Glycopolymer Grafted Silica Gel as Chromatographic Packing Materials

Abstract
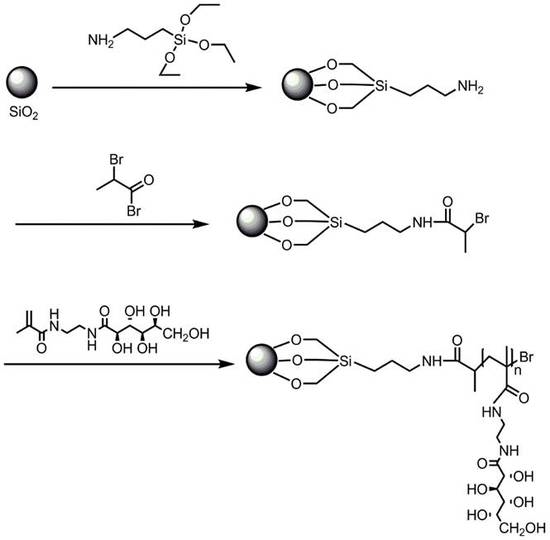
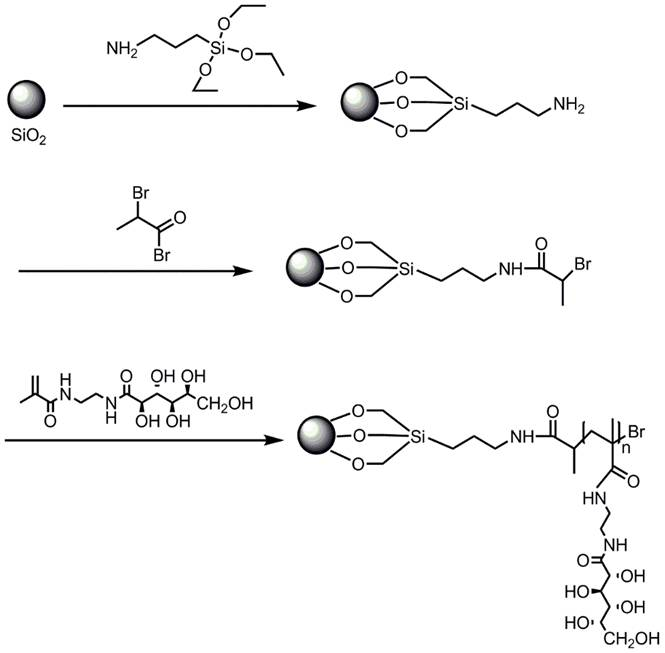
:
1. Introduction
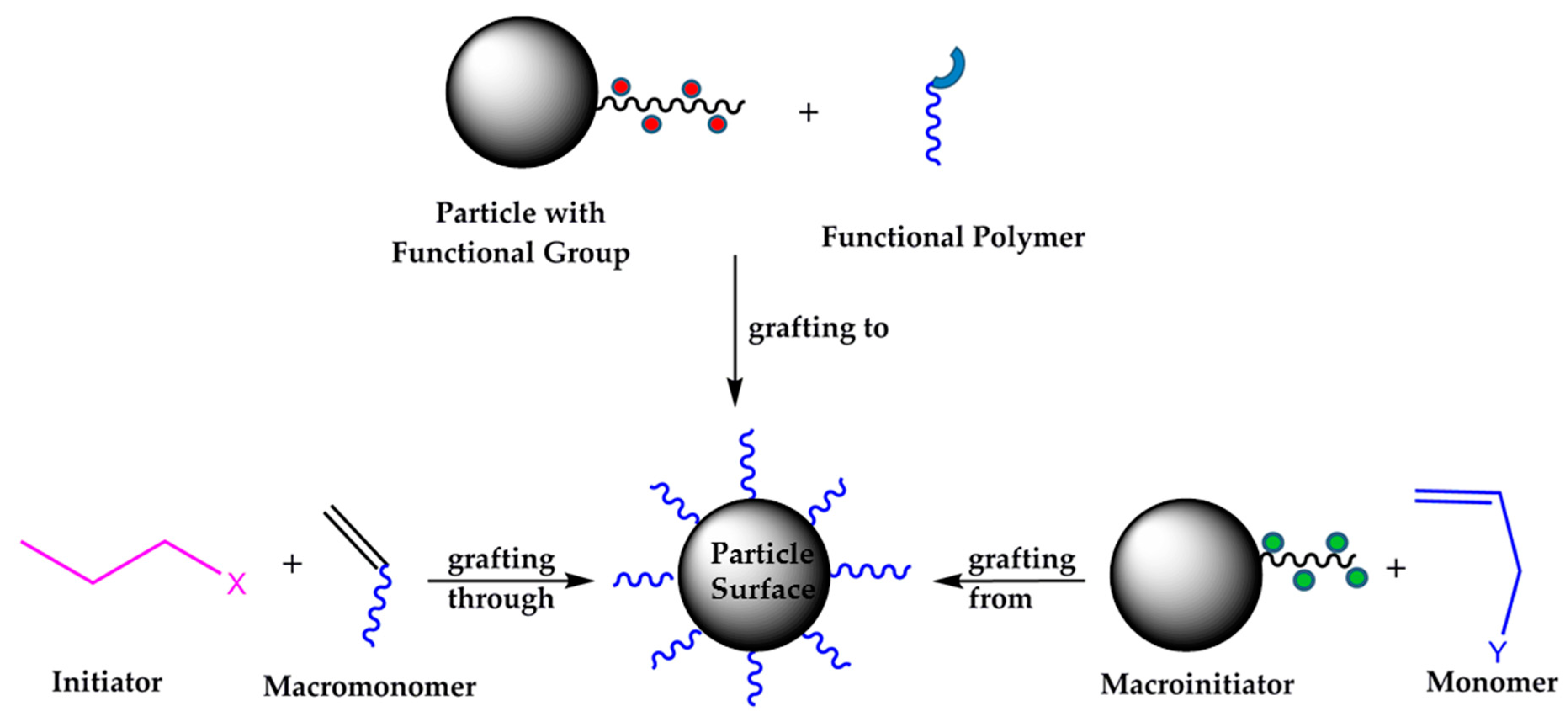
2. Results and Discussion
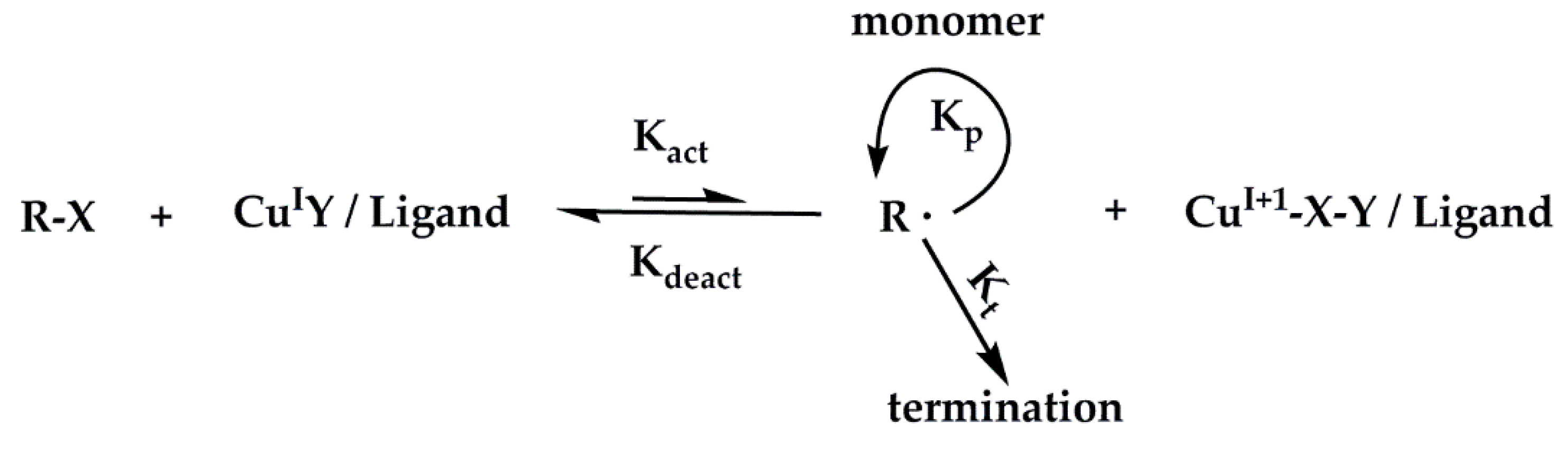
2.1. SiO2-g-poly (2-gluconamidoethyl methacrylamide)
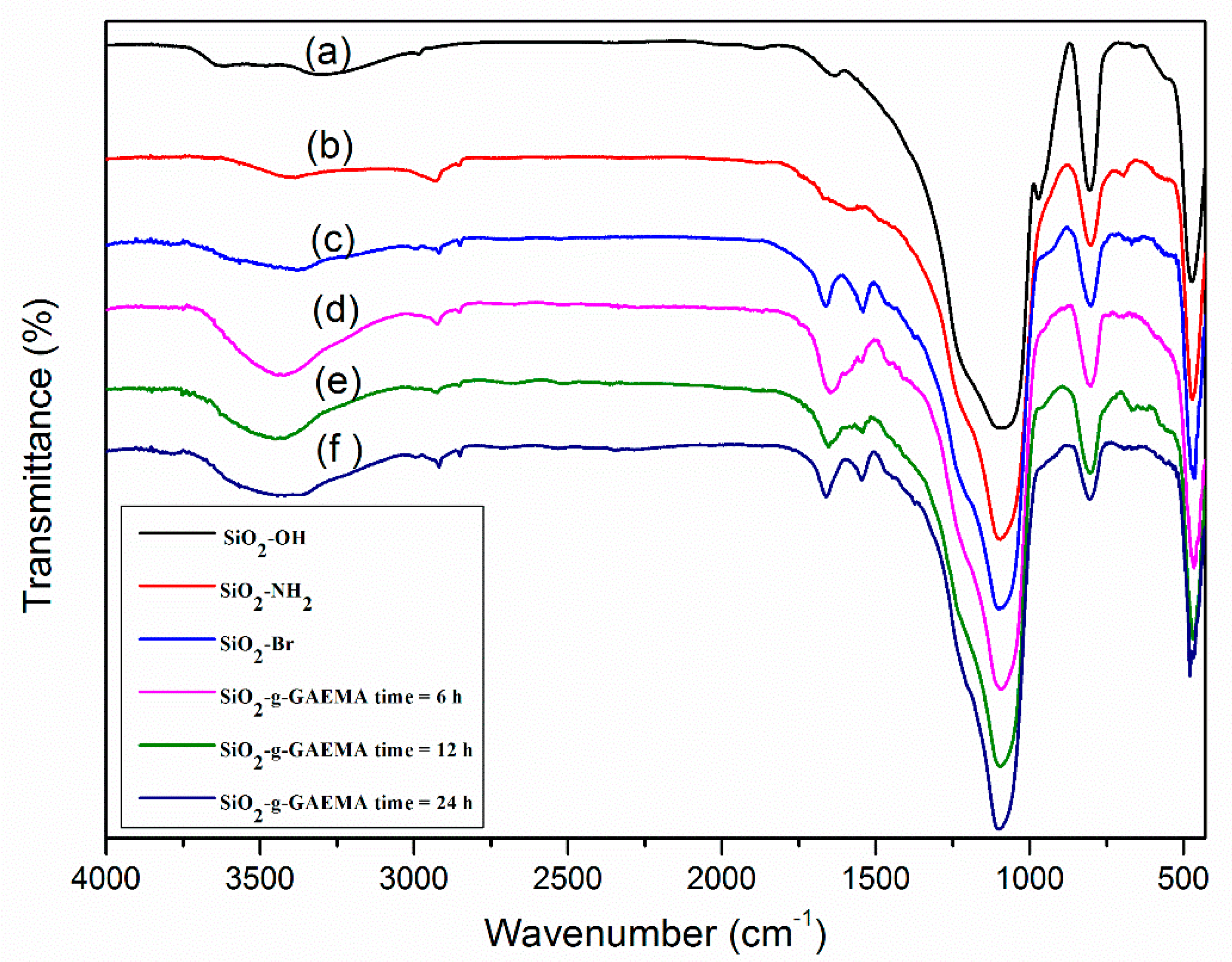
2.2. IR Analysis
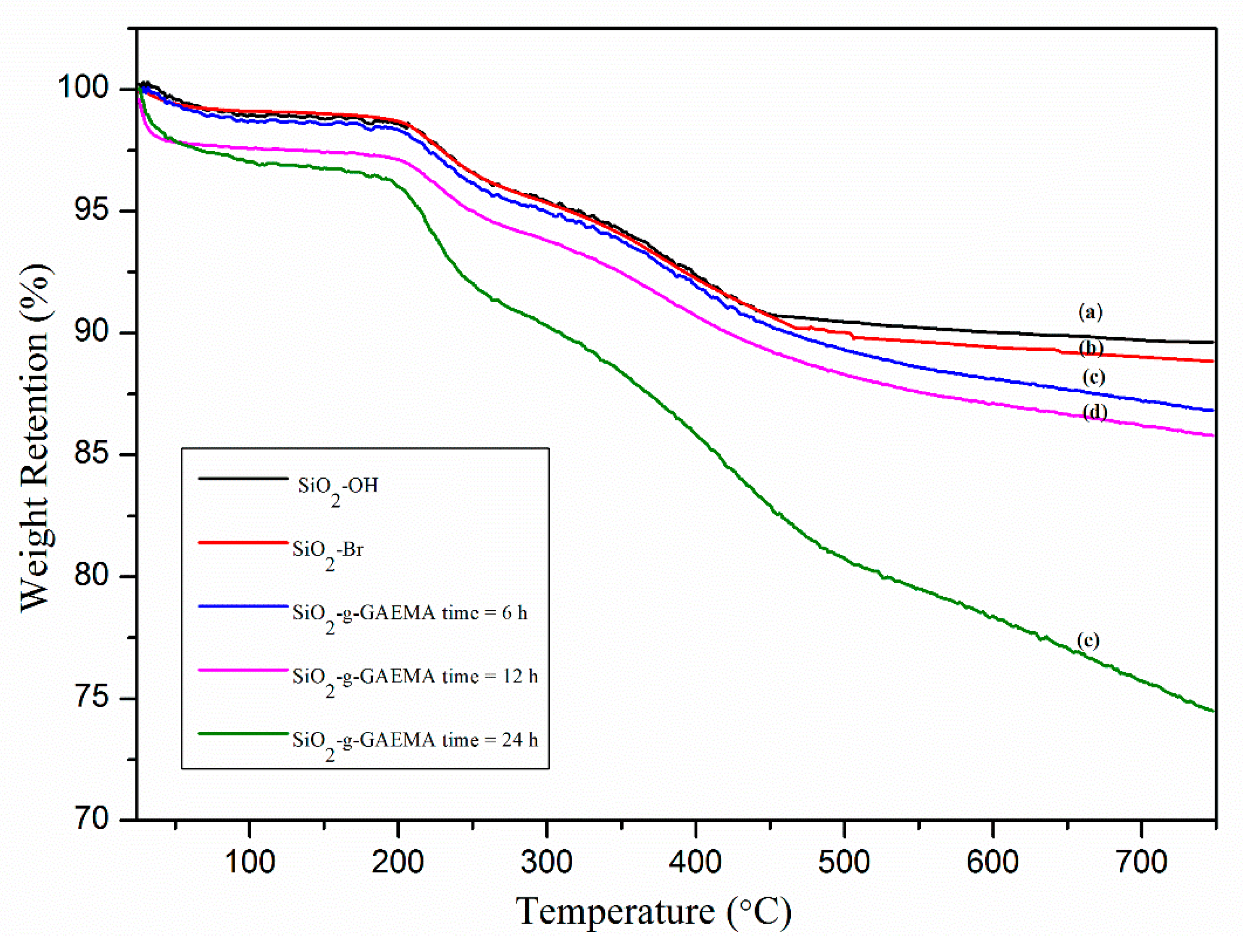
2.3. TGA Analysis
2.4. Characterization of GAEMA Grafted Silica Particles
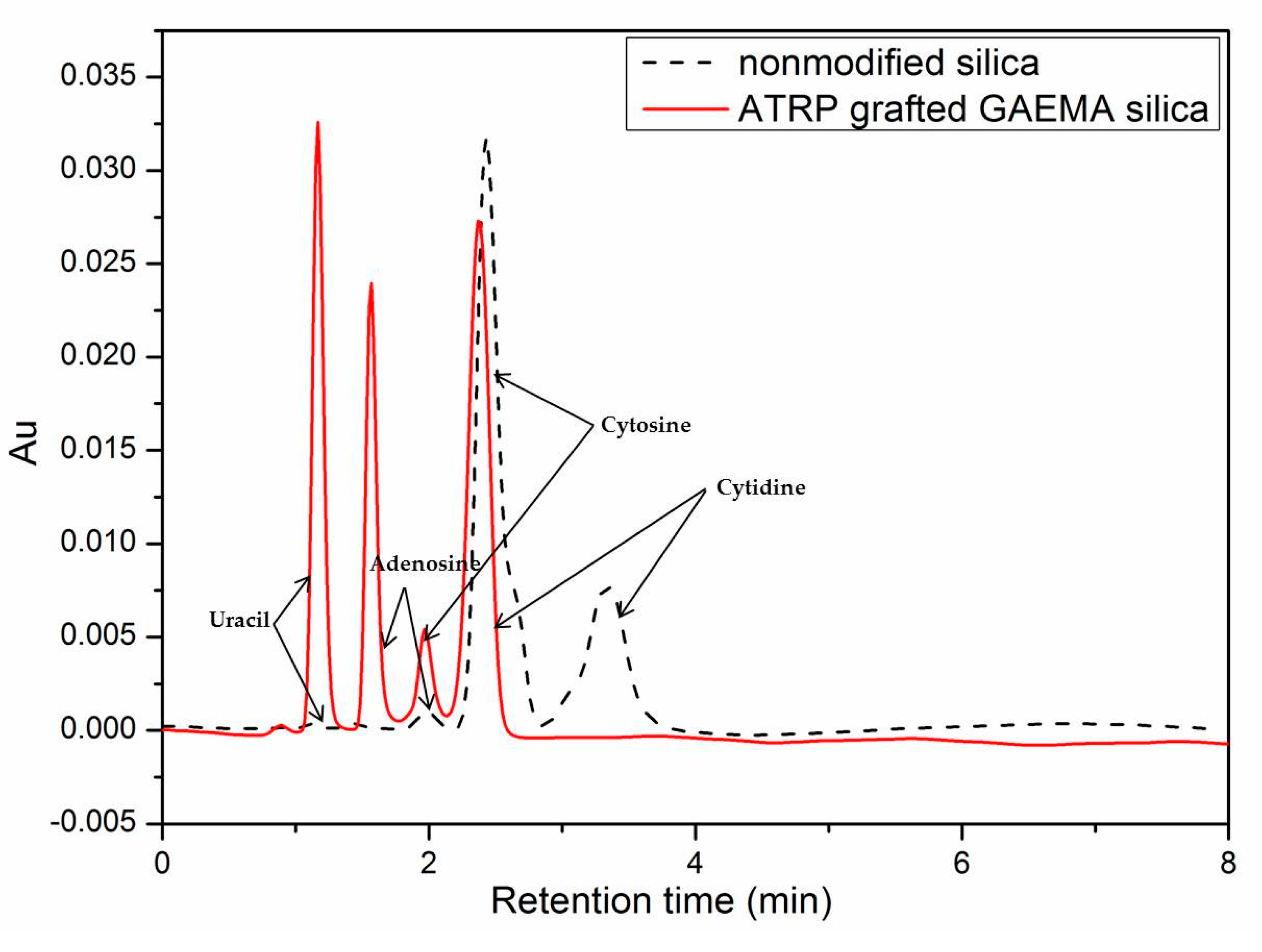
2.5. Chromatographic Performance of SiO2-g-GAEMA
3. Materials and Methods
3.1. Instrumentation
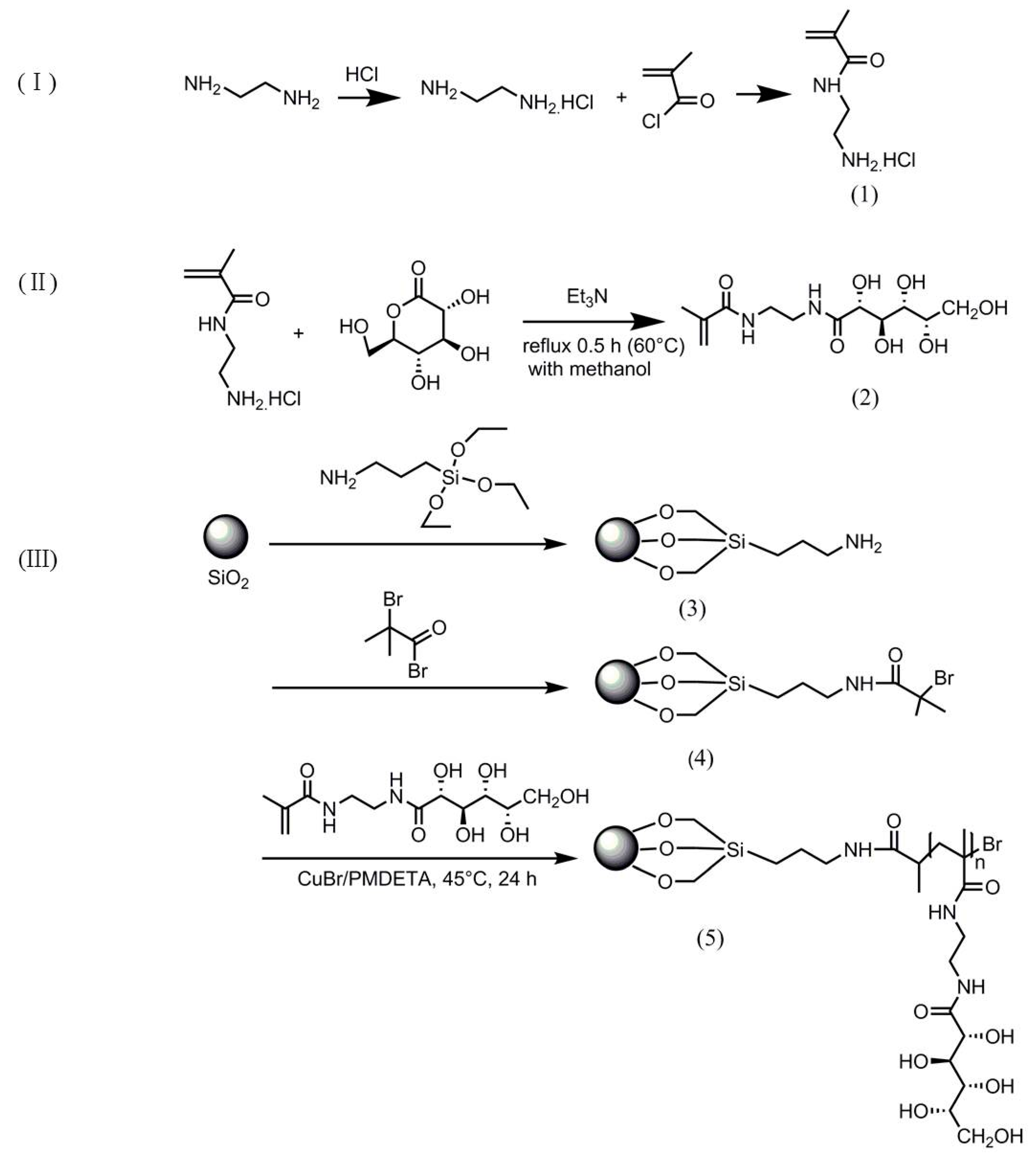
3.2. Synthesisof 2-Aminoethyl Methacrylamide Hydrochloride (AEMA, Scheme 2 (1))
3.3. Synthesis of 2-Gluconamidoethyl Methacrylamide (GAEMA, Scheme 2 (2))
3.4. Synthesis of APTES-Modified Porous Silica with Surface Amino Group (SiO2-NH2, Scheme 2 (3))
3.5. Preparation of ATRP Initiator-Immobilized Silica Particles (SiO2-Br, Scheme 2 (4))
3.6. Synthesis of SiO2-g-poly (2-gluconamidoethyl methacrylamide) Hybrid Microspheres via Surface-Grafted ATRP (Scheme 2 (5))
3.7. Calculation of Grafting Ratio of Sugar-Containing Polymer on Silica Gel
3.8. Column Packing and Chromatographic Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATRP | Atom transfer radical polymerization |
| AEMA | 2-Aminoethyl methacrylamide hydrochloride |
| GAEMA | 2-Gluconamidoethyl Methacrylamide |
| SiO2-g-GAEMA | SiO2-g-poly (2-gluconamidoethyl methacrylamide) |
| PMDETA | 1,1,4,7,7-pentamethyldiethylenetriamine |
References
- David, A.; Kopeckova, P.J.; Rubinstein, A. The role of galactose, lactose, and galactose valency in the biorecognition of N-(2-hydroxypropyl)methacrylamide copolymers by human colon adenocarcinoma cells. Pharm. Res. 2002, 19, 1114. [Google Scholar] [CrossRef] [PubMed]
- Murata, J.-I.; Ohya, Y.; Ouchi, T. Possibility of application of quaternary chitosan having pendant galactose residues as gene delivery tool. Carbohydr. Polym. 1996, 29, 69–74. [Google Scholar] [CrossRef]
- Liu, F.; Wu, Y.; Bai, L.; Peng, X.; Zhang, H.; Zhang, Y.; An, P.; Wang, S.; Ma, G.; Ba, X. Facile preparation of hyperbranched glycopolymers via an AB3* inimer promoted by a hydroxy/cerium(iv) redox process. Polym. Chem. 2018, 9, 5024–5031. [Google Scholar] [CrossRef]
- Katagiri, K.; Takasu, A.; Higuchi, M. Synthesis of Glycopolymer Containing Cell-Penetrating Peptides as Inducers of Recombinant Protein Expression under the Control of Lactose Operator/Repressor Systems. Biomacromolecules 2016, 17, 1902–1908. [Google Scholar] [CrossRef] [PubMed]
- Narain, R.; Armes, S.P. Synthesis of low polydispersity, controlled-structure sugar methacrylate polymers under mild conditions without protecting group chemistry. Chem. Commun. 2002, 23, 2776–2777. [Google Scholar] [CrossRef]
- Kitano, H.; Mai, K.; Naoki Kanayama, A.; Ohno, K. Interfacial Recognition of Sugars by Novel Boronic Acid-Carrying Amphiphiles Prepared with a Lipophilic Radical Initiator. Langmuir 1998, 14, 27091–27096. [Google Scholar] [CrossRef]
- Zhou, W.-J.; Wilson, M.E.; Kurth, M.J.; Hsieh, Y.-L.; Krochta, J.M.; Shoemaker, C.F. Synthesis and Properties of a Novel Water-Soluble Lactose-Containing Polymer and Its Cross-Linked Hydrogel. Macromolecules 1997, 30, 7063–7068. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, B.H.; Lee, S.; Kim, H.J.; Dorgan, J.R. Enhanced interfacial adhesion, mechanical, and thermal properties of natural flour-filled biodegradable polymer bio-composites. J. Therm. Anal. Calorim. 2011, 104, 331–338. [Google Scholar] [CrossRef]
- Yang, Q.; Xu, Z.K.; Hu, M.X.; Li, J.J.; Wu, J. Novel sequence for generating glycopolymer tethered on a membrane surface. Langmuir 2005, 21, 10717–10723. [Google Scholar] [CrossRef]
- Morandi, G.; Heath, L.; Thielemans, W. Cellulose Nanocrystals Grafted with Polystyrene Chains through Surface-Initiated Atom Transfer Radical Polymerization (SI-ATRP). Langmuir 2009, 25, 8280–8286. [Google Scholar] [CrossRef]
- Alpert, A.J. Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. J. Chromatogr. A 1990, 499, 177–196. [Google Scholar] [CrossRef]
- Guo, Z.; Lei, A.; Zhang, Y.; Xu, Q.; Xue, X.; Zhang, F.; Liang, X. “Click saccharides”: Novel separation materials for hydrophilic interaction liquid chromatography. Chem. Commun. 2007, 28, 2491–2493. [Google Scholar] [CrossRef] [PubMed]
- Rathnasekara, R.; El Rassi, Z. Polar silica-based stationary phases. Part II—Neutral silica stationary phases with surface bound maltose and sorbitol for hydrophilic interaction liquid chromatography. J. Chromatogr. A 2017, 1508, 24–32. [Google Scholar] [CrossRef]
- Huang, H.; Jin, Y.; Xue, M.; Yu, L.; Fu, Q.; Ke, Y.; Chu, C.; Liang, X. A novel click chitooligosaccharide for hydrophilic interaction liquid chromatography. Chem. Commun. 2009, 45, 6973–6975. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Kawaguchi, S. Poly(macromonomers): Homo- and Copolymerization; Springer: Berlin/Heidelberg, Germany, 1999; Volume 142, pp. 129–178. [Google Scholar]
- Yamada, K.; Miyazaki, M.; Ohno, K.; Fukuda, T.; Minoda, M. Atom Transfer Radical Polymerization of Poly(vinyl ether) Macromonomers. Macromolecules 1999, 32, 290–293. [Google Scholar] [CrossRef]
- Cheng, G.; Böker, A.; Zhang, M.; Krausch, G.; Muller, A.H.E. Amphiphilic Cylindrical Core-Shell Brushes via a “Grafting From” Process Using ATRP. Macromolecules 2001, 34, 6883–6888. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Gaynor, S.; Wang, J.S. Controlled Radical Polymerizations: The Use of Alkyl Iodides in Degenerative Transfer. Macromolecules 1995, 28, 2093–2095. [Google Scholar] [CrossRef]
- Gonzato, C.; Semsarilar, M.; Jones, E.R.; Li, F.; Krooshof, G.J.; Wyman, P.; Mykhaylyk, O.O.; Tuinier, R.; Armes, S.P. Rational synthesis of low-polydispersity block copolymer vesicles in concentrated solution via polymerization-induced self-assembly. J. Am. Chem. Soc. 2014, 136, 11100–11106. [Google Scholar] [CrossRef]
- And, R.N.; Armes, S.P. Direct Synthesis and Aqueous Solution Properties of Well-Defined Cyclic Sugar Methacrylate Polymers. Macromolecules 2003, 36, 4675–4678. [Google Scholar]
- Singleton, D.A.; Nowlan, D.T.; Jahed, N.; Matyjaszewski, K. Isotope Effects and the Mechanism of Atom Transfer Radical Polymerization. Macromolecules 2003, 36, 8609–8616. [Google Scholar] [CrossRef]
- Macquarrie, D.J. Direct preparation of organically modified MCM-type materials. Preparation and characterisation of aminopropyl?MCM and 2-cyanoethyl?MCM. Chem. Commun. 1996, 16, 1961–1962. [Google Scholar] [CrossRef]
- Yu, L.; Zhang, Y.; Wang, Y.; Zhang, H.; Liu, J. High flux, positively charged loose nanofiltration membrane by blending with poly (ionic liquid) brushes grafted silica spheres. J. Hazard. Mater. 2015, 287, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tang, J.; Jia, Z.; Li, X.; Xiao, Z. Grafting of amphiphilic polymers containing quaternary ammonium group on SiO2 surface via surface-initiated ATRP. J. Polym. Res. 2012, 19, 9804. [Google Scholar] [CrossRef]
- Huang, H.; Chuang, W.; Sharma, R.C.; Hsu, C.; Lin, K.; Hu, C. Molecular elimination of Br2 in 248 nm photolysis of bromoform probed by using cavity ring-down absorption spectroscopy. J. Chem. Phys. 2004, 121, 5253–5260. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Huang, W.; Miller, M.D.; Baker, G.L.; Bruening, M.L. Kinetics of surface-initiated atom transfer radical polymerization. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 386–394. [Google Scholar] [CrossRef]
- Kimata, K.; Iwaguchi, K.; Onishi, S.; Jinno, K.; Eksteen, R.; Hosoya, K.; Araki, M.; Tanaka, N. Chromatographic Characterization of Silica C18 Packing Materials. Correlation between a Preparation Method and Retention Behavior of Stationary Phase. J. Chromatogr. Sci. 1989, 27, 721–728. [Google Scholar] [CrossRef]
- Klejdus, B.; Petrlová, J.; Potěšil, D.; Adam, V.; Mikelová, R.; Vacek, J.; Kizek, R.; Kubáň, V. Simultaneous determination of water- and fat-soluble vitamins in pharmaceutical preparations by high-performance liquid chromatography coupled with diode array detection. Anal. Chim. Acta 2004, 520, 57–67. [Google Scholar] [CrossRef]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals; Pergamon Press: Oxford, UK, 1988; p. iv. [Google Scholar]
- Chan, G.Y.N.; Jhingran, A.G.; Kambouris, P.A.; Looney, M.G.; Solomon, D.H. Approaches to the controlled formation of network polymers: 1. Synthesis and evaluation of monomers with vinyl differentiation. Polymer 1998, 39, 5781–5787. [Google Scholar] [CrossRef]
- Liu, Q.; Li, W.; Wang, H.; Newby, B.M.; Cheng, F.; Liu, L. Amino Acid-Based Zwitterionic Polymer Surfaces Highly Resist Long-Term Bacterial Adhesion. Langmuir 2016, 32, 7866–7874. [Google Scholar] [CrossRef]
- Deng, Z.; Bouchékif, H.; Babooram, K.; Housni, A.; Choytun, N.; Narain, R. Facile synthesis of controlled-structure primary amine-based methacrylamide polymers via the reversible addition-fragmentation chain transfer process. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 4984–4996. [Google Scholar] [CrossRef]
- Lowe, A.B.; Billingham, N.C.; Armes, S.P. Synthesis and Properties of Low-Polydispersity Poly(sulfopropylbetaine)s and Their Block Copolymers. Macromolecules 1999, 32, 2141–2148. [Google Scholar] [CrossRef]
- Niu, Y.; Ma, M.; Gong, Y.; Wang, Y.; Gong, B. Synthesis of chlorogenic acid imprinted chromatographic packing by surface-initiated atom transfer radical polymerization and its application. Chem. Res. Chin. Univ. 2014, 30, 855–862. [Google Scholar] [CrossRef]
- Roohi, F.; Magdalena Titirici, M. Thin thermo-responsive polymer films onto the pore system of chromatographic beads via reversible addition–fragmentation chain transfer polymerization. New J. Chem. 2008, 32, 1409. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Content of element | ||
|---|---|---|---|
| N | C | H | |
| Si-OH | 0.030 | 1.003 | 0.300 |
| Si-NH2 | 2.750 | 4.906 | 0.994 |
| Si-Br | 1.775 | 5.070 | 0.910 |
| Si-g-GAEMA (reaction time = 6 h) | 0.937 | 5.250 | 0.302 |
| Si-g-GAEMA (reaction time = 12 h) | 0.890 | 7.560 | 0.357 |
| Si-g-GAEMA (reaction time = 24 h) | 2.413 | 11.523 | 0.553 |
| Sample | Reaction time (h) | Grafted (mg/m2) |
|---|---|---|
| 1 | 6 | 0.50 |
| 2 | 12 | 0.78 |
| 3 | 24 | 1.36 |
| Column | Retention time (min) | N (plates/m) | Symmetry factor |
|---|---|---|---|
| SiO2-OH | 2.687 | 3960 | 0.75 |
| SiO2-g-GAEMA | 2.380 | 8258 | 0.98 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, G.; Luo, X.; Sun, X.; Wang, W.; Shou, Q.; Liang, X.; Liu, H. Glycopolymer Grafted Silica Gel as Chromatographic Packing Materials. Int. J. Mol. Sci. 2019, 20, 10. https://doi.org/10.3390/ijms20010010
Ma G, Luo X, Sun X, Wang W, Shou Q, Liang X, Liu H. Glycopolymer Grafted Silica Gel as Chromatographic Packing Materials. International Journal of Molecular Sciences. 2019; 20(1):10. https://doi.org/10.3390/ijms20010010
Chicago/Turabian StyleMa, Gaoqi, Xitao Luo, Xitong Sun, Weiyan Wang, Qinghui Shou, Xiangfeng Liang, and Huizhou Liu. 2019. "Glycopolymer Grafted Silica Gel as Chromatographic Packing Materials" International Journal of Molecular Sciences 20, no. 1: 10. https://doi.org/10.3390/ijms20010010
APA StyleMa, G., Luo, X., Sun, X., Wang, W., Shou, Q., Liang, X., & Liu, H. (2019). Glycopolymer Grafted Silica Gel as Chromatographic Packing Materials. International Journal of Molecular Sciences, 20(1), 10. https://doi.org/10.3390/ijms20010010