Peptides as Therapeutic Agents for Inflammatory-Related Diseases


Abstract
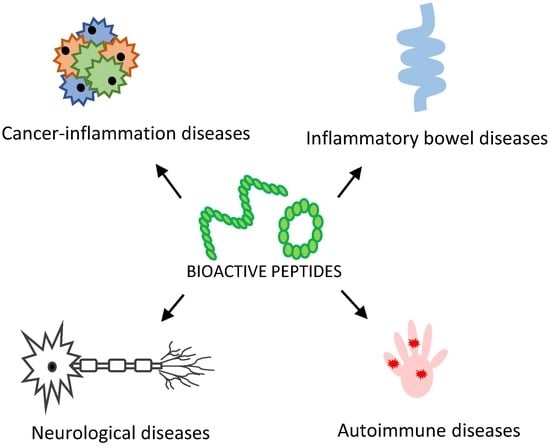
:
1. Introduction
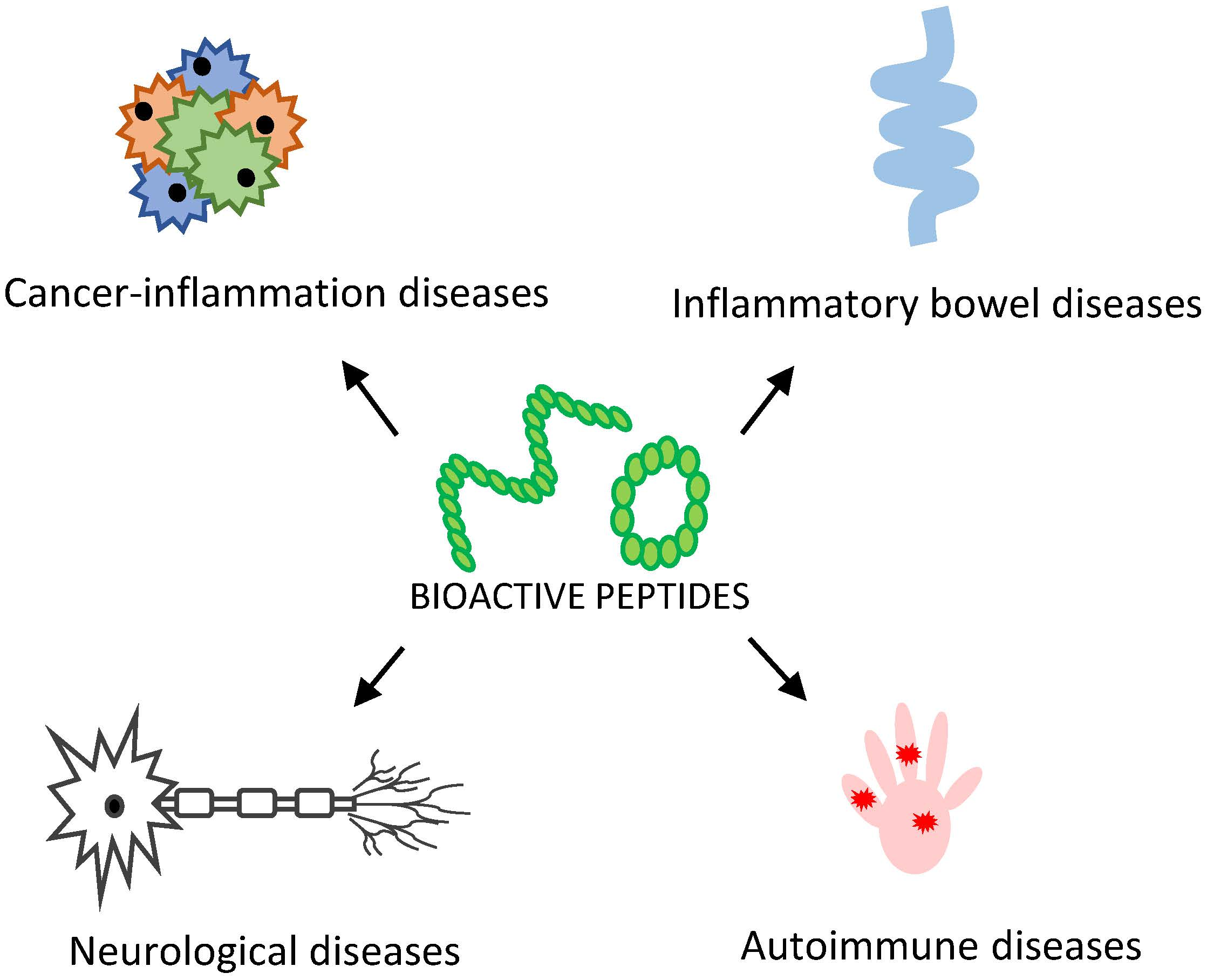
2. Peptides and Peptidomimetics as Modulators of Inflammation
2.1. Cancer-Inflammation Diseases
2.1.1. SOCS1/SOCS3 Peptidomimetics
SOCS1 Mimetics
SOCS3 Mimetics
2.1.2. Aminopeptidase N-term Inhibition in Cancer
2.2. Inflammatory Bowel Diseases (IBD)
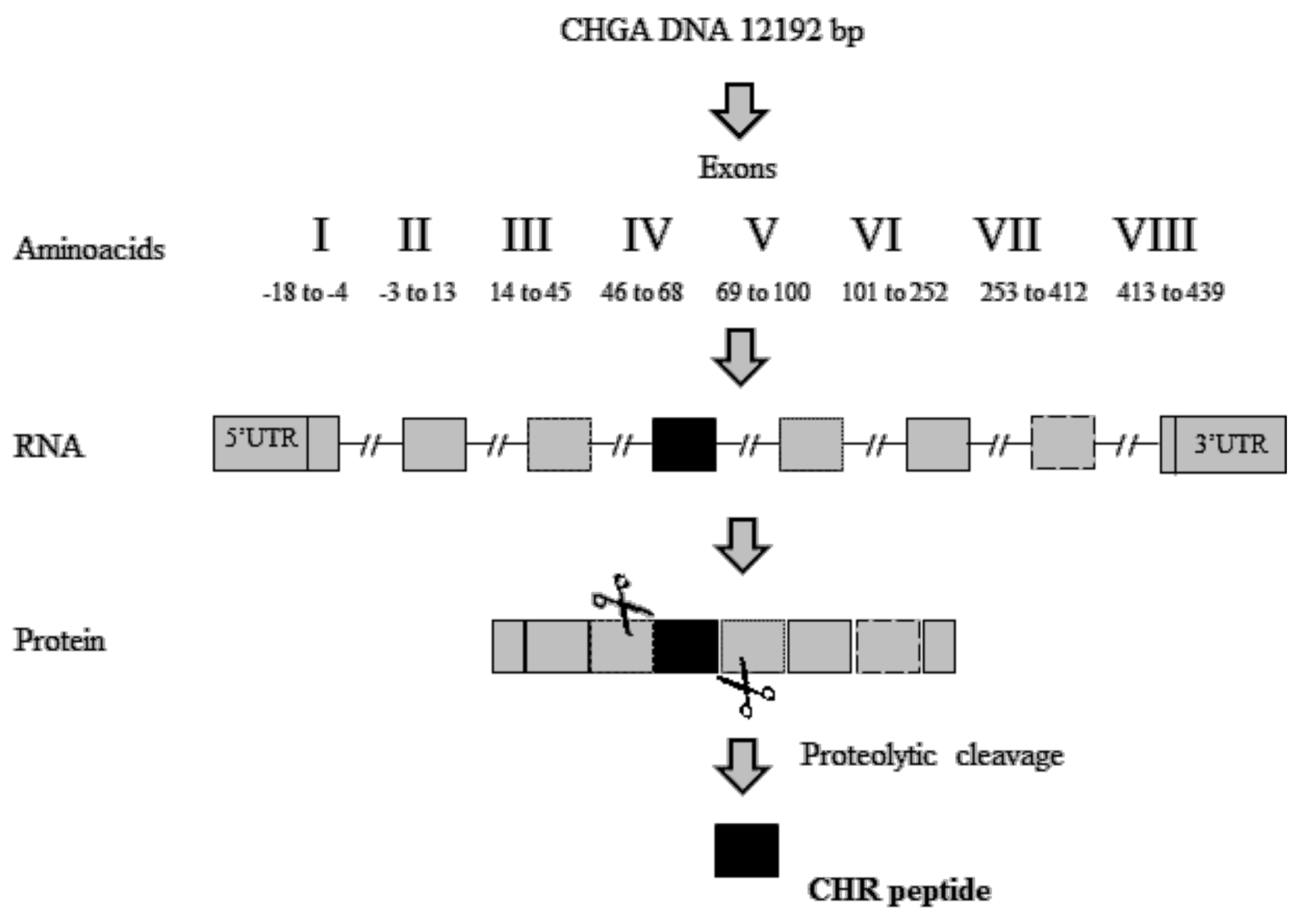
2.2.1. Chromofungin (CHR: CHGA47–66)
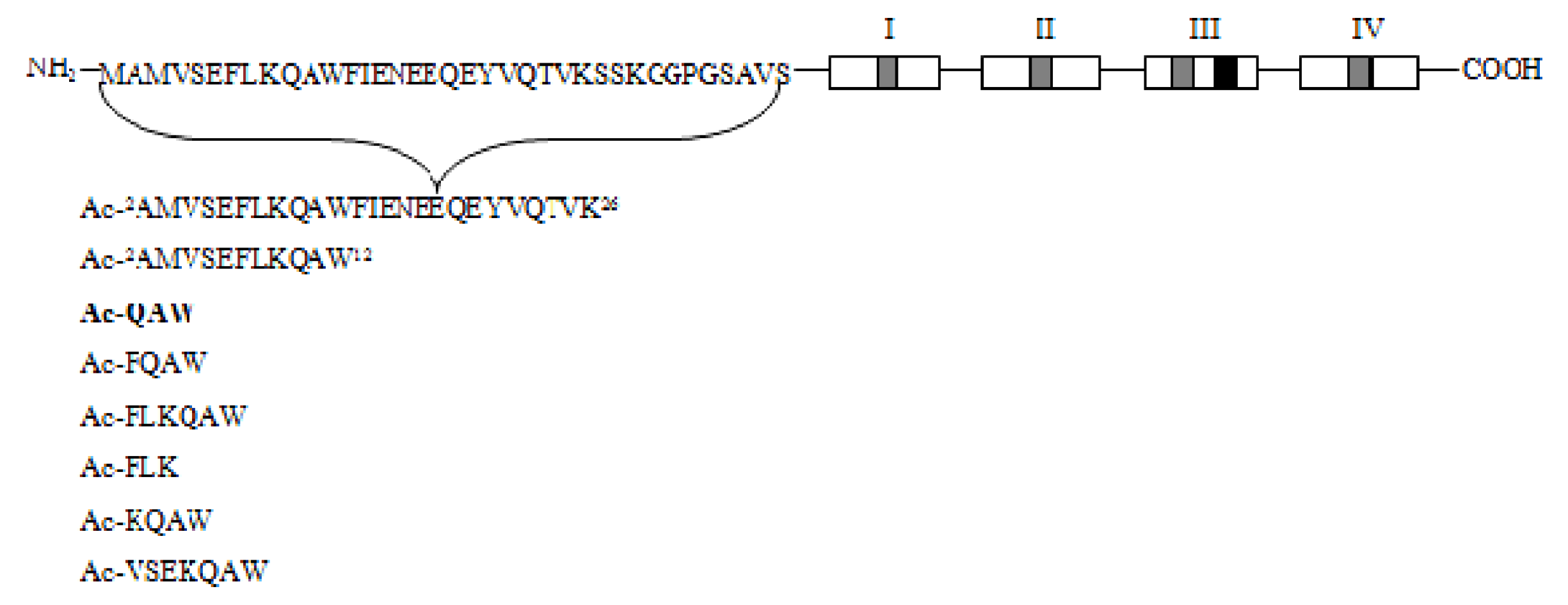
2.2.2. MC-12 Derived Peptides
2.3. Autoimmune Disease
2.3.1. IL-15 Antagonist Peptide
2.3.2. Cyclotide [T20K]kalata B1 in Multiple Sclerosis
2.4. Neurological Diseases
2.4.1. Neural Cell Adhesion Molecule (NCAM)-Derived Mimetic Peptide and Demyelinating Neurological Diseases
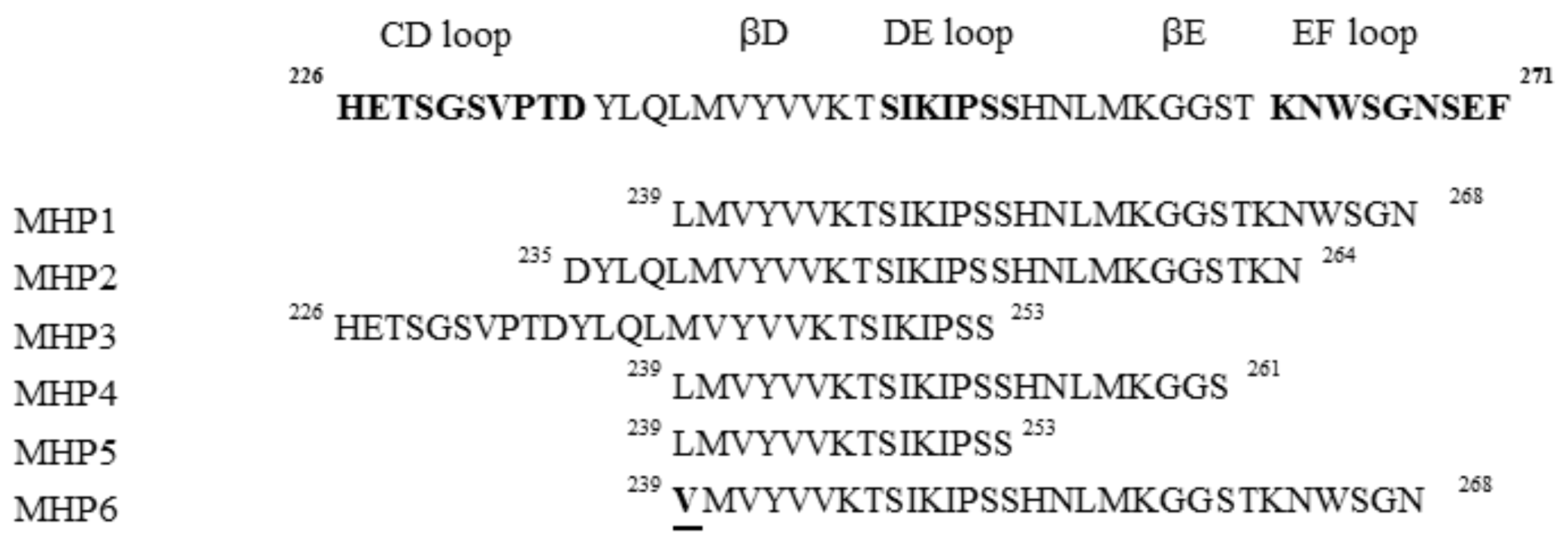
2.4.2. Microglial Healing Peptide 1(MHP1) in Ischemic Stroke
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wu, Y.; Antony, S.; Meitzler, J.L.; Doroshow, J.H. Molecular mechanisms underlying chronic inflammation-associated cancers. Cancer Lett. 2014, 345, 164–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calder, P.C.; Albers, R.; Antoine, J.M.; Blum, S.; Bourdet-Sicard, R.; Ferns, G.A.; Folkerts, G.; Friedmann, P.S.; Frost, G.S.; Guarner, F.; et al. Inflammatory disease processes and interactions with nutrition. Br. J. Nutr. 2009, 101, S1–S45. [Google Scholar] [CrossRef] [PubMed]
- Okin, D.; Medzhitov, R. Evolution of inflammatory diseases. Curr. Biol. 2012, 22, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Alam, Q.; Alam, M.Z.; Mushtaq, G.; Damanhouri, G.A.; Rasool, M.; Kamal, M.A.; Haque, A. Inflammatory Process in Alzheimer's and Parkinson's Diseases: Central Role of Cytokines. Curr. Pharm. Des. 2016, 22, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Korniluk, A.; Koper, O.; Kemona, H.; Dymicka-Piekarska, V. From inflammation to cancer. Irish J. Med. Sci. 2017, 186, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Ouzounova, M.; Lee, E.; Piranlioglu, R.; El Andaloussi, A.; Kolhe, R.; Demirci, M.F.; Marasco, D.; Asm, I.; Chadli, A.; Hassan, K.A.; et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat. Commun. 2017, 8, 14979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- La Manna, S.; Scognamiglio, P.L.; Di Natale, C.; Leone, M.; Mercurio, F.A.; Malfitano, A.M.; Cianfarani, F.; Madonna, S.; Caravella, S.; Albanesi, C.; et al. Characterization of linear mimetic peptides of Interleukin-22 from dissection of protein interfaces. Biochimie 2017, 138, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Delgado, M. Endogenous anti-inflammatory neuropeptides and pro-resolving lipid mediators: A new therapeutic approach for immune disorders. J. Cell. Mol. Med. 2008, 12, 1830–1847. [Google Scholar] [CrossRef] [PubMed]
- Perretti, M.; Chiang, N.; La, M.; Fierro, I.M.; Marullo, S.; Getting, S.J.; Solito, E.; Serhan, C.N. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat. Med. 2002, 8, 1296–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, U.; Durr, R.; Koch, J. Peptides as drugs: From screening to application. Curr. Pharm. Biotechnol. 2013, 14, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Flowers, L.O.; Johnson, H.M.; Mujtaba, M.G.; Ellis, M.R.; Haider, S.M.; Subramaniam, P.S. Characterization of a peptide inhibitor of Janus kinase 2 that mimics suppressor of cytokine signaling 1 function. J. Immunol. 2004, 172, 7510–7518. [Google Scholar] [CrossRef] [PubMed]
- Waiboci, L.W.; Ahmed, C.M.; Mujtaba, M.G.; Flowers, L.O.; Martin, J.P.; Haider, M.I.; Johnson, H.M. Both the suppressor of cytokine signaling 1 (SOCS-1) kinase inhibitory region and SOCS-1 mimetic bind to JAK2 autophosphorylation site: Implications for the development of a SOCS-1 antagonist. J. Immunol. 2007, 178, 5058–5068. [Google Scholar] [CrossRef] [PubMed]
- Doti, N.; Scognamiglio, P.L.; Madonna, S.; Scarponi, C.; Ruvo, M.; Perretta, G.; Albanesi, C.; Marasco, D. New mimetic peptides of the kinase-inhibitory region (KIR) of SOCS1 through focused peptide libraries. Biochem. J. 2012, 443, 231–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Manna, S.; Lopez-Sanz, L.; Leone, M.; Brandi, P.; Scognamiglio, P.L.; Morelli, G.; Novellino, E.; Gomez-Guerrero, C.; Marasco, D. Structure-activity studies of peptidomimetics based on kinase-inhibitory region of suppressors of cytokine signaling 1. Biopolymers 2017. [Google Scholar] [CrossRef] [PubMed]
- La Manna, S.; Lee, E.; Ouzounova, M.; Di Natale, C.; Novellino, E.; Merlino, A.; Korkaya, H.; Marasco, D. Mimetics of Suppressor of cytokine signalling 3: Novel potential therapeutics in triple breast cancer. Int. J. Cancer. 2018. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Chen, L.; Winter, M.B.; Lin, Y.L.; Yang, Y.; Shapovalova, M.; Smith, P.M.; Liu, C.; Li, F.; LeBeau, A.M. The Rational Design of Therapeutic Peptides for Aminopeptidase N using a Substrate-Based Approach. Sci. Rep. 2017, 7, 1424. [Google Scholar] [CrossRef] [PubMed]
- Eissa, N.; Hussein, H.; Kermarrec, L.; Grover, J.; Metz-Boutigue, M.E.; Bernstein, C.N.; Ghia, J.E. Chromofungin Ameliorates the Progression of Colitis by Regulating Alternatively Activated Macrophages. Front. Immunol. 2017, 8, 1131. [Google Scholar] [CrossRef] [PubMed]
- Cobos Caceres, C.; Bansal, P.S.; Navarro, S.; Wilson, D.; Don, L.; Giacomin, P.; Loukas, A.; Daly, N.L. An engineered cyclic peptide alleviates symptoms of inflammation in a murine model of inflammatory bowel disease. J. Biol. Chem. 2017, 292, 10288–10294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, A.; Cabrales, A.; Reyes, O.; Gerónimo, H.; Rodríguez, Y.; Garay, H.; Arrieta, C.; Silva, R.; Guillén, G. Identification of an interleukin-15 antagonist peptide that binds to IL-15Rα. Biotecnología Aplicada 2008, 25, 320–324. [Google Scholar]
- Grundemann, C.; Thell, K.; Lengen, K.; Garcia-Kaufer, M.; Huang, Y.H.; Huber, R.; Craik, D.J.; Schabbauer, G.; Gruber, C.W. Cyclotides Suppress Human T-Lymphocyte Proliferation by an Interleukin 2-Dependent Mechanism. PLoS ONE 2013, 8, e68016. [Google Scholar] [CrossRef] [PubMed]
- Zellinger, C.; Salvamoser, J.D.; Seeger, N.; Russmann, V.; Potschka, H. Impact of the neural cell adhesion molecule-derived peptide FGL on seizure progression and cellular alterations in the mouse kindling model. ACS Chem. Neurosci. 2014, 5, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Kurinami, H.; Shimamura, M.; Nakagami, H.; Shimizu, H.; Koriyama, H.; Kawano, T.; Wakayama, K.; Mochizuki, H.; Rakugi, H.; Morishita, R. A Novel Therapeutic Peptide as a Partial Agonist of RANKL in Ischemic Stroke. Sci. Rep. 2016, 6, 38062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, X.; Gestwicki, J.E. Inhibitors of protein-protein interactions (PPIs): An analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 2018, 44, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Ciemny, M.; Kurcinski, M.; Kamel, K.; Kolinski, A.; Alam, N.; Schueler-Furman, O.; Kmiecik, S. Protein-peptide docking: Opportunities and challenges. Drug Discov. Today 2018, 23, 1530–1537. [Google Scholar] [CrossRef] [PubMed]
- Scognamiglio, P.L.; Morelli, G.; Marasco, D. Synthetic and structural routes for the rational conversion of peptides into small molecules. Methods Mol. Biol. 2015, 1268, 159–193. [Google Scholar] [PubMed]
- Marasco, D.; Scognamiglio, P.L. Identification of inhibitors of biological interactions involving intrinsically disordered proteins. Int. J. Mol. Sci. 2015, 16, 7394–7412. [Google Scholar] [CrossRef] [PubMed]
- Milroy, L.G.; Grossmann, T.N.; Hennig, S.; Brunsveld, L.; Ottmann, C. Modulators of protein-protein interactions. Chem. Rev. 2014, 114, 4695–4748. [Google Scholar] [CrossRef] [PubMed]
- Mahlapuu, M.; Hakansson, J.; Ringstad, L.; Bjorn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef] [PubMed]
- Roviello, G.N.; Vicidomini, C.; Costanzo, V.; Roviello, V. Nucleic acid binding and other biomedical properties of artificial oligolysines. Int. J. Nanomed. 2016, 11, 5897–5904. [Google Scholar] [CrossRef] [PubMed]
- Roviello, G.N.; Musumeci, D.; Roviello, V. Cationic peptides as RNA compaction agents: A study on the polyA compaction activity of a linear alpha, epsilon-oligo-L-lysine. Int. J. Pharm. 2015, 485, 244–248. [Google Scholar] [CrossRef] [PubMed]
- O'Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; He, M.; Chen, T.; Liu, Y.; Tian, Y.L.; Wu, Y.L.; Zhao, Y.; Shen, Y.; Yuan, Z.Y. Multiple roles of SOCS proteins: Differential expression of SOCS1 and SOCS3 in atherosclerosis. Int. J. Mol. Med. 2013, 31, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, N.J.; Murphy, J.M.; Lucet, I.S.; Nicola, N.A.; Babon, J.J. Regulation of Janus kinases by SOCS proteins. Biochem. Soc. Trans. 2013, 41, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Liau, N.P.D.; Laktyushin, A.; Lucet, I.S.; Murphy, J.M.; Yao, S.; Whitlock, E.; Callaghan, K.; Nicola, N.A.; Kershaw, N.J.; Babon, J.J. The molecular basis of JAK-STAT inhibition by SOCS1. Nat. Commun. 2018, 9, 1558. [Google Scholar] [CrossRef] [PubMed]
- Shen-Orr, S.S.; Furman, D.; Kidd, B.A.; Hadad, F.; Lovelace, P.; Huang, Y.W.; Rosenberg-Hasson, Y.; Mackey, S.; Grisar, F.A.; Pickman, Y.; et al. Defective Signaling in the JAK-STAT Pathway Tracks with Chronic Inflammation and Cardiovascular Risk in Aging Humans. Cell Syst. 2016, 3, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, C.M.; Dabelic, R.; Bedoya, S.K.; Larkin, J., 3rd; Johnson, H.M. A SOCS1/3 Antagonist Peptide Protects Mice Against Lethal Infection with Influenza A Virus. Front. Immunol. 2015, 6, 574. [Google Scholar] [CrossRef] [PubMed]
- Balabanov, R.; Strand, K.; Goswami, R.; McMahon, E.; Begolka, W.; Miller, S.D.; Popko, B. Interferon-gamma-oligodendrocyte interactions in the regulation of experimental autoimmune encephalomyelitis. J. Neurosci. 2007, 27, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Ouzounova, M.; Quraishi, A.A.; Davis, A.; Tawakkol, N.; Clouthier, S.G.; Malik, F.; Paulson, A.K.; D'Angelo, R.C.; Korkaya, S.; et al. SOCS3-mediated regulation of inflammatory cytokines in PTEN and p53 inactivated triple negative breast cancer model. Oncogene 2015, 34, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Linossi, E.M.; Babon, J.J.; Hilton, D.J.; Nicholson, S.E. Suppression of cytokine signaling: The SOCS perspective. Cytokine Growth Factor Rev. 2013, 24, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trengove, M.C.; Ward, A.C. SOCS proteins in development and disease. Am. J. Clin. Exp. Immunol. 2013, 2, 1–29. [Google Scholar] [PubMed]
- Flowers, L.O.; Subramaniam, P.S.; Johnson, H.M. A SOCS-1 peptide mimetic inhibits both constitutive and IL-6 induced activation of STAT3 in prostate cancer cells. Oncogene 2005, 24, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, C.M.; Dabelic, R.; Waiboci, L.W.; Jager, L.D.; Heron, L.L.; Johnson, H.M. SOCS-1 mimetics protect mice against lethal poxvirus infection: Identification of a novel endogenous antiviral system. J. Virol. 2009, 83, 1402–1415. [Google Scholar] [CrossRef] [PubMed]
- Mujtaba, M.G.; Flowers, L.O.; Patel, C.B.; Patel, R.A.; Haider, M.I.; Johnson, H.M. Treatment of mice with the suppressor of cytokine signaling-1 mimetic peptide, tyrosine kinase inhibitor peptide, prevents development of the acute form of experimental allergic encephalomyelitis and induces stable remission in the chronic relapsing/remitting form. J. Immunol. 2005, 175, 5077–5086. [Google Scholar] [PubMed]
- Madonna, S.; Scarponi, C.; Morelli, M.; Sestito, R.; Scognamiglio, P.L.; Marasco, D.; Albanesi, C. SOCS3 inhibits the pathological effects of IL-22 in non-melanoma skin tumor-derived keratinocytes. Oncotarget 2017, 8, 24652–24667. [Google Scholar] [CrossRef] [PubMed]
- Madonna, S.; Scarponi, C.; Doti, N.; Carbone, T.; Cavani, A.; Scognamiglio, P.L.; Marasco, D.; Albanesi, C. Therapeutical potential of a peptide mimicking the SOCS1 kinase inhibitory region in skin immune responses. Eur. J. Immunol. 2013, 43, 1883–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, G.; Eickholt, B.J.; Maison, P.; Prinjha, R.; Walsh, F.S.; Doherty, P. A complementary peptide approach applied to the design of novel semaphorin/neuropilin antagonists. J. Neurochem. 2005, 92, 1180–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decaffmeyer, M.; Lins, L.; Charloteaux, B.; VanEyck, M.H.; Thomas, A.; Brasseur, R. Rational design of complementary peptides to the betaAmyloid 29-42 fusion peptide: An application of PepDesign. Biochim. Biophys. Acta 2006, 1758, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, C.M.; Larkin, J., 3rd; Johnson, H.M. SOCS1 Mimetics and Antagonists: A Complementary Approach to Positive and Negative Regulation of Immune Function. Front. Immunol. 2015, 6, 183. [Google Scholar] [CrossRef] [PubMed]
- Recio, C.; Oguiza, A.; Lazaro, I.; Mallavia, B.; Egido, J.; Gomez-Guerrero, C. Suppressor of cytokine signaling 1-derived peptide inhibits Janus kinase/signal transducers and activators of transcription pathway and improves inflammation and atherosclerosis in diabetic mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1953–1960. [Google Scholar] [CrossRef] [PubMed]
- Recio, C.; Lazaro, I.; Oguiza, A.; Lopez-Sanz, L.; Bernal, S.; Blanco, J.; Egido, J.; Gomez-Guerrero, C. Suppressor of Cytokine Signaling-1 Peptidomimetic Limits Progression of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Marasco, D.; Perretta, G.; Sabatella, M.; Ruvo, M. Past and Future Perspectives of Synthetic Peptide Libraries. Curr. Protein Pept. Sci. 2008, 9, 447–467. [Google Scholar] [CrossRef] [PubMed]
- Humet, M.; Carbonell, T.; Masip, I.; Sanchez-Baeza, F.; Mora, P.; Canton, E.; Gobernado, M.; Abad, C.; Perez-Paya, E.; Messeguer, A. A positional scanning combinatorial library of peptoids as a source of biological active molecules: Identification of antimicrobials. J. Comb. Chem. 2003, 5, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Madonna, S.; Scarponi, C.; Sestito, R.; Doti, N.; Carbone, T.; Nasorri, F.; Marasco, D.; Cavani, A.; Albanesi, C. Mimetic peptides of suppressor of cytokine signaling (SOCS)1 impair inflammatory responses of epidermal keratinocytes in vitro and in a mouse skin model of allergic contact dermatitis. J. Investig. Dermatol. 2011, 131, S11. [Google Scholar]
- Korkaya, H.; Kim, G.I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D'Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell. 2012, 47, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Inagaki-Ohara, K.; Kondo, T.; Ito, M.; Yoshimura, A. SOCS, inflammation, and cancer. Jakstat 2013, 2, e24053. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, N.J.; Murphy, J.M.; Liau, N.P.; Varghese, L.N.; Laktyushin, A.; Whitlock, E.L.; Lucet, I.S.; Nicola, N.A.; Babon, J.J. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat. Struct. Mol. Biol. 2013, 20, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Mucha, A.; Drag, M.; Dalton, J.P.; Kafarski, P. Metallo-aminopeptidase inhibitors. Biochimie 2010, 92, 1509–1529. [Google Scholar] [CrossRef] [PubMed]
- Drinkwater, N.; Lee, J.; Yang, W.; Malcolm, T.R.; McGowan, S. M1 aminopeptidases as drug targets: Broad applications or therapeutic niche? FEBS J. 2017, 284, 1473–1488. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Nakajima, Y.; Onohara, Y.; Takeo, M.; Nakashima, K.; Matsubara, F.; Ito, T.; Yoshimoto, T. Crystal structure of aminopeptidase N (proteobacteria alanyl aminopeptidase) from Escherichia coli and conformational change of methionine 260 involved in substrate recognition. J. Biol. Chem. 2006, 281, 33664–33676. [Google Scholar] [CrossRef] [PubMed]
- Wickstrom, M.; Larsson, R.; Nygren, P.; Gullbo, J. Aminopeptidase N (CD13) as a target for cancer chemotherapy. Cancer Sci. 2011, 102, 501–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyal, U.; Liu, Y.; Bhatta, A.K. Dermatologic manifestations of inflammatory bowel disease: A review. Discov. Med. 2018, 25, 225–233. [Google Scholar] [PubMed]
- Bain, C.C.; Mowat, A.M. Macrophages in intestinal homeostasis and inflammation. Immunol. Rev. 2014, 260, 102–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karrasch, T.; Jobin, C. NF-kappaB and the intestine: Friend or foe? Inflamm. Bowel. Dis. 2008, 14, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.P.; Cheng, Y.; Mahata, S.K.; Corti, A.; Tota, B. Chromogranin A and derived peptides in health and disease. J. Mol. Neurosci. 2012, 48, 347–356. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, M.A.; Ghinassi, B.; Izzicupo, P.; Manzoli, L.; Di Baldassarre, A. Biological function and clinical relevance of chromogranin A and derived peptides. Endocr. Connect. 2014, 3, R45–R54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eissa, N.; Hussein, H.; Kermarrec, L.; Elgazzar, O.; Metz-Boutigue, M.H.; Bernstein, C.N.; Ghia, J.E. Chromofungin (CHR: CHGA47-66) is downregulated in persons with active ulcerative colitis and suppresses pro-inflammatory macrophage function through the inhibition of NF-kappaB signaling. Biochem. Pharmacol. 2017, 145, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.L.; Koh, T.J. Macrophage phenotypes during tissue repair. J. Leukoc. Biol. 2013, 93, 875–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal. Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Grzanka, A.; Misiolek, M.; Golusinski, W.; Jarzab, J. Molecular mechanisms of glucocorticoids action: Implications for treatment of rhinosinusitis and nasal polyposis. Eur. Arch. Oto-Rhino-L 2011, 268, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, N.; Zhu, C.; Zhou, D.; Nie, T.; Go, M.F.; Richards, R.J.; Rigas, B. MC-12, an annexin A1-based peptide, is effective in the treatment of experimental colitis. PLoS ONE 2012, 7, e41585. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, M.; Petretto, A.; Vaglio, A.; Santucci, L.; Candiano, G.; Ghiggeri, G.M. Annexin A1 and Autoimmunity: From Basic Science to Clinical Applications. Int. J. Mol. Sci. 2018, 19, 1348. [Google Scholar] [CrossRef] [PubMed]
- Scannell, M.; Flanagan, M.B.; deStefani, A.; Wynne, K.J.; Cagney, G.; Godson, C.; Maderna, P. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J. Immunol. 2007, 178, 4595–4605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Huang, L.; Zhao, W.; Rigas, B. Annexin 1 induced by anti-inflammatory drugs binds to NF-kappaB and inhibits its activation: Anticancer effects in vitro and in vivo. Cancer Res. 2010, 70, 2379–2388. [Google Scholar] [CrossRef] [PubMed]
- Villadsen, L.S.; Schuurman, J.; Beurskens, F.; Dam, T.N.; Dagnaes-Hansen, F.; Skov, L.; Rygaard, J.; Voorhorst-Ogink, M.M.; Gerritsen, A.F.; van Dijk, M.A.; et al. Resolution of psoriasis upon blockade of IL-15 biological activity in a xenograft mouse model. J. Clin. Investig. 2003, 112, 1571–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giri, J.G.; Kumaki, S.; Ahdieh, M.; Friend, D.J.; Loomis, A.; Shanebeck, K.; DuBose, R.; Cosman, D.; Park, L.S.; Anderson, D.M. Identification and cloning of a novel IL-15 binding protein that is structurally related to the alpha chain of the IL-2 receptor. EMBO J. 1995, 14, 3654–3663. [Google Scholar] [CrossRef] [PubMed]
- Abadie, V.; Jabri, B. IL-15: A central regulator of celiac disease immunopathology. Immunol. Rev. 2014, 260, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Savio, A.S.; Acosta, O.R.; Perez, H.G.; Alvarez, Y.R.; Chico, A.; Ojeda, M.O.; Aguero, C.A.; Estevez, M.; Nieto, G.G. Enhancement of the inhibitory effect of an IL-15 antagonist peptide by alanine scanning. J. Pept. Sci. 2012, 18, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Alvarez, Y.; Cabrales-Rico, A.; Perera-Pintado, A.; Prats-Capote, A.; Garay-Perez, H.E.; Reyes-Acosta, O.; Perez-Garcia, E.; Chico-Capote, A.; Santos-Savio, A. In vitro and in vivo characterization of an interleukin-15 antagonist peptide by metabolic stability, Tc-99m-labeling, and biological activity assays. J. Pept. Sci. 2018, 24, e3078. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.K.; Xu, W.D.; Leng, R.X.; Liang, Y.; Liu, Y.Y.; Fang, X.Y.; Feng, C.C.; Li, R.; Cen, H.; Pan, H.F.; et al. Therapeutic potential of IL-15 in rheumatoid arthritis. Hum. Immunol. 2015, 76, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, S.M.; Sando, L.; Rosengren, K.J.; Wang, C.K.; Colgrave, M.L.; Daly, N.L.; Craik, D.J. Alanine scanning mutagenesis of the prototypic cyclotide reveals a cluster of residues essential for bioactivity. J. Biol. Chem. 2008, 283, 9805–9813. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Colgrave, M.L.; Clark, R.J.; Kotze, A.C.; Craik, D.J. Lysine-scanning mutagenesis reveals an amendable face of the cyclotide kalata B1 for the optimization of nematocidal activity. J. Biol. Chem. 2010, 285, 10797–10805. [Google Scholar] [CrossRef] [PubMed]
- Thell, K.; Hellinger, R.; Sahin, E.; Michenthaler, P.; Gold-Binder, M.; Haider, T.; Kuttke, M.; Liutkeviciute, Z.; Goransson, U.; Grundemann, C.; et al. Oral activity of a nature-derived cyclic peptide for the treatment of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 3960–3965. [Google Scholar] [CrossRef] [PubMed]
- Gadani, S.P.; Cronk, J.C.; Norris, G.T.; Kipnis, J. IL-4 in the Brain: A Cytokine To Remember. J. Immunol. 2012, 189, 4213–4219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, A.; Aiello, C.; Grieco, P.; Marasco, D. Targeting “Undruggable” Proteins: Design of Synthetic Cyclopeptides. Curr. Med. Chem. 2016, 23, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Kiselyov, V.V.; Skladchikova, G.; Hinsby, A.M.; Jensen, P.H.; Kulahin, N.; Soroka, V.; Pedersen, N.; Tsetlin, V.; Poulsen, F.M.; Berezin, V.; et al. Structural basis for a direct interaction between FGFR1 and NCAM and evidence for a regulatory role of ATP. Structure 2003, 11, 691–701. [Google Scholar] [CrossRef]
- Secher, T.; Novitskaia, V.; Berezin, V.; Bock, E.; Glenthoj, B.; Klementiev, B. A neural cell adhesion molecule-derived fibroblast growth factor receptor agonist, the FGL-peptide, promotes early postnatal sensorimotor development and enhances social memory retention. Neuroscience 2006, 141, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Aonurm-Helm, A.; Berezin, V.; Bock, E.; Zharkovsky, A. NCAM-mimetic, FGL peptide, restores disrupted fibroblast growth factor receptor (FGFR) phosphorylation and FGFR mediated signaling in neural cell adhesion molecule (NCAM)-deficient mice. Brain Res. 2010, 1309, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Downer, E.J.; Cowley, T.R.; Lyons, A.; Mills, K.H.; Berezin, V.; Bock, E.; Lynch, M.A. A novel anti-inflammatory role of NCAM-derived mimetic peptide, FGL. Neurobiol. Aging 2010, 31, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Asua, D.; Bougamra, G.; Calleja-Felipe, M.; Morales, M.; Knafo, S. Peptides Acting as Cognitive Enhancers. Neuroscience 2018, 370, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Lakhan, S.E.; Kirchgessner, A.; Hofer, M. Inflammatory mechanisms in ischemic stroke: Therapeutic approaches. J. Transl. Med. 2009, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Doti, N.; Reuther, C.; Scognamiglio, P.L.; Dolga, A.M.; Plesnila, N.; Ruvo, M.; Culmsee, C. Inhibition of the AIF/CypA complex protects against intrinsic death pathways induced by oxidative stress. Cell Death Dis. 2014, 5, e993. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, M.; Nakagami, H.; Osako, M.K.; Kurinami, H.; Koriyama, H.; Zhengda, P.; Tomioka, H.; Tenma, A.; Wakayama, K.; Morishita, R. OPG/RANKL/RANK axis is a critical inflammatory signaling system in ischemic brain in mice. Proc. Natl. Acad. Sci. USA 2014, 111, 8191–8196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, C.A.; Warren, J.T.; Wang, M.W.H.; Teitelbaum, S.L.; Fremont, D.H. RANKL Employs Distinct Binding Modes to Engage RANK and the Osteoprotegerin Decoy Receptor. Structure 2012, 20, 1971–1982. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Wakabayashi, K.; Ubukata, O.; Hayashi, S.; Okada, F.; Hata, T. Crystal structure of the extracellular domain of mouse RANK ligand at 2.2-A resolution. J. Biol. Chem. 2002, 277, 6631–6636. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.F.; Oey, I.; Bremer, P.; Carne, A.; Silcock, P. Bioactive peptides derived from egg proteins: A review. Crit. Rev. Food Sci. Nutr. 2017, 13, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Tornesello, A.L.; Buonaguro, L.; Tornesello, M.L.; Buonaguro, F.M. New Insights in the Design of Bioactive Peptides and Chelating Agents for Imaging and Therapy in Oncology. Molecules 2017, 22, 1282. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
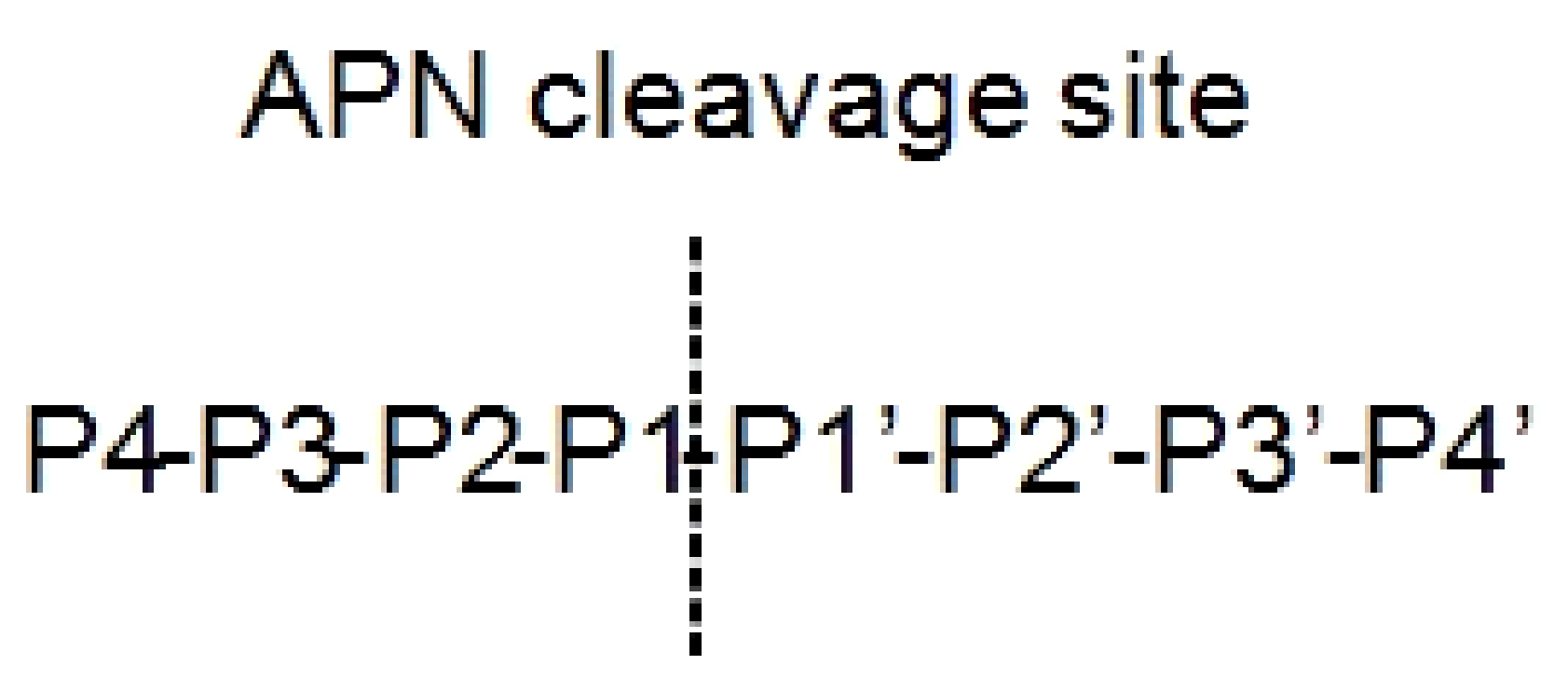{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequences | Activity | Diseases | References |
|---|---|---|---|---|
| Tkip | WLVFFVIFYFFR | Inhibition of JAK-STAT pathway | inflammatory disease, Autoimmune encephalitis | [13] |
| SOCS1-KIR | DTHFRTFRSHSDYRRI | [14] | ||
| PS-5 | DTC(Acm)RQTFRSH | Type-1 skin, cardiovascular diseases | [15] | |
| Cyclic PS5 |  | [16] | ||
| Linear PS5 Nal1 | AcDTC(Acm)RQTNalRSH | [16] | ||
| Cyclic PS5 Nal1 |  | [16] | ||
| KIRESS | LKTFSSKSEYQLVVNAVRKLQESG | Triple.negativ-e breast cancer | [17] | |
| cyc-LHSPW |  | Inhibition of APN | Neuroendocrine prostate cancer | [18] |
| Chromofungin (CHR: CHGA47-66) | RILSILRHQNLLKELQDLAL | Regulation of alternatively activated macrophages | Inflammatory bowel disease (UC) | [19] |
| Bi-cyc-MC-12 |  | Inhibition of NF-κB expression | Inflammatory bowel disease | [20] |
| [K6T]P8 peptide | KVTAMTCFLL | Inhibition of IL-15R | Rheumatoid Arthritis | [21] |
| Cyclotide [T20K]kalata B1 |  | Inhibition lymphocyte proliferation | Multiple sclerosis | [22] |
| FGL | EVYVVAENQQGKSKA | Stimulate the secretion of IL-4 | Demyelinating Neurological Diseases | [23] |
| MHP1 | LMVYVVKTSIKIPSSHNLMKGGSTKNWSGN | Inhibition of LPS-induced cytokine | Ischemical stroke | [24] |
| Name | KD (μM) |
|---|---|
| KIR | 2.03 |
| ESS | >>1000 |
| KIRESS | 1.86 |
| Sequence | IC50 (μM) | Ki (μM) |
|---|---|---|
| NorHSPW | 6.5 | - |
| AHSPW | 9.4 | - |
| LHSPW | 10.6 | - |
| Cyc-LHSPW | - | 24.7 |
| Name | Sequence | % Peptide Remaining in Serum |
|---|---|---|
| SFTI-1 |  | ≈ 100 |
| Bi-cyc-MC-12 |  | ≈ 100 |
| Mono-cyc-MC-12 (n) |  | ≈ 60 |
| Mono-cyc -MC-12 (p) |  | ≈ 60 |
| Mono-cyc -MC-12 (l2) |  | ≈ 60 |
| Name | Sequence | IC50 (µM) |
|---|---|---|
| P8 | KVTAMKCFLL | 130 |
| P8 dimer | KVTAMKCFLLdimer | 24 |
| [K1A]P8 | AVTAMKCFLL | ND |
| [V2A]P8 | KATAMKCFLL | 130 |
| [T3A]P8 | KVAAMKCFLL | 130 |
| [M5A]P8 | KVTAAKCFLL | 130 |
| [K6A]P8 | KVTAMACFLL | ND |
| [C7A]P8 | KVTAMKAFLL | inactive |
| [F8A]P8 | KVTAMKCALL | inactive |
| [L9A]P8 | KVTAMKCFAL | 200 |
| [L10A]P8 | KVTAMKCFLA | 260 |
| [C7S]P8 | KVTAMKSFLL | inactive |
| [K6E]P8 | KVTAMECFLL | inactive |
| [K6T]P8 | KVTAMTCFLL | 24.6 |
| [K6T]P8 dimer | KVTAMTCFLLdimer | 8.0 |
| Peptide | IC50 (µM) Lymphocytes (PBMCs) | IC50 (µM) Purified T-cells |
|---|---|---|
| native kalata B1 | 2.9 | 2.4 |
| [T8K] | Inactive | - |
| [V10A] | Inactive | - |
| [V10K] | Inactive | - |
| [G18K] | 4.4 | 3.2 |
| [T20K] | 1.9 | 2.7 |
| [N29K] | 3.2 | 2.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Manna, S.; Di Natale, C.; Florio, D.; Marasco, D. Peptides as Therapeutic Agents for Inflammatory-Related Diseases. Int. J. Mol. Sci. 2018, 19, 2714. https://doi.org/10.3390/ijms19092714
La Manna S, Di Natale C, Florio D, Marasco D. Peptides as Therapeutic Agents for Inflammatory-Related Diseases. International Journal of Molecular Sciences. 2018; 19(9):2714. https://doi.org/10.3390/ijms19092714
Chicago/Turabian StyleLa Manna, Sara, Concetta Di Natale, Daniele Florio, and Daniela Marasco. 2018. "Peptides as Therapeutic Agents for Inflammatory-Related Diseases" International Journal of Molecular Sciences 19, no. 9: 2714. https://doi.org/10.3390/ijms19092714
APA StyleLa Manna, S., Di Natale, C., Florio, D., & Marasco, D. (2018). Peptides as Therapeutic Agents for Inflammatory-Related Diseases. International Journal of Molecular Sciences, 19(9), 2714. https://doi.org/10.3390/ijms19092714