STAT3 in Breast Cancer Onset and Progression: A Matter of Time and Context



Abstract
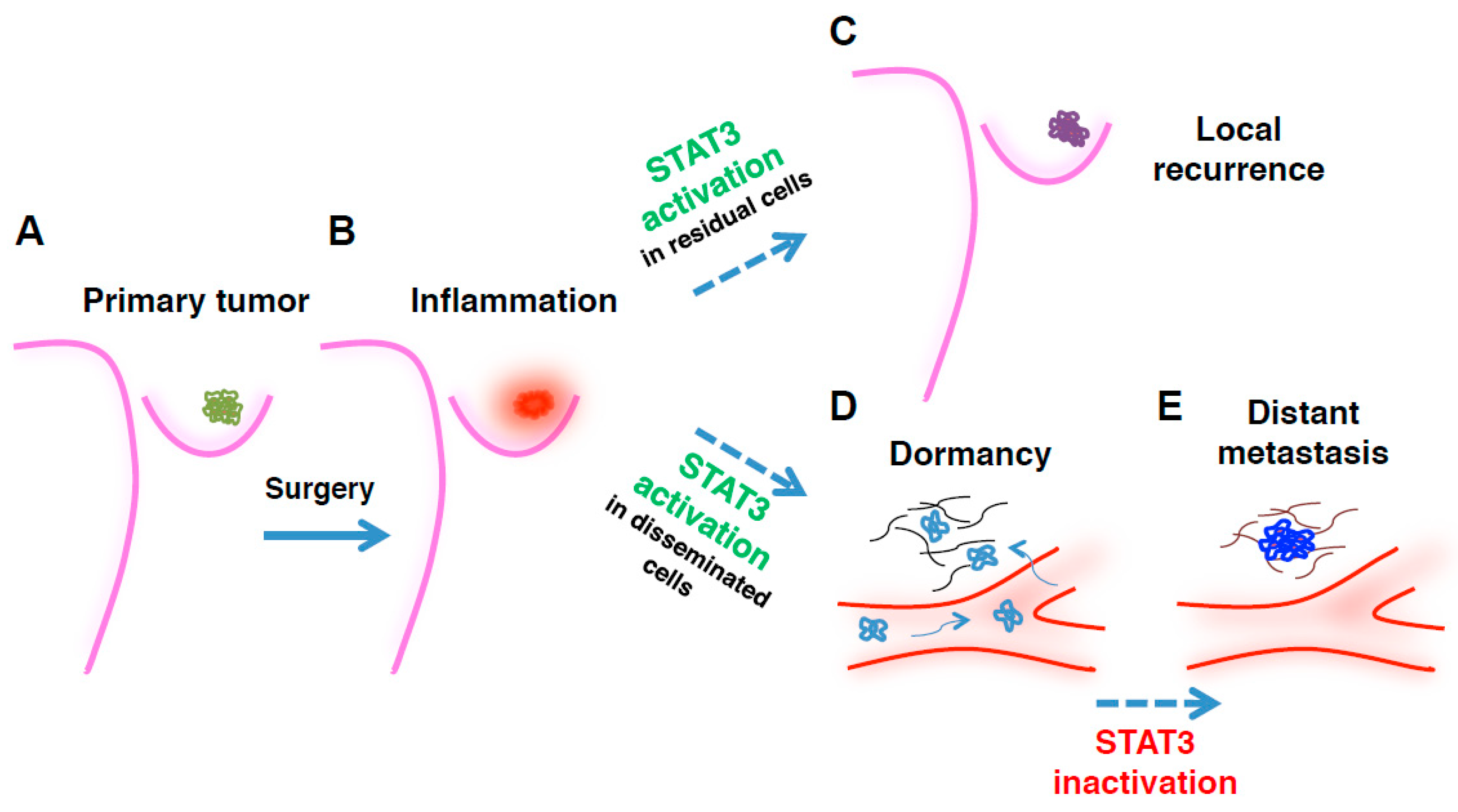
:1. Introduction
2. STAT3 in Anti-Cancer Strategies
3. STAT3 in Normal Mammary Gland and Development
4. STAT3 in Surgery-Induced Inflammation and Breast Cancer Local Recurrence
5. STAT3 in Breast Cancer Distant Metastases
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Avalle, L.; Camporeale, A.; Camperi, A.; Poli, V. STAT3 in cancer: A double edged sword. Cytokine 2017, 98, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.S.; Sikka, S.; Surana, R.; Dai, X.; Zhang, J.; Kumar, A.P.; Tan, B.K.; Sethi, G.; Bishayee, A. Targeting the STAT3 signaling pathway in cancer: Role of synthetic and natural inhibitors. Biochim. Biophys. Acta 2014, 1845, 136–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J. Signal transducers and activators of transcription as regulators of growth, apoptosis and breast development. Breast Cancer Res. BCR 2000, 2, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Hossain, D.M.S.; Dos Santos, C.; Zhang, Q.; Kozlowska, A.; Liu, H.; Gao, C.; Moreira, D.; Swiderski, P.; Jozwiak, A.; Kline, J.; et al. Leukemia cell-targeted STAT3 silencing and TLR9 triggering generate systemic antitumor immunity. Blood 2014, 123, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Eyking, A.; Ey, B.; Rünzi, M.; Roig, A.I.; Reis, H.; Schmid, K.W.; Gerken, G.; Podolsky, D.K.; Cario, E. Toll-like receptor 4 variant D299G induces features of neoplastic progression in Caco-2 intestinal cells and is associated with advanced human colon cancer. Gastroenterology 2011, 141, 2154–2165. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Deng, J.; Kujawski, M.; Yang, C.; Liu, Y.; Herrmann, A.; Kortylewski, M.; Horne, D.; Somlo, G.; Forman, S.; et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 2010, 16, 1421–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, K.; Watson, C.J. The multifaceted role of STAT3 in mammary gland involution and breast cancer. Int. J. Mol. Sci. 2018, 19, 1695. [Google Scholar] [CrossRef] [PubMed]
- Segatto, I.; Berton, S.; Sonego, M.; Massarut, S.; Perin, T.; Piccoli, E.; Colombatti, A.; Vecchione, A.; Baldassarre, G.; Belletti, B. Surgery-induced wound response promotes stem-like and tumor-initiating features of breast cancer cells, via STAT3 signaling. Oncotarget 2014, 5, 6267–6279. [Google Scholar] [CrossRef] [PubMed]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell 2017, 32, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Priego, N.; Zhu, L.; Monteiro, C.; Mulders, M.; Wasilewski, D.; Bindeman, W.; Doglio, L.; Martínez, L.; Martínez-Saez, E.; Ramón y Cajal, S.; et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat. Med. 2018, 24, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Laudisi, F.; Cherubini, F.; Monteleone, G.; Stolfi, C. STAT3 Interactors as potential therapeutic targets for cancer treatment. Int. J. Mol. Sci. 2018, 19, 1787. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J. STAT proteins as novel targets for cancer drug discovery. Expert Opin. Ther. Targets 2004, 8, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Lee, S.H.; Han, S.W.; Kim, M.J.; Kim, T.M.; Kim, T.Y.; Heo, D.S.; Yuasa, M.; Yanagihara, Y.; Bang, Y.J.; et al. Phase I study of OPB-31121, an oral STAT3 inhibitor, in patients with advanced solid tumors. Cancer Res. Treat. 2015, 47, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.L.; Soo, R.A.; Tan, D.S.; Lee, S.C.; Lim, J.S.; Marban, P.C.; Kong, L.R.; Lee, Y.J.; Wang, L.Z.; Thuya, W.L.; et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann. Oncol. 2015, 26, 998–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, S.; Gooding, W.E.; Grandis, J.R. In vivo antitumor efficacy of STAT3 blockade using a transcription factor decoy approach: Implications for cancer therapy. Oncogene 2005, 24, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.S.; Sano, S.; Kiguchi, K.; Anders, J.; Komazawa, N.; Takeda, J.; DiGiovanni, J. Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J. Clin. Investig. 2004, 114, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Odate, S.; Veschi, V.; Yan, S.; Lam, N.; Woessner, R.; Thiele, C.J. Inhibition of STAT3 with the generation 2.5 antisense oligonucleotide, AZD9150, decreases neuroblastoma tumorigenicity and increases chemosensitivity. Clin. Cancer Res. 2017, 23, 1771–1784. [Google Scholar] [CrossRef] [PubMed]
- Verdura, S.; Cuyàs, E.; Llorach-Parés, L.; Pérez-Sánchez, A.; Micol, V.; Nonell-Canals, A.; Joven, J.; Valiente, M.; Sánchez-Martínez, M.; Bosch-Barrera, J.; et al. Silibinin is a direct inhibitor of STAT3. Food Chem. Toxicol. 2018, 116, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, I.; Quaglino, E.; Maritano, D.; Pannellini, T.; Riera, L.; Cavallo, F.; Joven, J.; Valiente, M.; Sánchez-Martínez, M.; Bosch-Barrera, J.; et al. Stat3 is required for anchorage-independent growth and metastasis but not for mammary tumor development downstream of the ErbB-2 oncogene. Mol. Carcinog. 2010, 49, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Furth, P.A. STAT signaling in different breast cancer sub-types. Mol. Cell. Endocrinol. 2014, 382, 612–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resemann, H.K.; Watson, C.J.; Lloyd-Lewis, B. The Stat3 paradox: A killer and an oncogene. Mol. Cell. Endocrinol. 2014, 382, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, F.-C.; Cheng, G.; Lin, J. Evaluation of potential Stat3-regulated genes in human breast cancer. Biochem. Biophys. Res. Commun. 2005, 335, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Ranger, J.J.; Levy, D.E.; Shahalizadeh, S.; Hallett, M.; Muller, W.J. Identification of a Stat3-dependent transcription regulatory network involved in metastatic progression. Cancer Res. 2009, 69, 6823–6830. [Google Scholar] [CrossRef] [PubMed]
- Philp, J.A.; Burdon, T.G.; Watson, C.J. Differential activation of STATs 3 and 5 during mammary gland development. FEBS Lett. 1996, 396, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Pensa, S.; Watson, C.J.; Poli, V. Stat3 and the inflammation/acute phase response in involution and breast cancer. J. Mammary Gland Biol. Neoplasia 2009, 14, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Calautti, E.; Avalle, L.; Poli, V. Psoriasis: A STAT3-Centric View. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Demicheli, R.; Valagussa, P.; Bonadonna, G. Does surgery modify growth kinetics of breast cancer micrometastases? Br. J. Cancer 2001, 85, 490–492. [Google Scholar] [CrossRef] [PubMed]
- Demicheli, R.; Retsky, M.W.; Hrushesky, W.J.; Baum, M. Tumor dormancy and surgery-driven interruption of dormancy in breast cancer: Learning from failures. Nat. Clin. Pract. Oncol. 2007, 4, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Baldassarre, G.; Nicoloso, M.S.; Schiappacassi, M.; Chimienti, E.; Belletti, B. Linking inflammation to cell cycle progression. Curr. Pharm. Des. 2004, 10, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.L.; Ballard-Barbash, R.; Bernstein, L.; Baumgartner, R.N.; Neuhouser, M.L.; Wener, M.H.; Baumgartner, K.B.; Gilliland, F.D.; Sorensen, B.E.; McTiernan, A.; et al. Elevated biomarkers of inflammation are associated with reduced survival among breast cancer patients. J. Clin. Oncol. 2009, 27, 3437–3444. [Google Scholar] [CrossRef] [PubMed]
- Segatto, I.; Berton, S.; Sonego, M.; Massarut, S.; D’Andrea, S.; Perin, T.; Fabris, L.; Armenia, J.; Rampioni, G.; Lovisa, S.; et al. Inhibition of breast cancer local relapse by targeting p70S6 kinase activity. J. Mol. Cell Biol. 2013, 5, 428–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segatto, I.; Berton, S.; Sonego, M.; Massarut, S.; Fabris, L.; Armenia, J.; Mileto, M.; Colombatti, A.; Vecchione, A.; Baldassarre, G.; et al. p70S6 kinase mediates breast cancer cell survival in response to surgical wound fluid stimulation. Mol. Oncol. 2014, 8, 766–780. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.; Finger, E.C.; Olcina, M.M.; Vilalta, M.; Aguilera, T.; Miao, Y.; Merkel, A.R.; Johnson, J.R.; Sterling, J.A.; Wu, J.Y.; et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol. 2016, 18, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.S.; Avivar-Valderas, A.; Estrada, Y.; Bragado, P.; Sosa, M.S.; Aguirre-Ghiso, J.A.; Segall, J.E. Dormancy signatures and metastasis in estrogen receptor positive and negative breast cancer. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Demicheli, R. Tumour dormancy: Findings and hypotheses from clinical research on breast cancer. Semin. Cancer Biol. 2001, 11, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Grabner, B.; Schramek, D.; Mueller, K.M.; Moll, H.P.; Svinka, J.; Hoffmann, T.; Bauer, E.; Blaas, L.; Hruschka, N.; Zboray, K.; et al. Disruption of STAT3 signalling promotes KRAS-induced lung tumorigenesis. Nat. Commun. 2015, 6, 6285. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Inhibitor | Indication | Study Phase | Status | NCT Identifier |
|---|---|---|---|---|
| AZD9150 IONIS-STAT3Rx (STAT3 antisense oligonucleotide) | NSCLC, advanced solid tumors | I/II | Recruiting | NCT03421353 |
| Advanced pancreatic cancer, NSCLC, and CRC | II | Recruiting | NCT02983578 | |
| Advanced/metastatic hepatocellular cancer | I | Completed | NCT01839604 | |
| DLBCL | I | Recruiting | NCT02549651 | |
| Advanced solid tumors, metastatic HNSCC | I/II | Recruiting | NCT02499328 | |
| Advanced tumors, DLBCL, lymphoma | I/II | Completed | NCT01563302 | |
| OPB-31121 (STAT3 SH2 domain) | Advanced cancer, solid tumors | I | Completed | NCT00955812 |
| Advanced solid tumors | I | Unknown | NCT00657176 | |
| Hepatocellular carcinoma | I/II | Completed | NCT01406574 | |
| OPB-51602 (STAT3 SH2 domain) | Advanced tumors | I | Completed | NCT01423903 |
| Multiple myeloma, NHL, AML, ALL, and CML | I | Completed | NCT01344876 | |
| Advanced solid tumors | I | Completed | NCT01184807 | |
| OPB-111077 (STAT3 phosphorylation) | Advanced tumors | I | Completed | NCT01711034 |
| Napabucasin DSP-0337 (STAT3 SH2 domain) | Metastatic pancreatic adenocarcinoma | III | Recruiting | NCT02993731 |
| Metastatic CRC | II | Not yet recruiting | NCT03647839 | |
| Advanced solid tumors | I | Not yet recruiting | NCT03416816 | |
| STAT3 DECOY (STAT3 response element) | HNSCC | Early I | Completed | NCT00696176 |
| TTI-101 (STAT3 SH2 domain) | Advanced tumors | I | Recruiting | NCT03195699 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segatto, I.; Baldassarre, G.; Belletti, B. STAT3 in Breast Cancer Onset and Progression: A Matter of Time and Context. Int. J. Mol. Sci. 2018, 19, 2818. https://doi.org/10.3390/ijms19092818
Segatto I, Baldassarre G, Belletti B. STAT3 in Breast Cancer Onset and Progression: A Matter of Time and Context. International Journal of Molecular Sciences. 2018; 19(9):2818. https://doi.org/10.3390/ijms19092818
Chicago/Turabian StyleSegatto, Ilenia, Gustavo Baldassarre, and Barbara Belletti. 2018. "STAT3 in Breast Cancer Onset and Progression: A Matter of Time and Context" International Journal of Molecular Sciences 19, no. 9: 2818. https://doi.org/10.3390/ijms19092818
APA StyleSegatto, I., Baldassarre, G., & Belletti, B. (2018). STAT3 in Breast Cancer Onset and Progression: A Matter of Time and Context. International Journal of Molecular Sciences, 19(9), 2818. https://doi.org/10.3390/ijms19092818