Widespread Expression of Hedgehog Pathway Components in a Large Panel of Human Tumor Cells and Inhibition of Tumor Growth by GANT61: Implications for Cancer Therapy


 and
and
Abstract
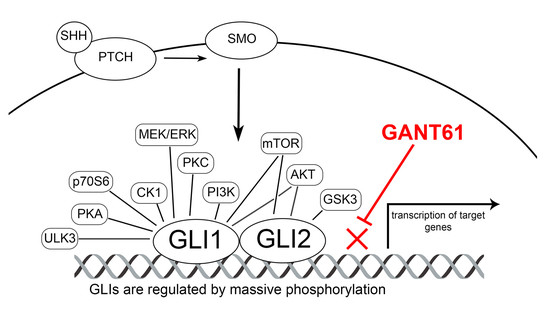
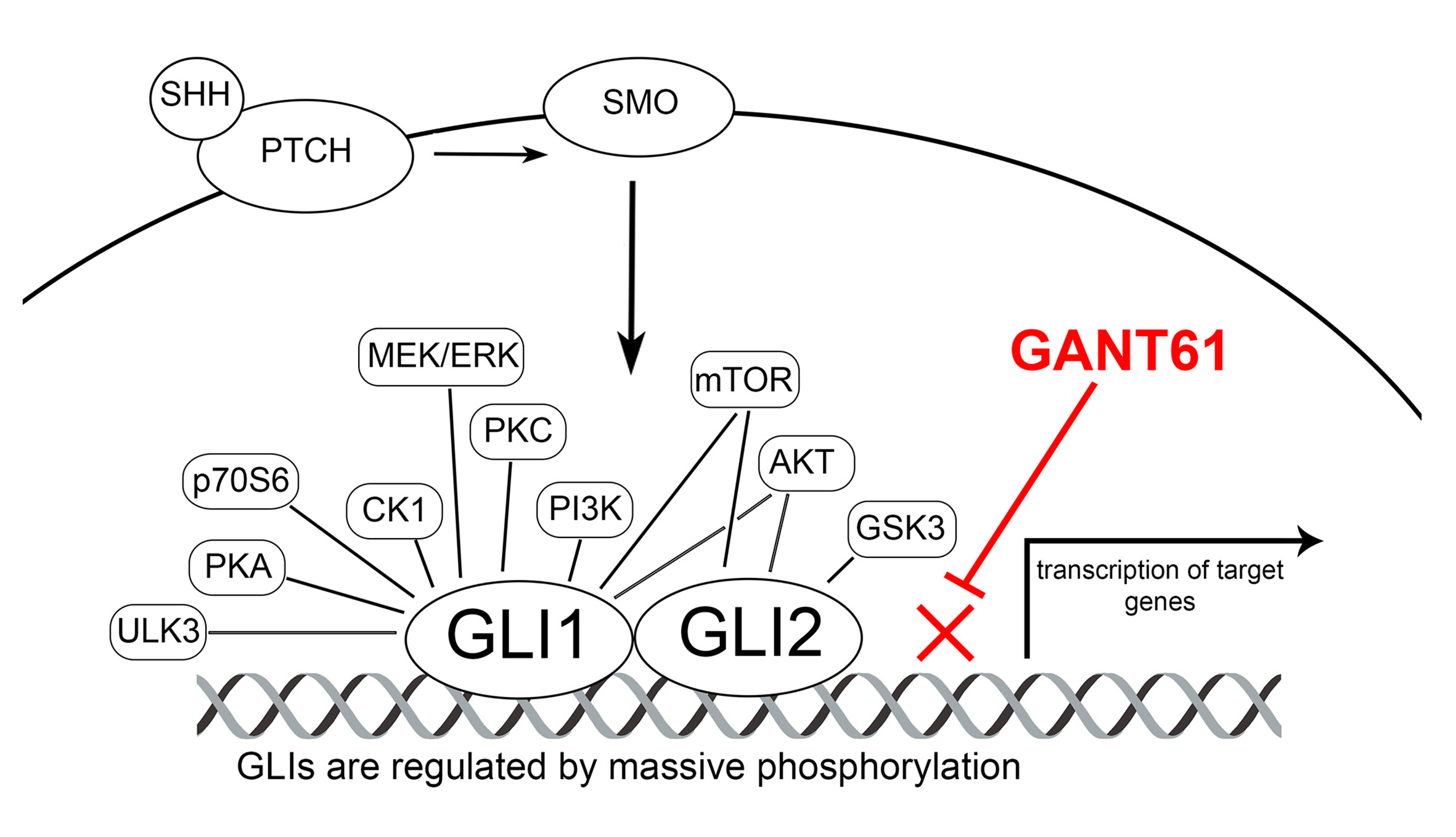
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
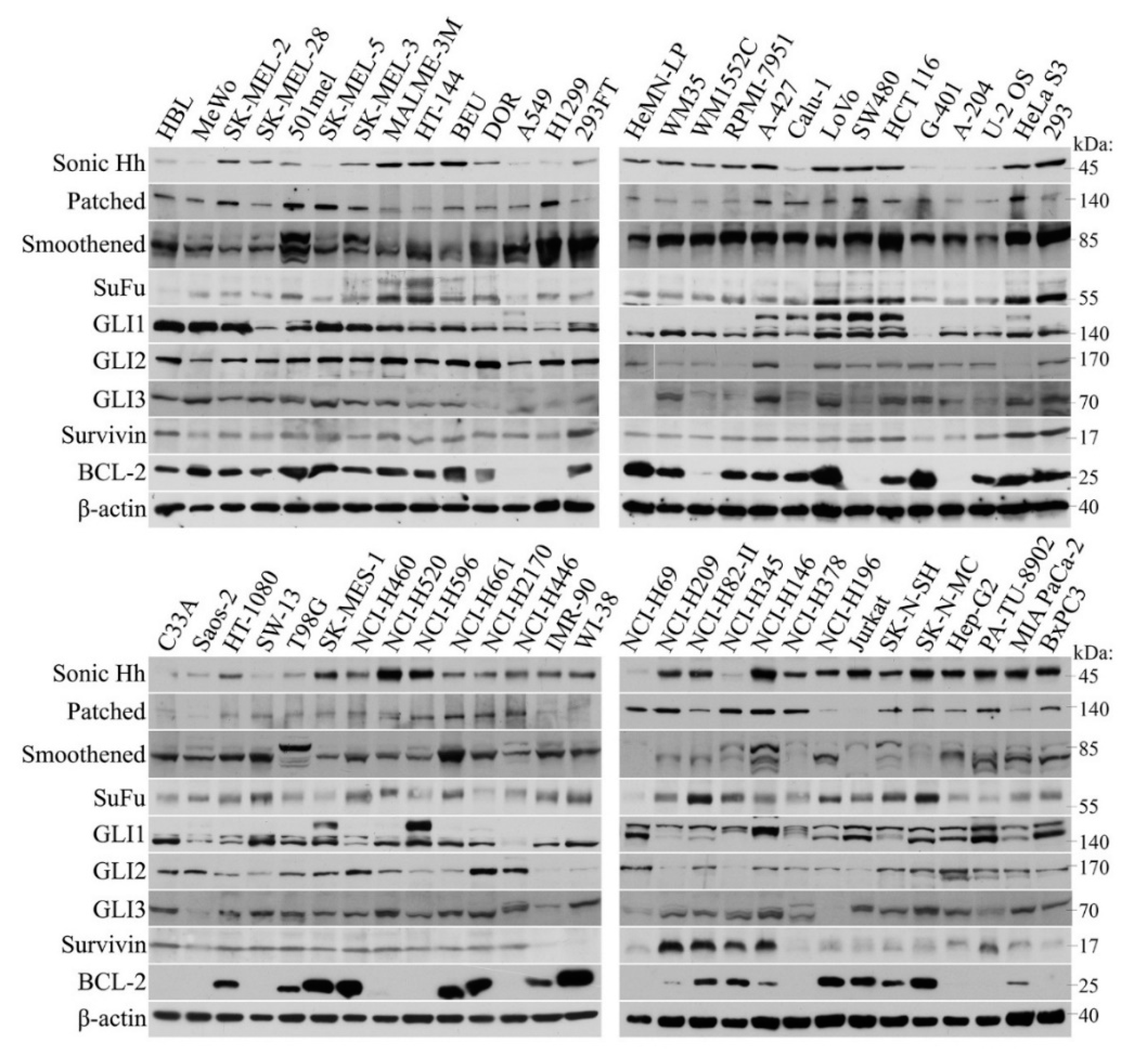
2.1. Broad Expression of HH Cascade Components in Human Tumor Cell Lines
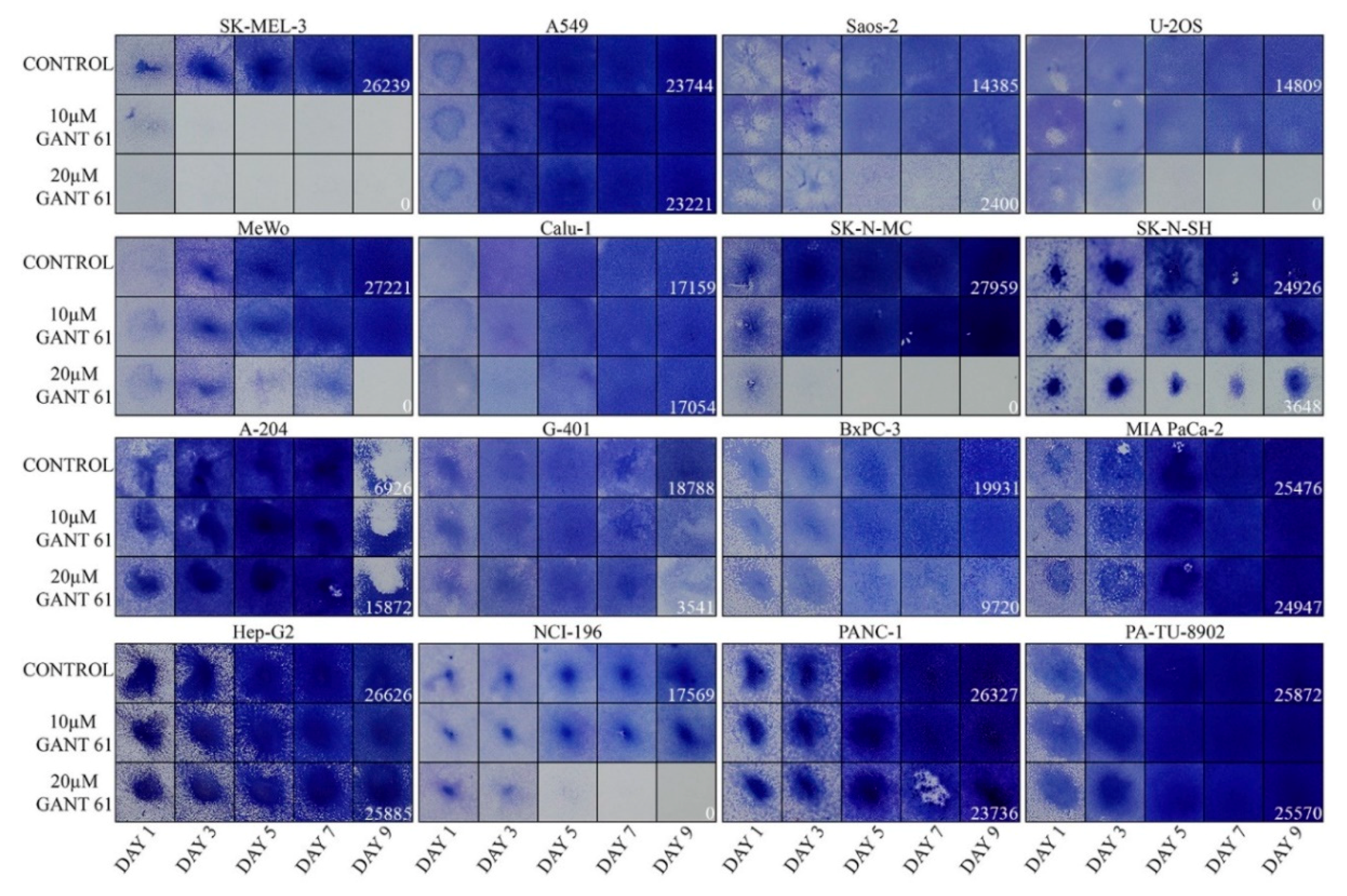
2.2. Inhibition of Cell Proliferation by GLI Inhibitor GANT61
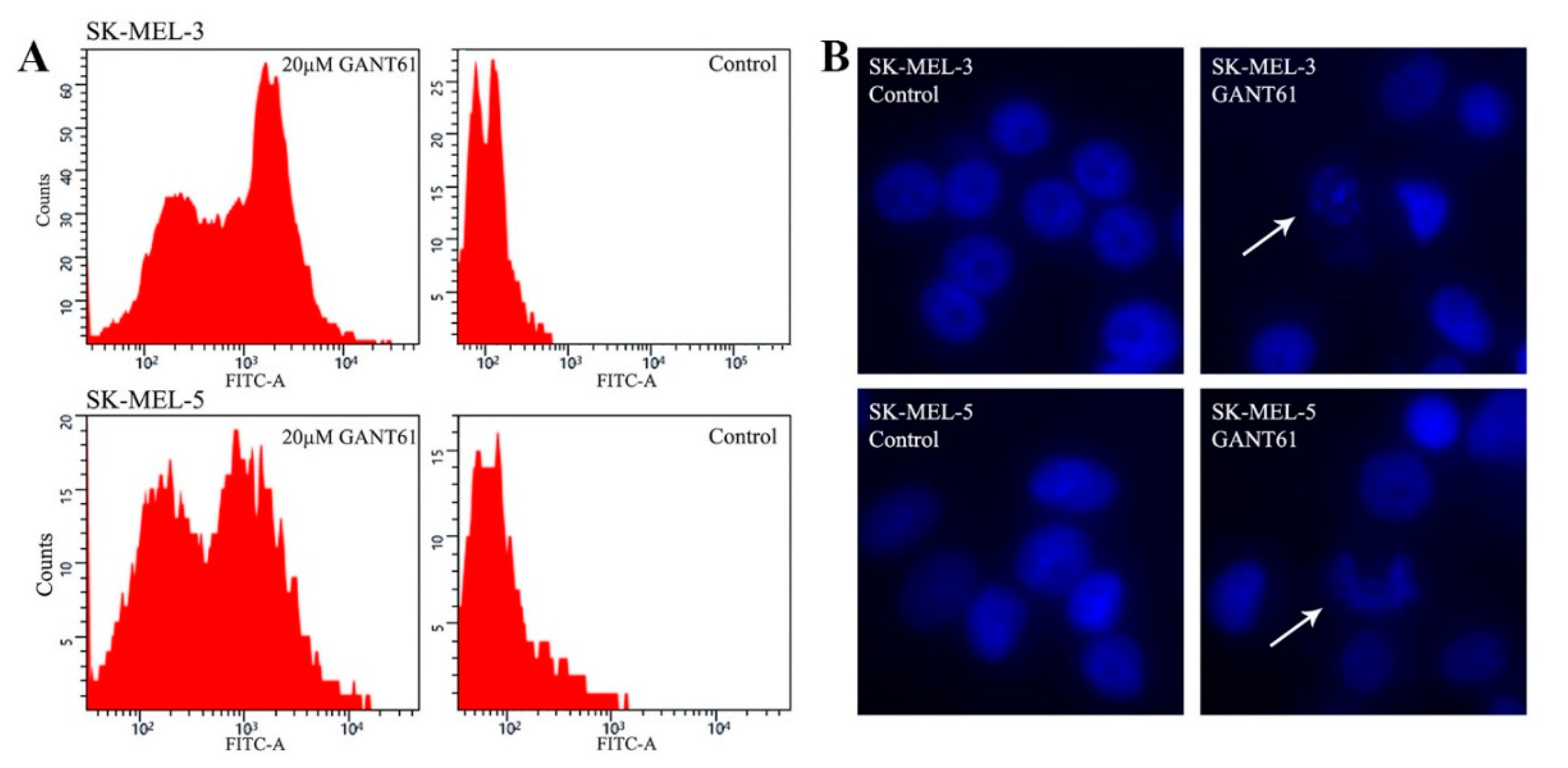
2.3. GANT61 Eradicates Tumor Cells through Apoptosis
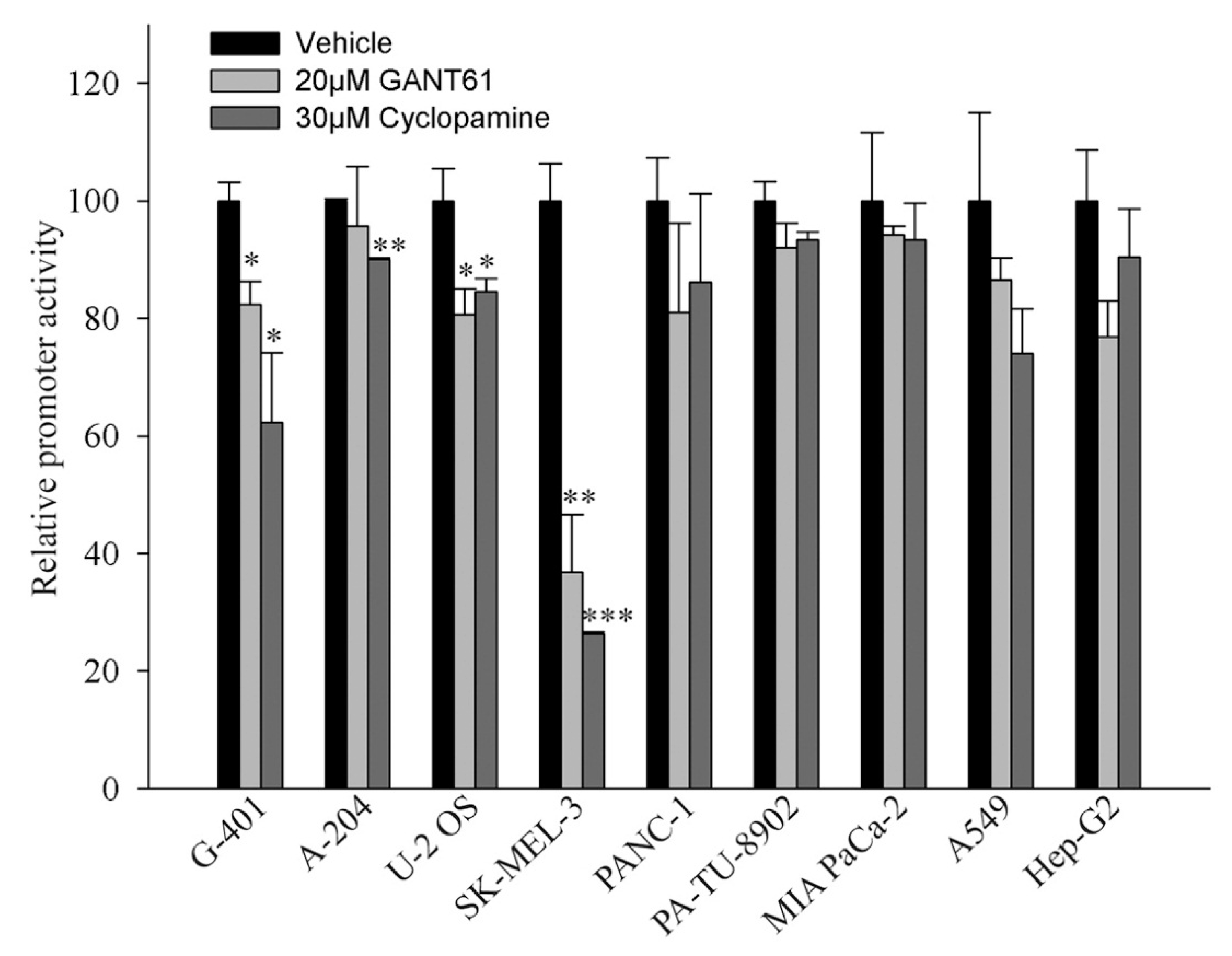
2.4. Activity of the Promoter Containing 12xGLI Consensus Site
3. Discussion
4. Materials and Methods
4.1. Cell Cultivation
4.2. Cell Lines
4.3. Western Blots
4.4. Proliferation Assays
4.5. Detection of Apoptosis
4.6. Microscopic Detection of Apoptotic Nuclei
4.7. Reporter Assays
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CSC | cancer stem cells |
| HRP | horseradish peroxidase |
| DAPI | 40,6-Diamidino-2-Phenylindole, Dihydrochloride |
| SMARCB1 | SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily b, member 1 |
| EMT | epithelial-to-mesenchymal transition |
| TUNEL | terminal deoxynucleotidyl transferase-mediated d-UTP Nick End Labeling |
| AR | androgen receptor |
| BCC | basal cell carcinoma |
| GLI | glioma family zinc finger protein |
| MITF | microphthalmia-associated trancription factor |
| HGF | hepatocyte growth factor |
| PTCH | patched |
| SMO | smoothened, frizzled class receptor |
| NSCLC | Non-small cell lung cancer |
| SCLC | Small cell lung cancer |
| SWI/SNF | SWItch/Sucrose Non-Fermentable |
| mTOR | mechanistic target of rapamycin |
| FITC | fluorescein isothiocyanate |
References
- Cohen, M.M., Jr. The hedgehog signaling network. Am. J. Med. Genet. A 2003, 123A, 5–28. [Google Scholar] [CrossRef] [PubMed]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog signal transduction network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.E.; Chiang, C. Hedgehog secretion and signal transduction in vertebrates. J. Biol. Chem. 2012, 287, 17905–17913. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Hallahan, A.R.; Pritchard, J.I.; Eberhart, C.G.; Watkins, D.N.; Chen, J.K.; Cooper, M.K.; Taipale, J.; Olson, J.M.; et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science 2002, 297, 1559–1561. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.E.; Chaudhry, A.; Farah, M.H.; Eberhart, C.G. Hedgehog signaling promotes medulloblastoma survival via Bc/II. Am. J. Pathol. 2007, 170, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Teglund, S.; Toftgard, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta 2010, 1805, 181–208. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chi, S.; Xie, J. Hedgehog signaling in skin cancers. Cell Signal. 2011, 23, 1235–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, T.C.; Weeraratne, S.D.; Pomeroy, S.L. Hedgehog-GLI Pathway in Medulloblastoma. J. Clin. Oncol. 2012, 30, 2154–2156. [Google Scholar] [CrossRef] [PubMed]
- Vlckova, K.; Ondrusova, L.; Vachtenheim, J.; Reda, J.; Dundr, P.; Zadinova, M.; Zakova, P.; Pouckova, P. Survivin, a novel target of the Hedgehog/GLI signaling pathway in human tumor cells. Cell Death Dis. 2016, 7, e2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Maitah, M.Y.; Ahmad, A.; Kong, D.; Bao, B.; Sarkar, F.H. Targeting the Hedgehog signaling pathway for cancer therapy. Expert Opin. Ther. Targets 2012, 16, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Chang, A.L.; Oro, A.E. Hedgehog pathway inhibition and the race against tumor evolution. J. Cell Biol. 2012, 199, 193–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Onishi, H.; Katano, M. Hedgehog signaling pathway as a therapeutic target in various types of cancer. Cancer Sci. 2011, 102, 1756–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, I.; Hashemi, S.N.; Duan, Z.H.; Shi, T. Aberrant signaling pathways in squamous cell lung carcinoma. Cancer Inform. 2011, 10, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Blanco, J.; Schilling, N.S.; Tokhunts, R.; Giambelli, C.; Long, J.; Liang, F.D.; Singh, S.; Black, K.E.; Wang, Z.; Galimberti, F.; et al. The Hedgehog processing pathway is required for NSCLC growth and survival. Oncogene 2013, 32, 2335–2345. [Google Scholar] [CrossRef] [PubMed]
- Justilien, V.; Walsh, M.P.; Ali, S.A.; Thompson, E.A.; Murray, N.R.; Fields, A.P. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell 2014, 25, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Tanaka, N. The Hedgehog Signaling Networks in Lung Cancer: The Mechanisms and Roles in Tumor Progression and Implications for Cancer Therapy. BioMed Res. Int. 2016, 2016, 7969286. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.N.; Berman, D.M.; Baylin, S.B. Hedgehog signaling: Progenitor phenotype in small-cell lung cancer. Cell Cycle 2003, 2, 196–198. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Martelotto, L.G.; Peifer, M.; Sos, M.L.; Karnezis, A.N.; Mahjoub, M.R.; Bernard, K.; Conklin, J.F.; Szczepny, A.; Yuan, J.; et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat. Med. 2011, 17, 1504–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauth, M.; Toftgard, R. Hedgehog signaling and pancreatic tumor development. Adv. Cancer Res. 2011, 110, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Moore, T.T.; Hattersley, M.M.; Scarpitti, M.; Yang, B.; Devereaux, E.; Ramachandran, V.; Arumugam, T.; Ji, B.; Logsdon, C.D.; et al. Inhibition of the Hedgehog pathway targets the tumor-associated stroma in pancreatic cancer. Mol. Cancer Res. 2012, 10, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Rodova, M.; Roy, S.K.; Sharma, J.; Singh, K.P.; Srivastava, R.K.; Shankar, S. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 2013, 330, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Ma, L.; Zhang, Z.; Liu, X.; Gao, H.; Zhuang, Y.; Yang, P.; Kornmann, M.; Tian, X.; Yang, Y. Hedgehog signaling regulates epithelial-mesenchymal transition in pancreatic cancer stem-like cells. J. Cancer 2016, 7, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; An, Y.; Wang, X.; Zha, W.; Li, X. Inhibition of the Hedgehog pathway induces autophagy in pancreatic ductal adenocarcinoma cells. Oncol. Rep. 2014, 31, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhou, Y.; Xie, C.; Wei, S.M.; Gan, H.; He, S.; Wang, F.; Xu, L.; Lu, J.; Dai, W.; et al. Genome-wide screening reveals an EMT molecular network mediated by Sonic Hedgehog-Gli1 signaling in pancreatic cancer cells. PLoS ONE 2012, 7, e43119. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz, I.A. Melanomas require Hedgehog-Gli signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [PubMed]
- Alexaki, V.I.; Javelaud, D.; Van Kempen, L.C.; Mohammad, K.S.; Dennler, S.; Luciani, F.; Hoek, K.S.; Juarez, P.; Goydos, J.S.; Fournier, P.J.; et al. GLI2-mediated melanoma invasion and metastasis. J. Natl. Cancer Inst. 2010, 102, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Vlckova, K.; Reda, J.; Ondrusova, L.; Krayem, M.; Ghanem, G.; Vachtenheim, J. GLI inhibitor GANT61 kills melanoma cells and acts in synergy with obatoclax. Int. J. Oncol. 2016, 49, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Ok, C.Y.; Singh, R.R.; Vega, F. Aberrant activation of the Hedgehog signaling pathway in malignant hematological neoplasms. Am. J. Pathol. 2012, 180, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, N.; Minami, Y.; Kakiuchi, S.; Kuwatsuka, Y.; Hayakawa, F.; Jamieson, C.; Kiyoi, H.; Naoe, T. Small-molecule Hedgehog inhibitor attenuates the leukemia-initiation potential of acute myeloid leukemia cells. Cancer Sci. 2016, 107, 1422–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakiuchi, S.; Minami, Y.; Miyata, Y.; Mizutani, Y.; Goto, H.; Kawamoto, S.; Yakushijin, K.; Kurata, K.; Matsuoka, H.; Minami, H. NANOG expression as a responsive biomarker during treatment with Hedgehog signal inhibitor in acute myeloid leukemia. Int. J. Mol. Sci. 2017, 18, 486. [Google Scholar] [CrossRef] [PubMed]
- Aberger, F.; Hutterer, E.; Sternberg, C.; del Burgo, P.J.; Hartmann, T.N. Acute myeloid leukemia—Strategies and challenges for targeting oncogenic Hedgehog/GLI signaling. Cell Commun. Signal. 2017, 15, 8. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.A.; Liao, Z.W.; Yamagata, N.; Pouliot, G.P.; Stevenson, K.E.; Neuberg, D.S.; Thorner, A.R.; Ducar, M.; Silverman, E.A.; Hunger, S.P.; et al. Hedgehog pathway mutations drive oncogenic transformation in high-risk T-cell acute lymphoblastic leukemia. Leukemia 2018. [Google Scholar] [CrossRef] [PubMed]
- Fei, D.L.; Sanchez-Mejias, A.; Wang, Z.; Flaveny, C.; Long, J.; Singh, S.; Rodriguez-Blanco, J.; Tokhunts, R.; Giambelli, C.; Briegel, K.J.; et al. Hedgehog signaling regulates bladder cancer growth and tumorigenicity. Cancer Res. 2012, 72, 4449–4458. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.; Clement, V.; Altaba, A. Therapeutic targeting of the Hedgehog-GLI pathway in prostate cancer. Cancer Res. 2005, 65, 2990–2992. [Google Scholar] [CrossRef] [PubMed]
- Thiyagarajan, S.; Bhatia, N.; Reagan-Shaw, S.; Cozma, D.; Thomas-Tikhonenko, A.; Ahmad, N.; Spiegelman, V.S. Role of GLI2 transcription factor in growth and tumorigenicity of prostate cells. Cancer Res. 2007, 67, 10642–10646. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Truong, S.; Nouri, M.; Moore, J.; Al Nakouzi, N.; Lubik, A.A.; Buttyan, R. Non-canonical activation of Hedgehog in prostate cancer cells mediated by the interaction of transcriptionally active androgen receptor proteins with Gli3. Oncogene 2018, 37, 2313–2325. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Hu, L.; Liu, Z.; Qin, Y.; Li, R.; Zhang, G.; Zhao, B.; Bi, C.; Lei, Y.; Bai, Y. Inhibition of Gli1- mediated prostate cancer cell proliferation by inhibiting the mTOR/S6K1 signaling pathway. Oncol. Lett. 2017, 14, 7970–7976. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Altaba, A. Hedgehog-Gli1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Takezaki, T.; Hide, T.; Takanaga, H.; Nakamura, H.; Kuratsu, J.; Kondo, T. Essential role of the Hedgehog signaling pathway in human glioma-initiating cells. Cancer Sci. 2011, 102, 1306–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagao-Kitamoto, H.; Nagata, M.; Nagano, S.; Kitamoto, S.; Ishidou, Y.; Yamamoto, T.; Nakamura, S.; Tsuru, A.; Abematsu, M.; Fujimoto, Y.; et al. GLI2 is a novel therapeutic target for metastasis of osteosarcoma. Int. J. Cancer 2015, 136, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Han, L.; Chen, Y.; He, F.; Sun, B.; Kamar, S.; Zhang, Y.; Yang, Y.; Wang, C.; Yang, Z. Hedgehog signalling in the tumourigenesis and metastasis of osteosarcoma, and its potential value in the clinical therapy of osteosarcoma. Cell Death Dis. 2018, 9, 701. [Google Scholar] [CrossRef] [PubMed]
- Satheesha, S.; Manzella, G.; Bovay, A.; Casanova, E.A.; Bode, P.K.; Belle, R.; Feuchtgruber, S.; Jaaks, P.; Dogan, N.; Koscielniak, E.; et al. Targeting Hedgehog signaling reduces self-renewal in embryonal rhabdomyosarcoma. Oncogene 2016, 35, 2020–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Wang, X.; Wan, J.; Li, T.; Gong, X.; Zhang, K.; Yi, L.; Xiang, Z.; Xu, M.; Cui, H. Sonic Hedgehog pathway is essential for neuroblastoma cell proliferation and tumor growth. Mol. Cell Biochem. 2012, 364, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Szkandera, J.; Kiesslich, T.; Haybaeck, J.; Gerger, A.; Pichler, M. Hedgehog signaling pathway in ovarian cancer. Int. J. Mol. Sci. 2013, 14, 1179–1196. [Google Scholar] [CrossRef] [PubMed]
- Levanat, S.; Sabol, M.; Musani, V.; Ozretic, P.; Trnski, D. Hedgehog signaling pathway as genetic and epigenetic target in ovarian tumors. Curr. Pharm. Des. 2017, 23, 73–94. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Yoon, J.W.; Xiao, X.; Dean, N.M.; Monia, B.P.; Marcusson, E.G. Selective down-regulation of glioma-associated oncogene 2 inhibits the proliferation of hepatocellular carcinoma cells. Cancer Res. 2007, 6, 73583–73593. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; Devecchio, J.; Shi, T.; Jones, J.; Agyeman, A.; Houghton, J.A. Hedgehog signaling drives cellular survival in human colon carcinoma cells. Cancer Res. 2011, 71, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.L.; Schumacher, D.; Staudte, S.; Steffen, A.; Haybaeck, J.; Keilholz, U.; Schweiger, C.; Golob-Schwarzl, N.; Mumberg, D.; Henderson, D.; et al. Non-canonical Hedgehog signaling is a positive regulator of the WNT pathway and is required for the survival of colon cancer stem cells. Cell Rep. 2017, 21, 2813–2828. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.S.; Jeng, C.J.; Sheen, I.S.; Wu, S.H.; Lu, S.J.; Wang, C.H.; Chang, C.F. Glioma-associated oncogene homolog inhibitors have the potential of suppressing cancer stem cells of breast cancer. Int. J. Mol. Sci. 2018, 19, 1375. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Tucker, J.A.; Khullar, S.; Samant, R.S.; Shevde, L.A. Hedgehog signaling in tumor cells facilitates osteoblast-enhanced osteolytic metastases. PLoS ONE 2012, 7, e34374. [Google Scholar] [CrossRef] [PubMed]
- Jagani, Z.; Mora-Blanco, E.L.; Sansam, C.G.; McKenna, E.S.; Wilson, B.; Chen, D.; Klekota, J.; Tamayo, P.; Nguyen, P.T.; Tolstorukov, M.; et al. Loss of the tumor suppressor SNF5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat. Med. 2010, 16, 1429–1433. [Google Scholar] [CrossRef]
- Riobo, N.A.; Lu, K.; Ai, X.; Haines, G.M.; Emerson, C.P., Jr. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4505–4510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauth, M.; Toftgard, R. Non-canonical activation of GLI transcription factors: Implications for targeted anti- cancer therapy. Cell Cycle 2007, 6, 2458–2463. [Google Scholar] [CrossRef] [PubMed]
- Shevde, L.A.; Samant, R.S. Nonclassical Hedgehog-Gli signaling and its clinical implications. Int. J. Cancer 2014, 135, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Po, A.; Silvano, M.; Miele, E.; Capalbo, C.; Eramo, A.; Salvati, V.; Todaro, M.; Besharat, Z.M.; Catanzaro, G.; Cucchi, D.; et al. Noncanonical GLI1 signaling promotes stemness features and in vivo growth in lung adenocarcinoma. Oncogene 2017, 36, 4641–4652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates Hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Q.; Yen, C.J.; Xia, W.; Izzo, J.G.; Lang, J.Y.; Li, C.W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Matsubara, S.; Ding, Q.; Tsukasa, K.; Yoshimitsu, M.; Kosai, K.; Takao, S. Efficient elimination of pancreatic cancer stem cells by hedgehog/GLI inhibitor GANT61 in combination with mTOR inhibition. Mol. Cancer 2016, 15, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desch, P.; Asslaber, D.; Kern, D.; Schnidar, H.; Mangelberger, D.; Alinger, B.; Stoecher, M.; Hofbauer, S.W.; Neureiter, D.; Tinhofer, I.; et al. Inhibition of GLI, but not Smoothened, induces apoptosis in chronic lymphocytic leukemia cells. Oncogene 2010, 29, 4885–4895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Han, C.; Lu, L.; Magliato, S.; Wu, T. Hedgehog signaling pathway regulates autophagy in human hepatocellular carcinoma cells. Hepatology 2013, 58, 995–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant Hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, Q.; Duan, W.; Liu, H.; Xu, H.; Wu, E. Paracrine sonic Hedgehog signaling derived from tumor epithelial cells: A key regulator in the pancreatic tumor microenvironment. Crit. Rev. Eukaryot. Gene Expr. 2012, 22, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Frias-Aldeguer, J.; Hermann, P.C.; Heeschen, C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle 2012, 11, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Saiyin, H.; Fu, D.; Li, J. Stroma—A double-edged sword in pancreatic cancer: A lesson from targeting stroma in pancreatic cancer with Hedgehog signaling inhibitors. Pancreas 2018, 47, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Rucki, A.A.; Foley, K.; Zhang, P.; Xiao, Q.; Kleponis, J.; Wu, A.A.; Sharma, R.; Mo, G.; Liu, A.; Van Eyk, J.; et al. Heterogeneous stromal signaling within the tumor microenvironment controls the metastasis of pancreatic cancer. Cancer Res. 2017, 77, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006, 12, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Vachtenheim, J.; Ondrusova, L. Microphthalmia-associated transcription factor expression levels in melanoma cells contribute to cell invasion and proliferation. Exp. Dermatol. 2015, 24, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Santini, R.; Vinci, M.C.; Pandolfi, S.; Penachioni, J.Y.; Montagnani, V.; Olivito, B.; Gattai, R.; Pimpinelli, N.; Gerlini, G.; Borgognoni, L.; et al. Hedgehog-Gli signaling drives self-renewal and tumorigenicity of human melanoma-initiating cells. Stem Cells 2012, 30, 1808–1818. [Google Scholar] [CrossRef] [PubMed]
- Vlckova, K.; Vachtenheim, J.; Reda, J.; Horak, P.; Ondrusova, L. Inducibly decreased MITF levels do not affect proliferation and phenotype switching but reduce differentiation of melanoma cells. J. Cell Mol. Med. 2018, 22, 2240–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Réda, J.; Vachtenheim, J.; Vlčková, K.; Horák, P.; Vachtenheim, J., Jr.; Ondrušová, L. Widespread Expression of Hedgehog Pathway Components in a Large Panel of Human Tumor Cells and Inhibition of Tumor Growth by GANT61: Implications for Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 2682. https://doi.org/10.3390/ijms19092682
Réda J, Vachtenheim J, Vlčková K, Horák P, Vachtenheim J Jr., Ondrušová L. Widespread Expression of Hedgehog Pathway Components in a Large Panel of Human Tumor Cells and Inhibition of Tumor Growth by GANT61: Implications for Cancer Therapy. International Journal of Molecular Sciences. 2018; 19(9):2682. https://doi.org/10.3390/ijms19092682
Chicago/Turabian StyleRéda, Jiri, Jiri Vachtenheim, Kateřina Vlčková, Pavel Horák, Jiri Vachtenheim, Jr., and Lubica Ondrušová. 2018. "Widespread Expression of Hedgehog Pathway Components in a Large Panel of Human Tumor Cells and Inhibition of Tumor Growth by GANT61: Implications for Cancer Therapy" International Journal of Molecular Sciences 19, no. 9: 2682. https://doi.org/10.3390/ijms19092682
APA StyleRéda, J., Vachtenheim, J., Vlčková, K., Horák, P., Vachtenheim, J., Jr., & Ondrušová, L. (2018). Widespread Expression of Hedgehog Pathway Components in a Large Panel of Human Tumor Cells and Inhibition of Tumor Growth by GANT61: Implications for Cancer Therapy. International Journal of Molecular Sciences, 19(9), 2682. https://doi.org/10.3390/ijms19092682