. 2005, 371, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, G.; Matsumura, Y.; Tadano, K.; Hashimoto, T.; Morimoto, S. Involvement of nitric oxide in endothelin ETB receptor-mediated inhibitory actions on antidiuresis and norepinephrine overflow induced by stimulation of renal nerves in anesthetized dogs. J. Cardiovasc. Pharmacol. 1997, 30, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Ritter, J.K.; Li, C.; Xia, M.; Poklis, J.L.; Lichtman, A.H.; Abdullah, R.A.; Dewey, W.L.; Li, P.L. Production and actions of the anandamide metabolite prostamide E2 in the renal medulla. J. Pharmacol. Exp. Ther. 2012, 342, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Stefano, G.B.; Esch, T.; Cadet, P.; Zhu, W.; Mantione, K.; Benson, H. Endocannabinoids as autoregulatory signaling molecules: Coupling to nitric oxide and a possible association with the relaxation response. Med. Sci. Monit. 2003, 9, RA63–RA75. [Google Scholar] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, A.; Dempsey, S.K.; Daneva, Z.; Azam, M.; Li, N.; Li, P.-L.; Ritter, J.K. Role of Nitric Oxide in the Cardiovascular and Renal Systems. Int. J. Mol. Sci. 2018, 19, 2605. https://doi.org/10.3390/ijms19092605
Ahmad A, Dempsey SK, Daneva Z, Azam M, Li N, Li P-L, Ritter JK. Role of Nitric Oxide in the Cardiovascular and Renal Systems. International Journal of Molecular Sciences. 2018; 19(9):2605. https://doi.org/10.3390/ijms19092605
Chicago/Turabian StyleAhmad, Ashfaq, Sara K. Dempsey, Zdravka Daneva, Maleeha Azam, Ningjun Li, Pin-Lan Li, and Joseph K. Ritter. 2018. "Role of Nitric Oxide in the Cardiovascular and Renal Systems" International Journal of Molecular Sciences 19, no. 9: 2605. https://doi.org/10.3390/ijms19092605
APA StyleAhmad, A., Dempsey, S. K., Daneva, Z., Azam, M., Li, N., Li, P. -L., & Ritter, J. K. (2018). Role of Nitric Oxide in the Cardiovascular and Renal Systems. International Journal of Molecular Sciences, 19(9), 2605. https://doi.org/10.3390/ijms19092605