Therapeutic Inhibition of Myc in Cancer. Structural Bases and Computer-Aided Drug Discovery Approaches

{kind=link}
{kind=link}
{kind=link}
{kind=link}
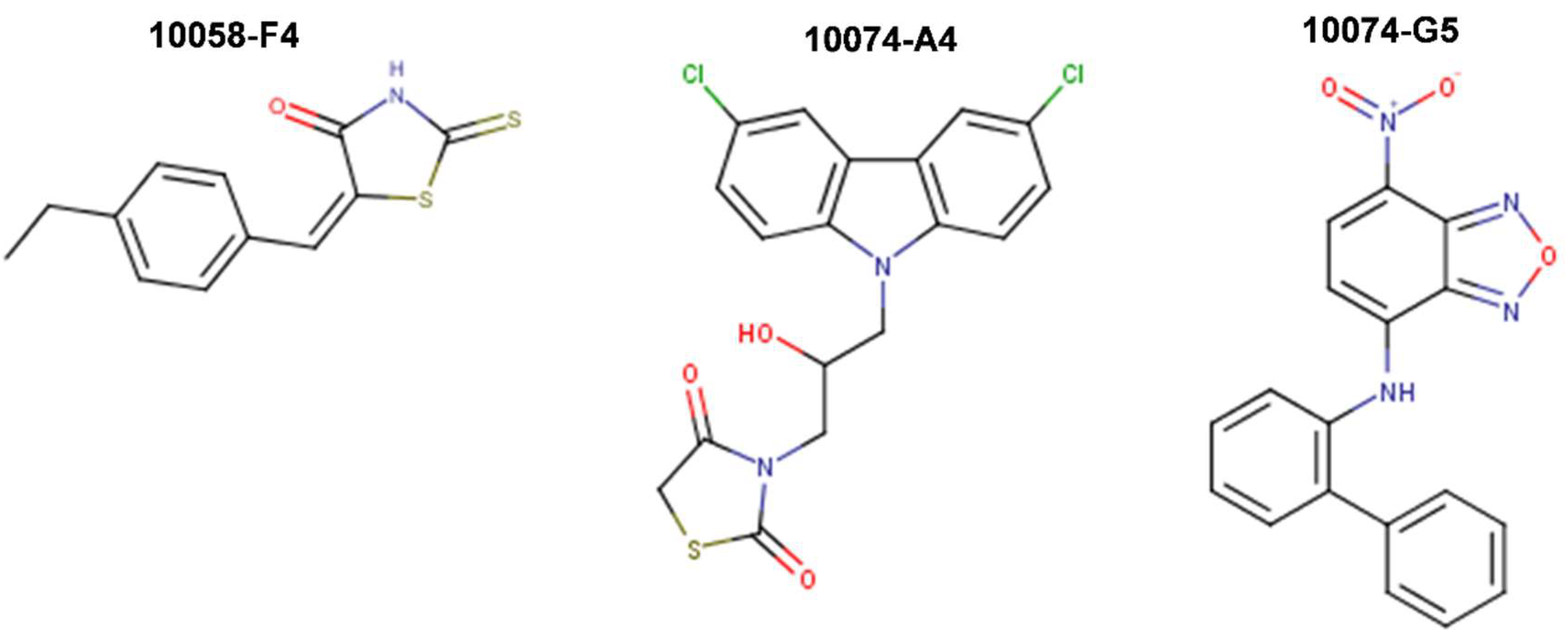{kind=link}
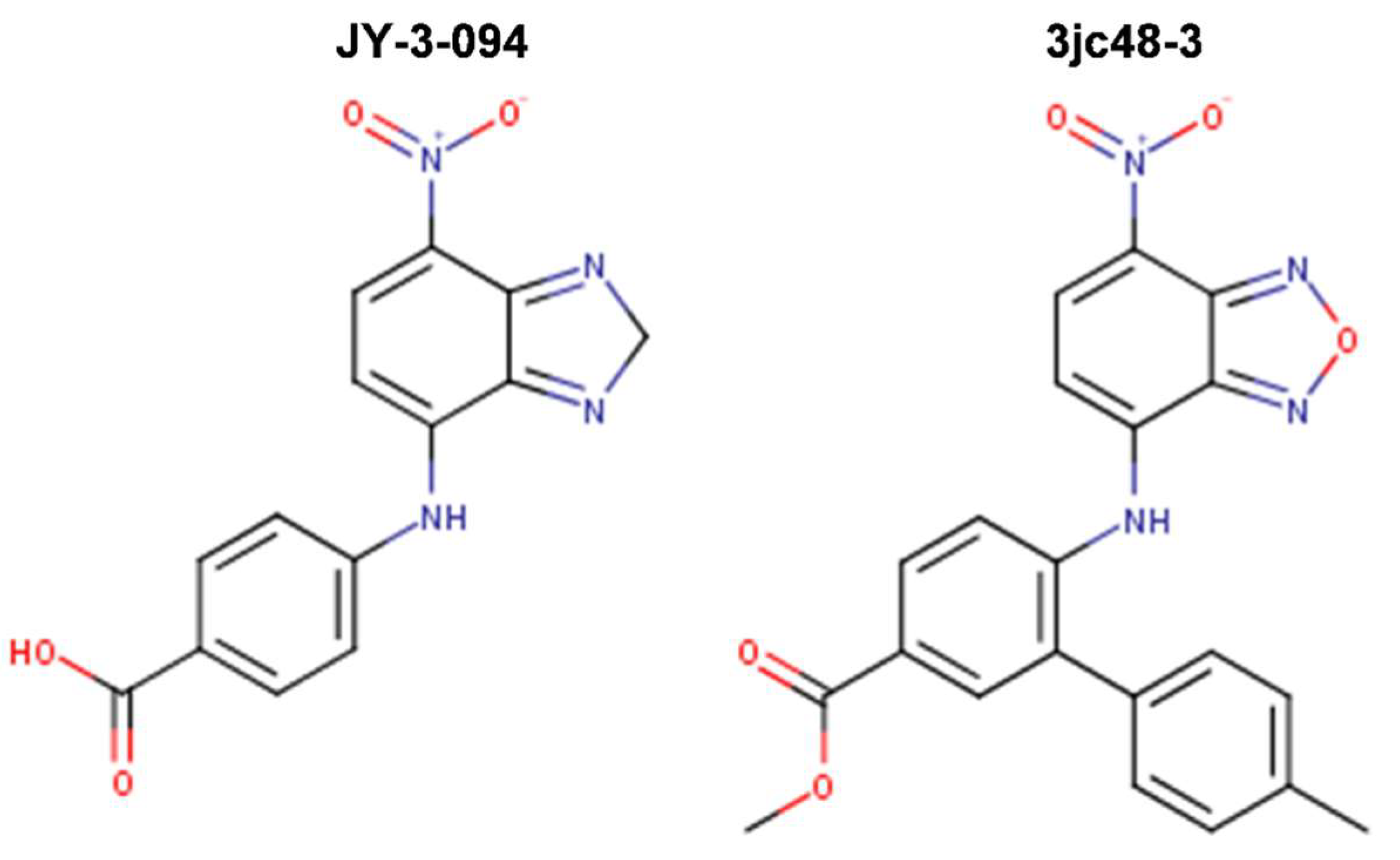{kind=link}
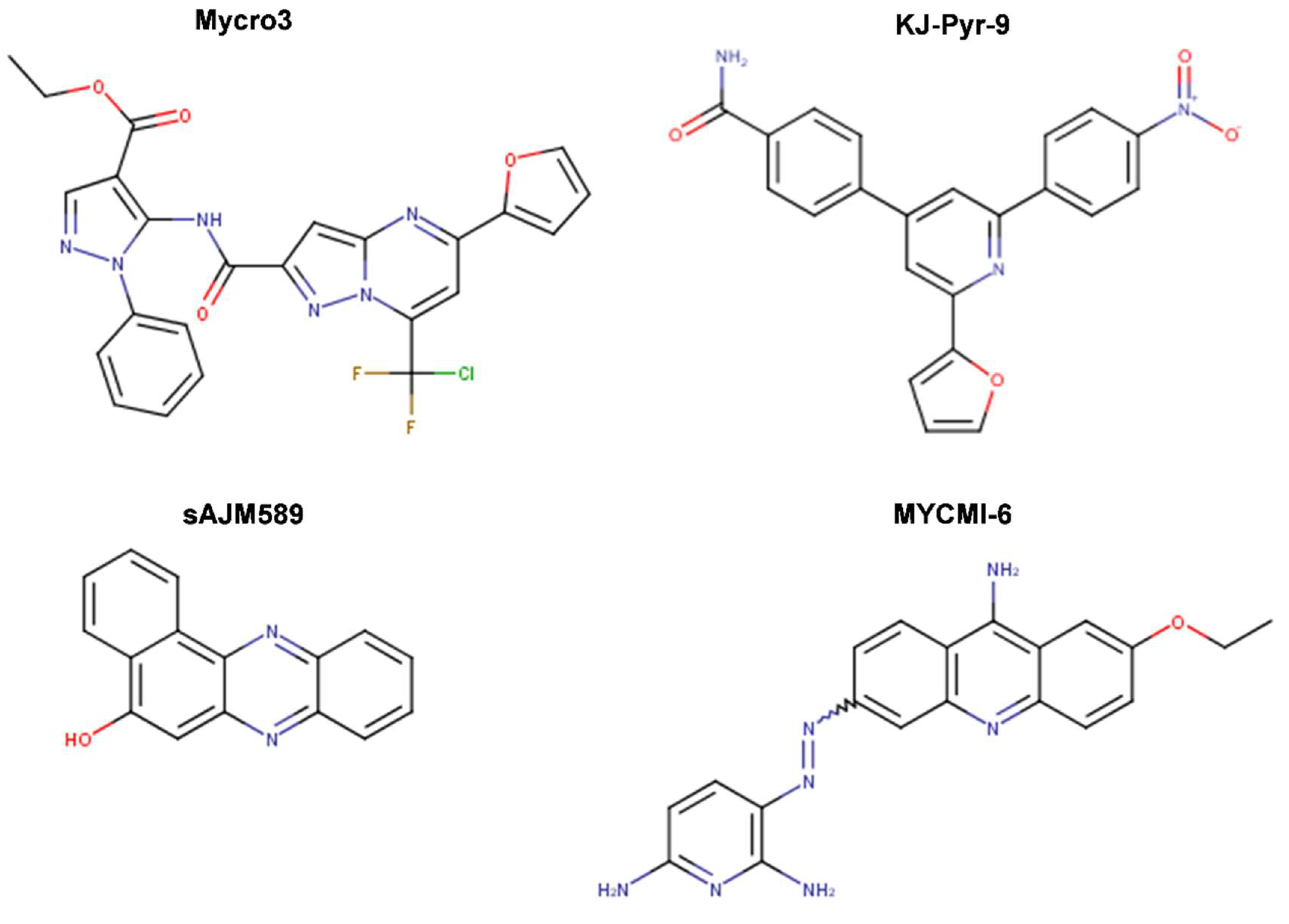{kind=link}
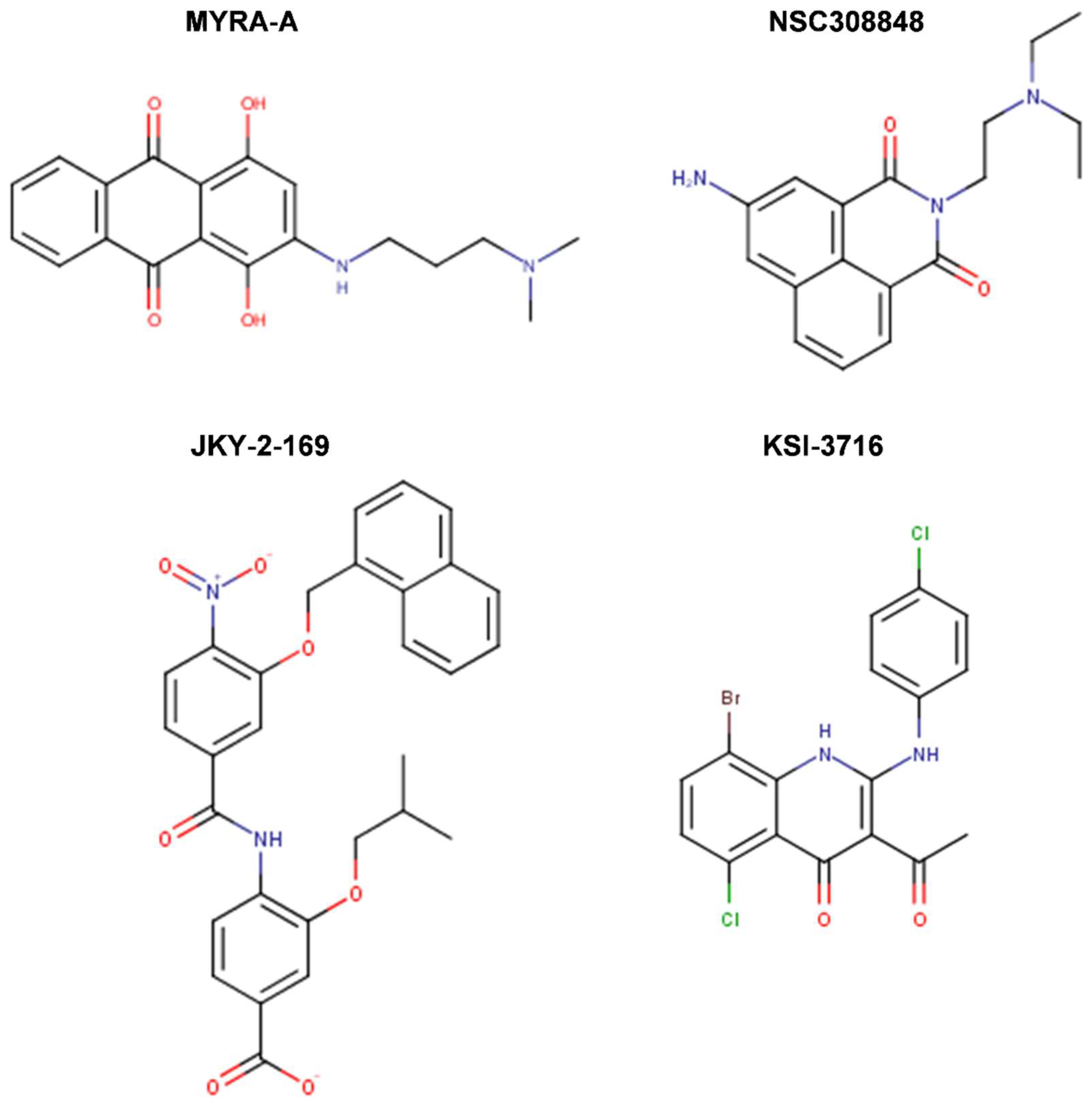{kind=link}
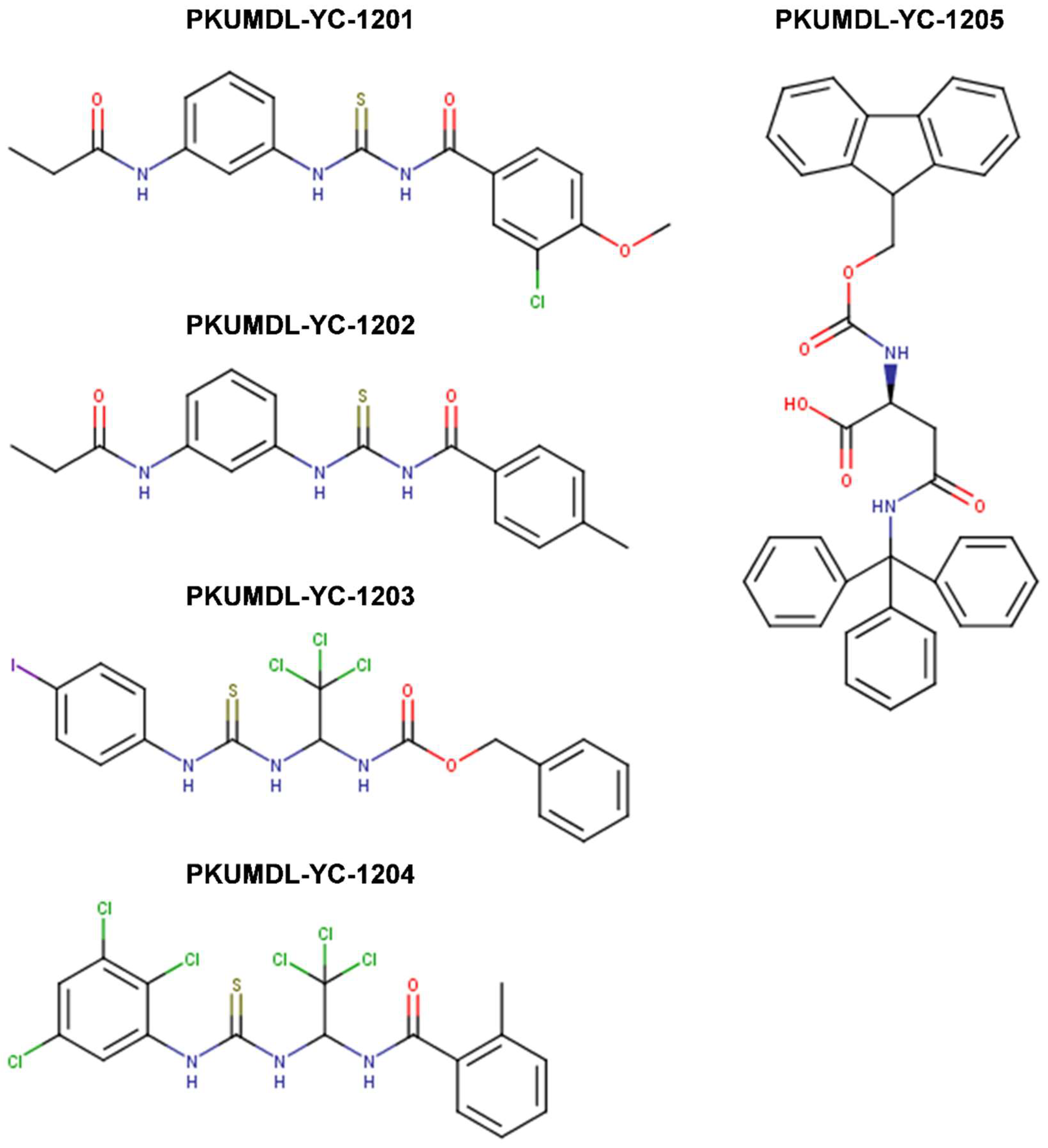{kind=link}
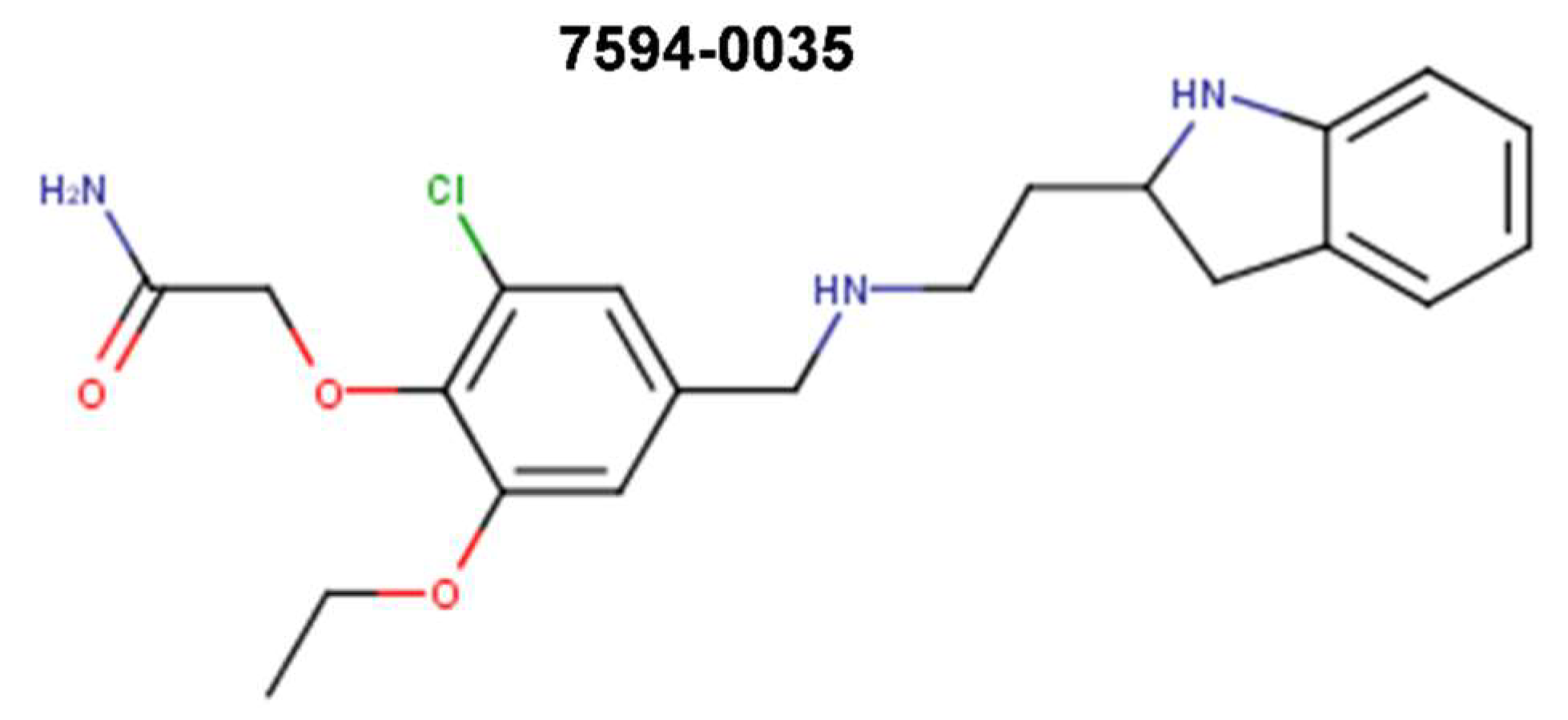{kind=link}
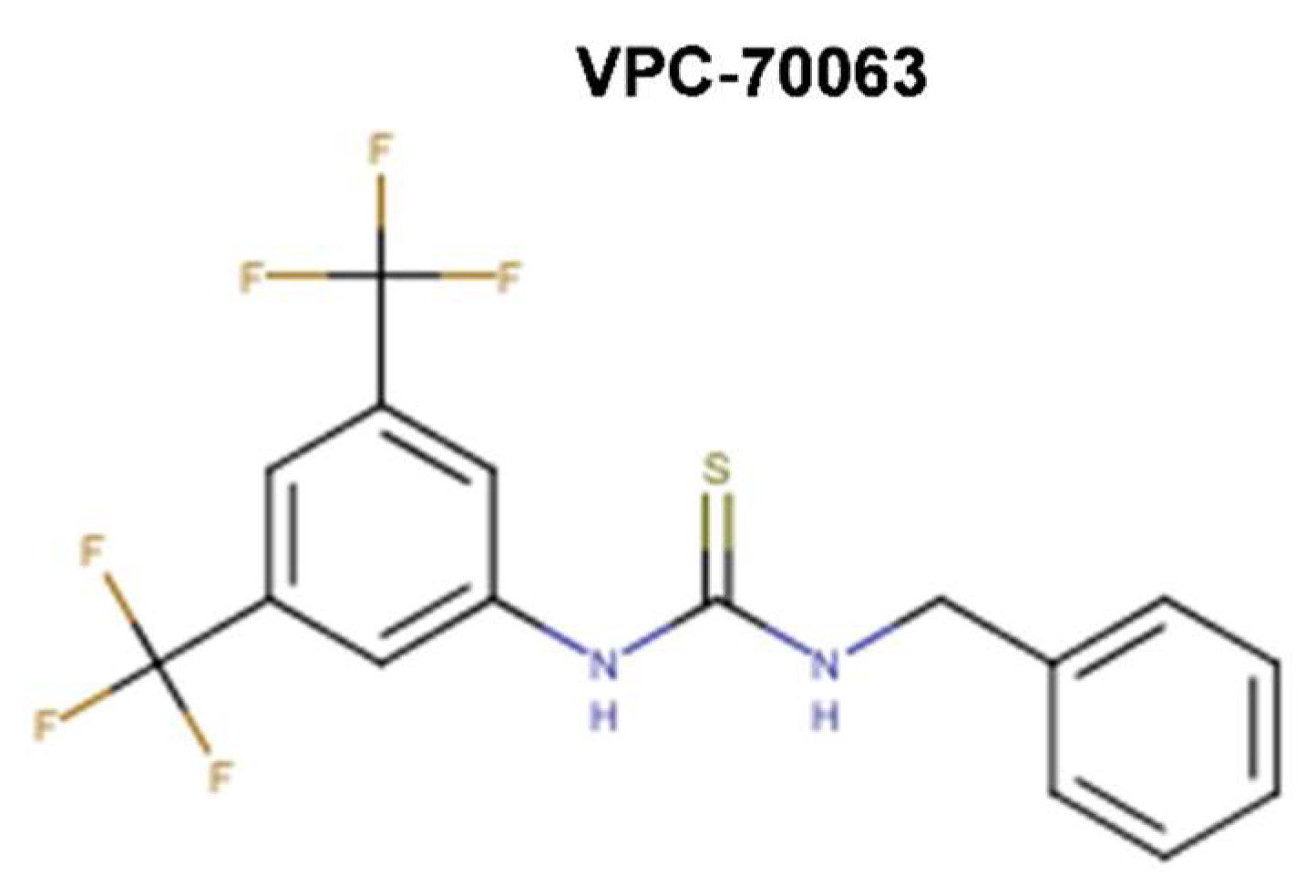{kind=link}
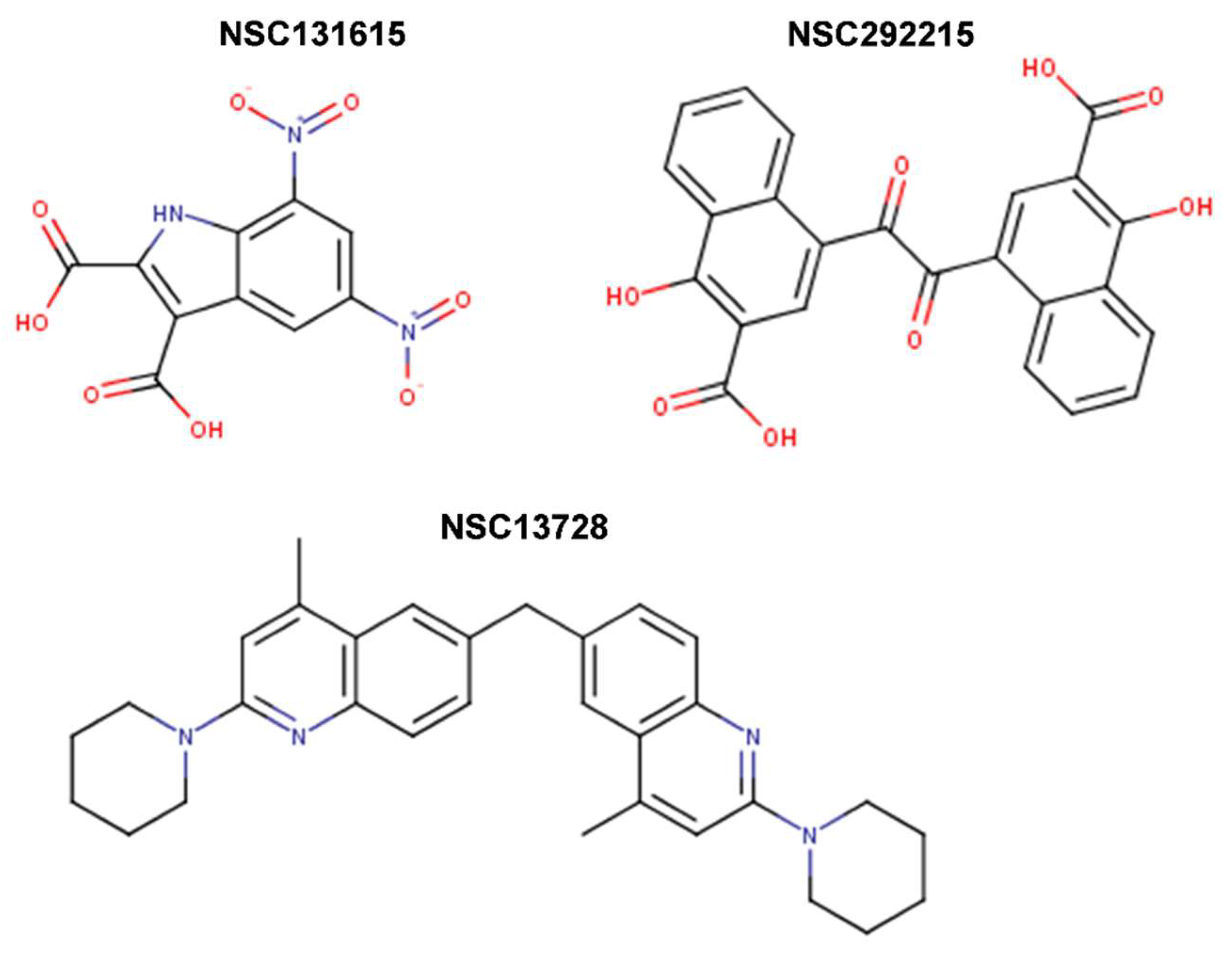{kind=link}
{kind=link}
{kind=link}
Abstract
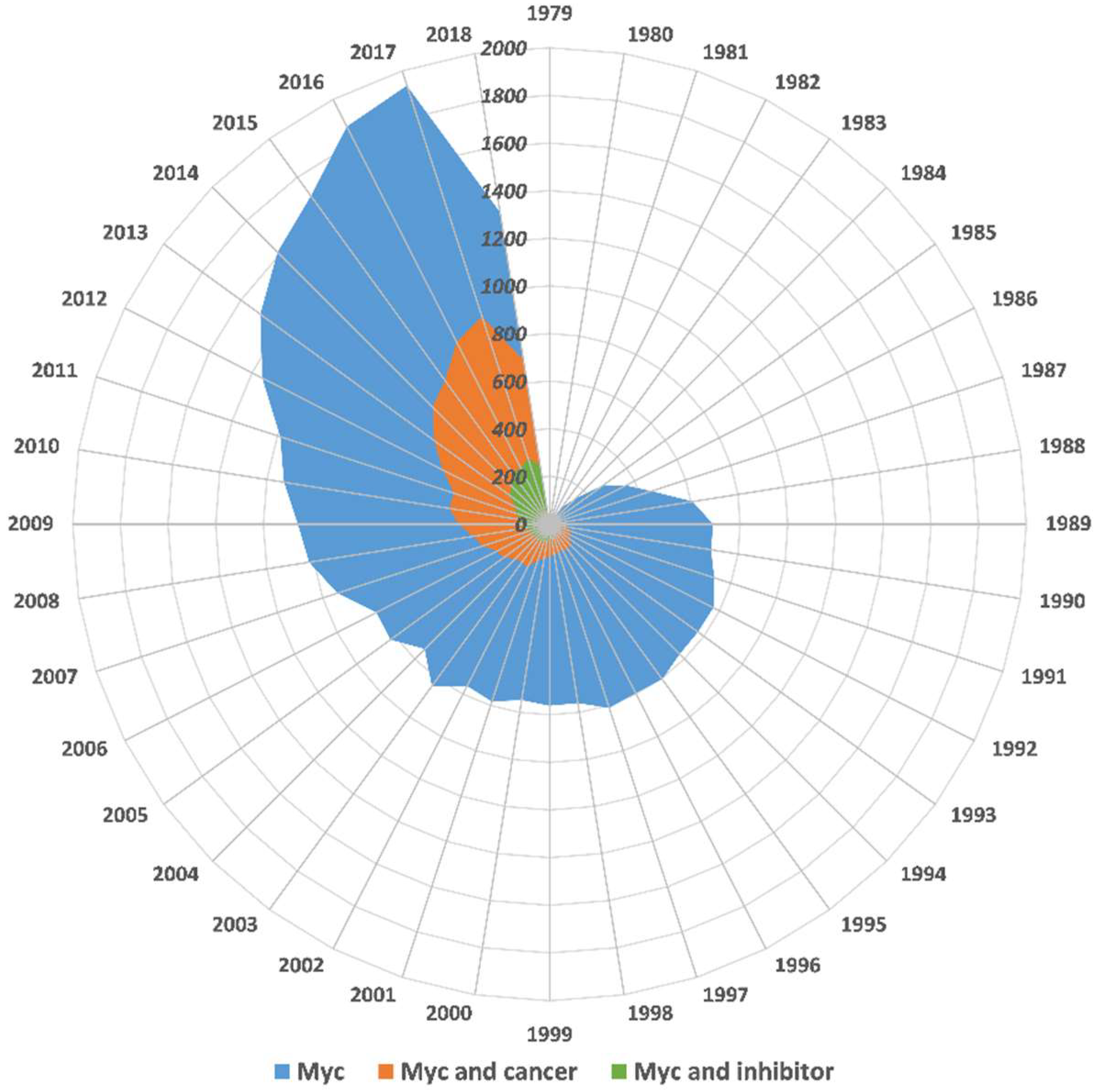
:1. Introduction
2. Myc Regulation. Structure-Function Relationships
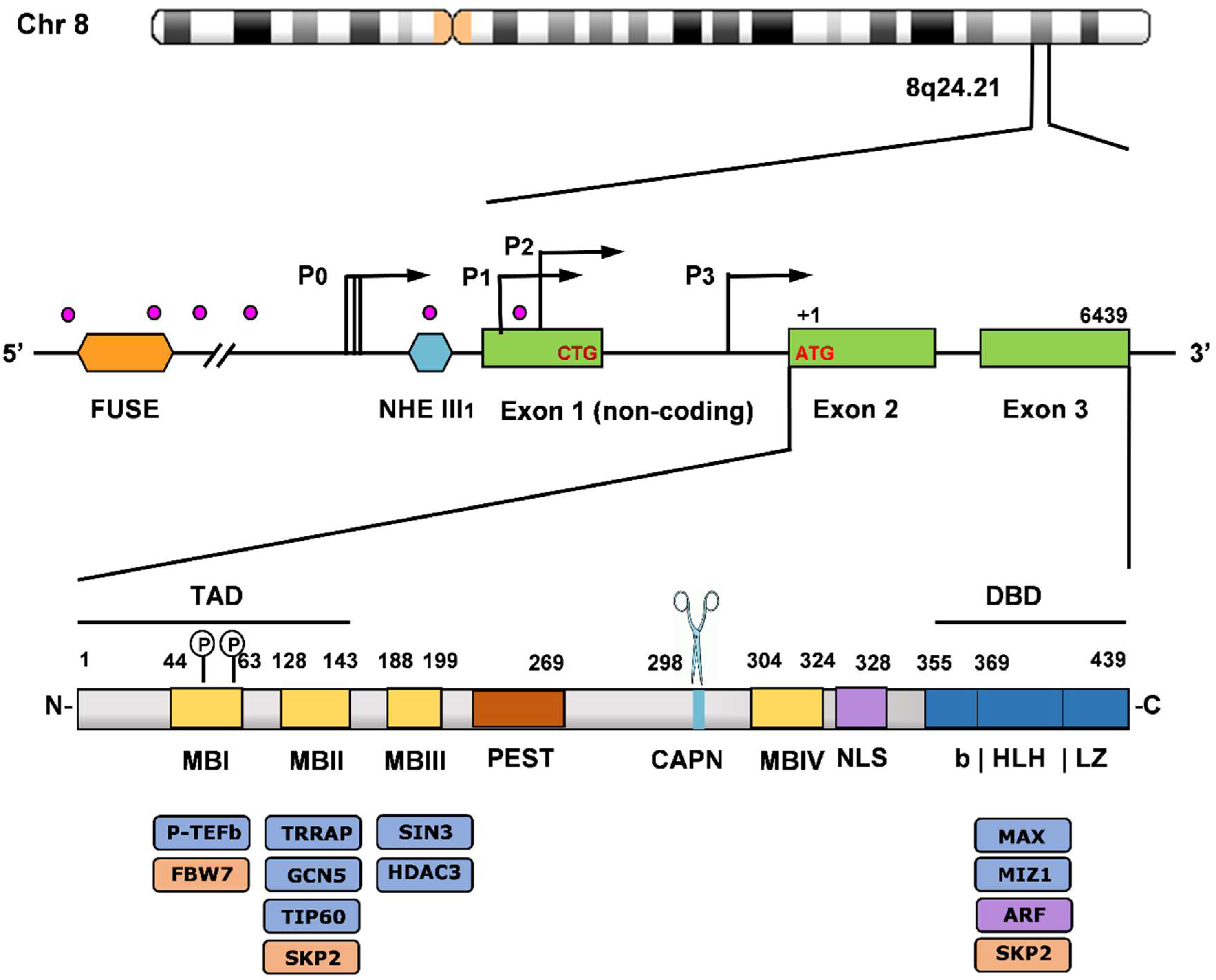
2.1. Structure of MYC Gene and Protein. Regulatory Sites and Functional Domains
2.1.1. MYC Gene Expression. Transcriptional and Posttranscriptional Control
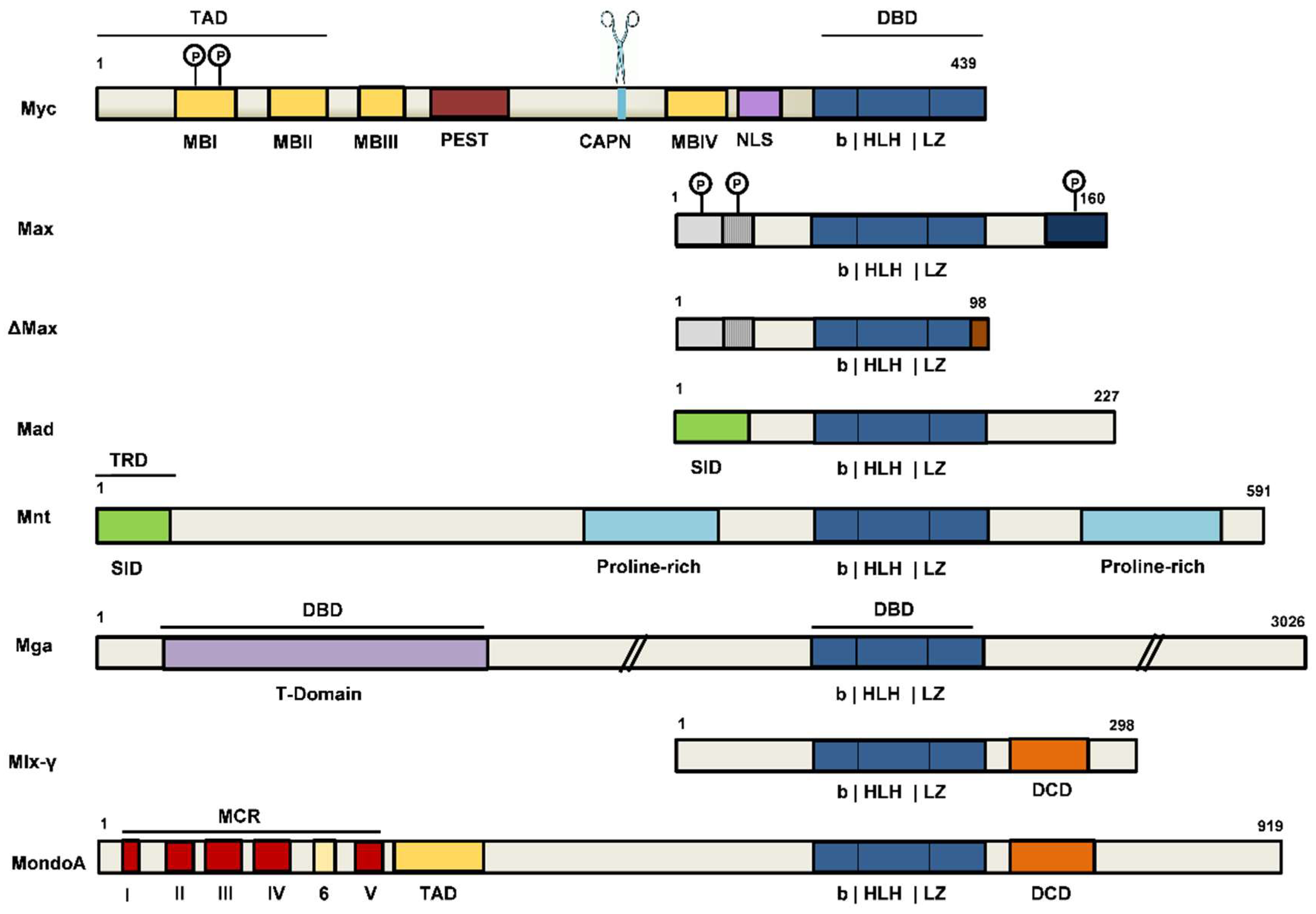
2.1.2. Myc and Max Protein Organization and Interactors. Translational and Posttranslational Control
2.2. The Extended Myc/Max/Mxd Network. The Players and Their Regulatory Functions
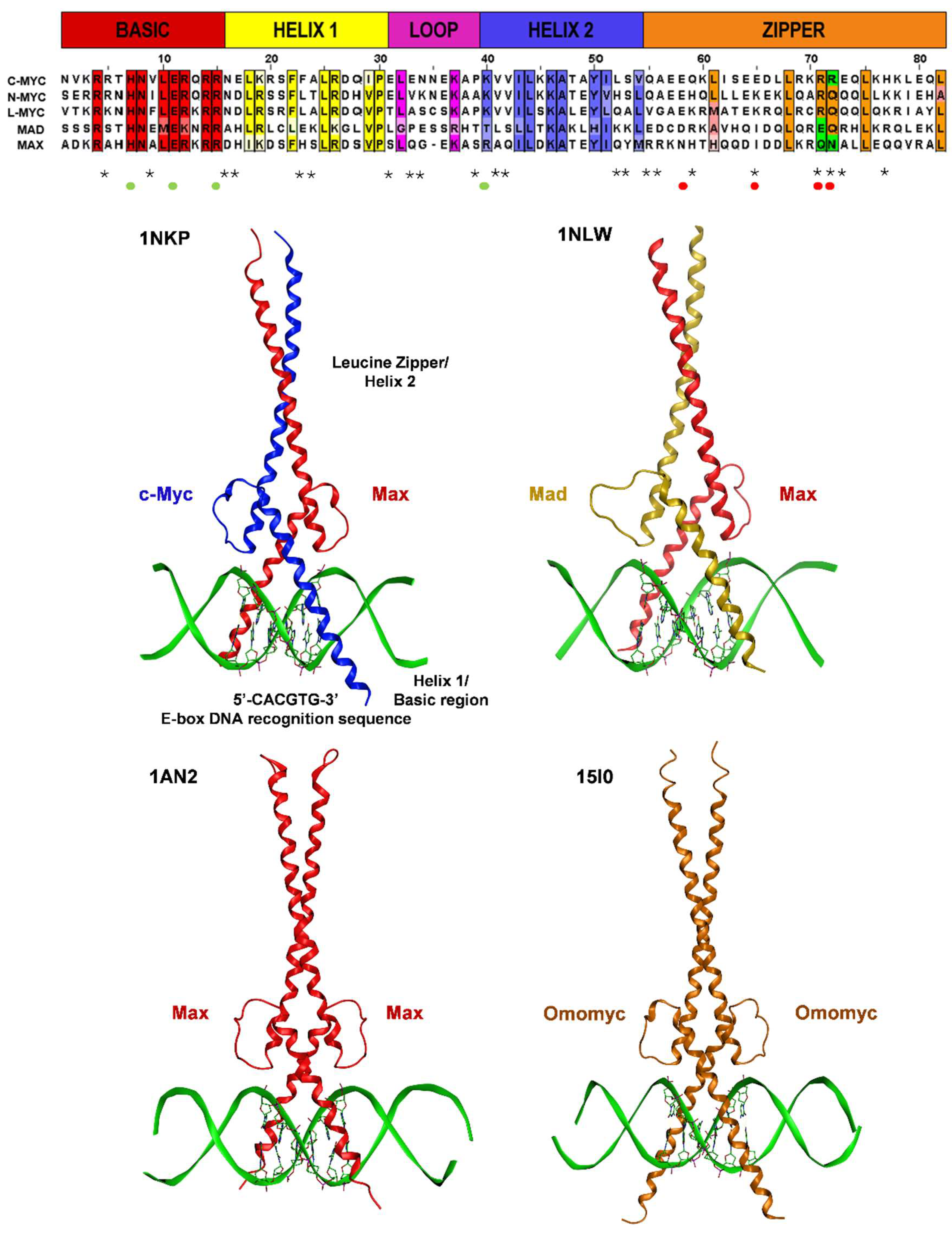
2.3. Structural Aspects of Protein–Protein and Protein-DNA Interactions within the Myc/Max/Mad Network and Beyond
3. Myc Targeting Approaches
3.1. Small-Molecule Myc-Max Inhibitors
- those that act by interfering with protein–protein interactions and block heterodimerization of Myc with Max; and,
- those that directly block Myc-Max binding to DNA.
3.1.1. Direct Myc-Max Protein–Protein Interactions Inhibitors
3.1.2. Direct Inhibition of Myc-Max Interaction with DNA
3.1.3. Computational Approaches toward Myc-Max Inhibition
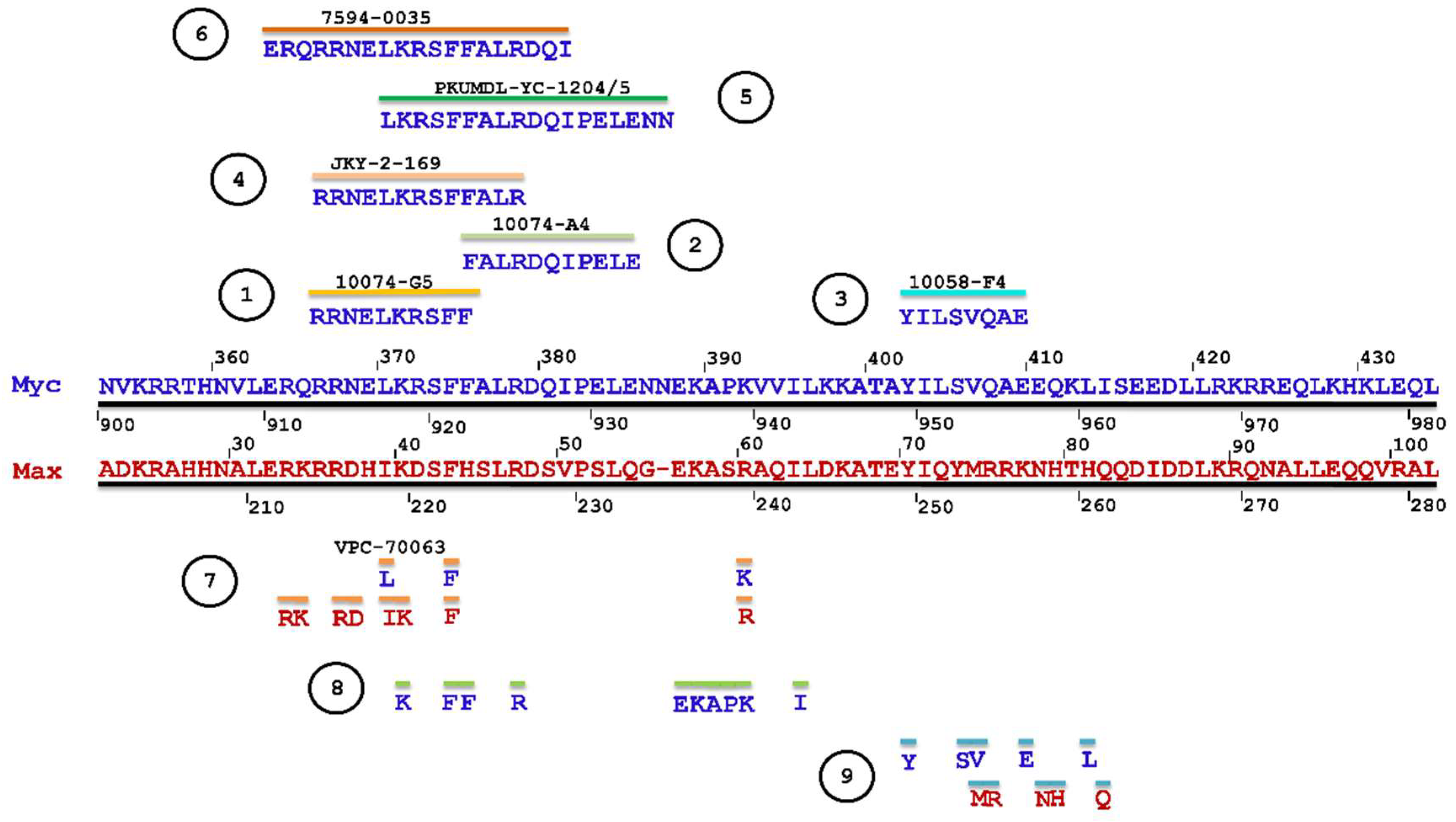
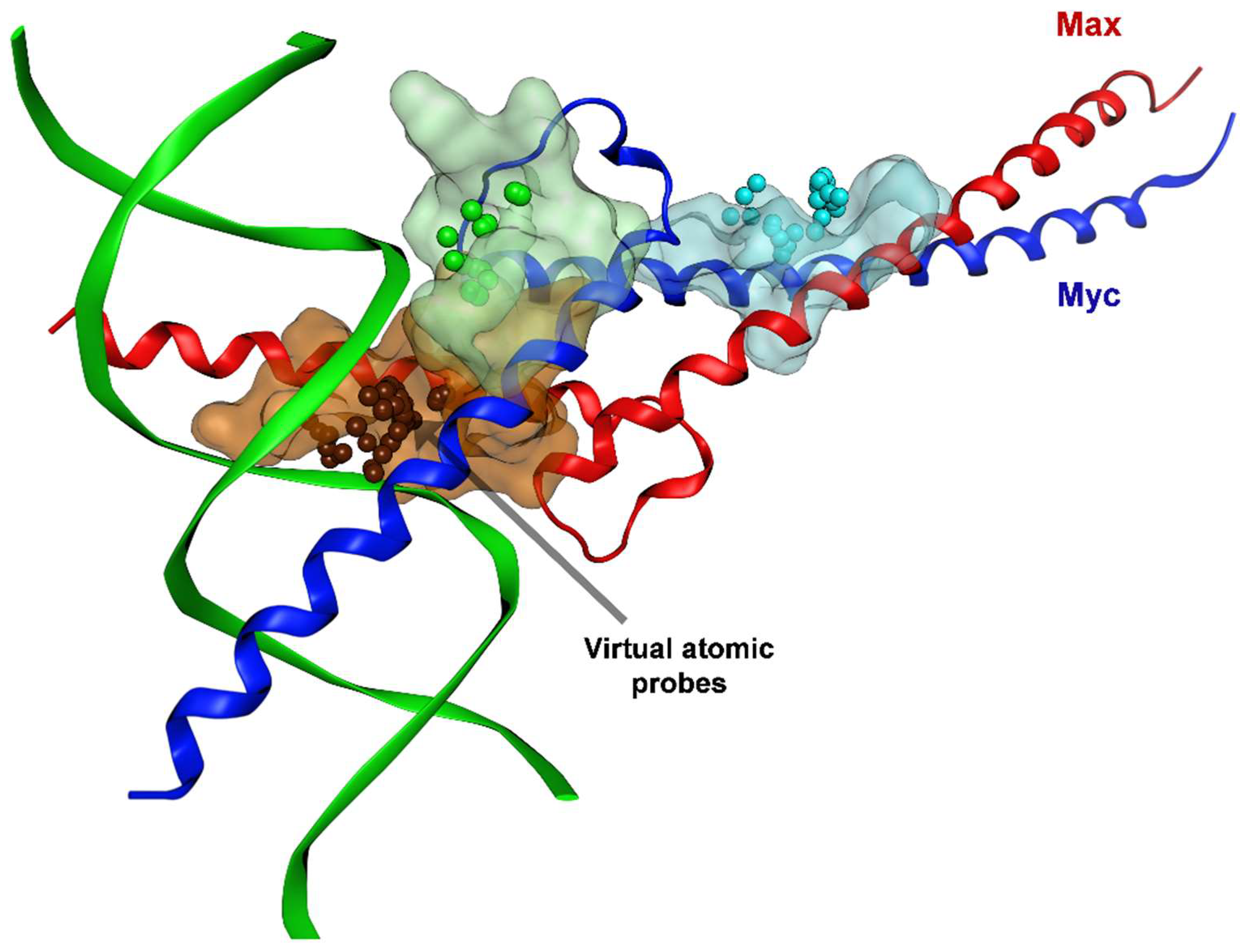
3.1.4. Binding Sites for Myc-Max Small-Molecule Inhibitors
3.2. Small-Molecule G-Quadruplex Stabilizers
4. Discussion and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC Deregulation in Primary Human Cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef] [PubMed]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef] [PubMed]
- Varmus, H.E. The molecular genetics of cellular oncogenes. Annu. Rev. Genet. 1984, 18, 553–612. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Alitalo, K.; Klempnauer, K.H.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983, 305, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Ellison, J.; Busch, M.; Rosenau, W.; Varmus, H.E.; Bishop, J.M. Enhanced expression of the human gene N-myc consequent to amplification of DNA may contribute to malignant progression of neuroblastoma. Proc. Natl. Acad. Sci. USA 1984, 81, 4940–4944. [Google Scholar] [CrossRef] [PubMed]
- Nau, M.M.; Brooks, B.J.; Battey, J.; Sausville, E.; Gazdar, A.F.; Kirsch, I.R.; McBride, O.W.; Bertness, V.; Hollis, G.F.; Minna, J.D. L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature 1985, 318, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Dang, C.V.; Resar, L.M.; Emison, E.; Kim, S.; Li, Q.; Prescott, J.E.; Wonsey, D.; Zeller, K. Function of the c-Myc oncogenic transcription factor. Exp. Cell Res. 1999, 253, 63–77. [Google Scholar] [CrossRef]
- Dang, C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harbor Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef]
- McMahon, S.B. MYC and the control of apoptosis. Cold Spring Harbor Perspect. Med. 2014, 4, a014407. [Google Scholar] [CrossRef]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sears, R.C. The life cycle of C-myc: From synthesis to degradation. Cell Cycle 2004, 3, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harbor Perspect. Med. 2014, 4, a014357. [Google Scholar] [CrossRef] [PubMed]
- Facchini, L.M.; Chen, S.; Marhin, W.W.; Lear, J.N.; Penn, L.Z. The Myc negative autoregulation mechanism requires Myc-Max association and involves the c-myc P2 minimal promoter. Mol. Cell. Biol. 1997, 17, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harbor Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef] [PubMed]
- Hann, S.R. MYC cofactors: Molecular switches controlling diverse biological outcomes. Cold Spring Harbor Perspect. Med. 2014, 4, a014399. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.B.; Helander, S.; Pilstal, R.; Hickman, K.A.; Lourenco, C.; Jurisica, I.; Raught, B.; Wallner, B.; Sunnerhagen, M.; Penn, L.Z. Myc and its interactors take shape. Biochim. Biophys. Acta 2015, 1849, 469–483. [Google Scholar] [CrossRef]
- Knoepfler, P.S.; Zhang, X.Y.; Cheng, P.F.; Gafken, P.R.; McMahon, S.B.; Eisenman, R.N. Myc influences global chromatin structure. EMBO J. 2006, 25, 2723–2734. [Google Scholar] [CrossRef] [Green Version]
- Blackwood, E.M.; Eisenman, R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef]
- Amati, B.; Brooks, M.W.; Levy, N.; Littlewood, T.D.; Evan, G.I.; Land, H. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell 1993, 72, 233–245. [Google Scholar] [CrossRef]
- Blackwell, T.K.; Huang, J.; Ma, A.; Kretzner, L.; Alt, F.W.; Eisenman, R.N.; Weintraub, H. Binding of myc proteins to canonical and noncanonical DNA sequences. Mol. Cell. Biol. 1993, 13, 5216–5224. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, D.; Anderton, B.; Goga, A. Taking on challenging targets: Making MYC druggable. Am. Soc. Clin. Oncol. Educ. Book Am. Soc. Clin. Oncol. Meet. 2014, e497–e502. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, F.; Yu, C.; Lai, L.; Liu, Z. Ligand clouds around protein clouds: A scenario of ligand binding with intrinsically disordered proteins. PLoS Computat. Biol. 2013, 9, e1003249. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Niu, X.; Jin, F.; Liu, Z.; Jin, C.; Lai, L. Structure-based Inhibitor Design for the Intrinsically Disordered Protein c-Myc. Sci. Rep. 2016, 6, 22298. [Google Scholar] [CrossRef] [Green Version]
- Ponzielli, R.; Katz, S.; Barsyte-Lovejoy, D.; Penn, L.Z. Cancer therapeutics: Targeting the dark side of Myc. Eur. J. Cancer 2005, 41, 2485–2501. [Google Scholar] [CrossRef] [PubMed]
- McKeown, M.R.; Bradner, J.E. Therapeutic strategies to inhibit MYC. Cold Spring Harbor Perspect. Med. 2014, 4, a014266. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Therapy 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, J.R.; Beaulieu, M.E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef]
- Koh, C.M.; Sabo, A.; Guccione, E. Targeting MYC in cancer therapy: RNA processing offers new opportunities. BioEssays News Rev. Mol. Cell. Dev. Biol. 2016, 38, 266–275. [Google Scholar] [CrossRef] [Green Version]
- Rickman, D.S.; Schulte, J.H.; Eilers, M. The Expanding World of N-MYC-Driven Tumors. Cancer Discov. 2018, 8, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Wierstra, I.; Alves, J. The c-myc promoter: Still MysterY and challenge. Adv. Cancer Res. 2008, 99, 113–333. [Google Scholar]
- Jones, T.R.; Cole, M.D. Rapid cytoplasmic turnover of c-myc mRNA: Requirement of the 3’ untranslated sequences. Mol. Cell. Biol. 1987, 7, 4513–4521. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.; Ross, J. Poly(A) shortening and degradation of the 3’ A+U-rich sequences of human c-myc mRNA in a cell-free system. Mol. Cell. Biol. 1988, 8, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, P.L.; Herrick, D.J.; Prokipcak, R.D.; Ross, J. Control of c-myc mRNA half-life in vitro by a protein capable of binding to a coding region stability determinant. Genes Dev. 1992, 6, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, P.; Mahaira, L.; Papadopoulou, A.; Teixeira, M.R.; Heim, S.; Andersen, J.A.; Evangelou, E.; Dafni, U.; Pandis, N.; Trangas, T. CRD-BP: A c-Myc mRNA stabilizing protein with an oncofetal pattern of expression. Anticancer Res. 2003, 23, 2179–2183. [Google Scholar] [PubMed]
- Bahram, F.; von der Lehr, N.; Cetinkaya, C.; Larsson, L.G. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood 2000, 95, 2104–2110. [Google Scholar]
- Gregory, M.A.; Hann, S.R. c-Myc proteolysis by the ubiquitin-proteasome pathway: Stabilization of c-Myc in Burkitt’s lymphoma cells. Mol. Cell. Biol. 2000, 20, 2423–2435. [Google Scholar] [CrossRef]
- Hemann, M.T.; Bric, A.; Teruya-Feldstein, J.; Herbst, A.; Nilsson, J.A.; Cordon-Cardo, C.; Cleveland, J.L.; Tansey, W.P.; Lowe, S.W. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 2005, 436, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Akhoondi, S.; Sun, D.; von der Lehr, N.; Apostolidou, S.; Klotz, K.; Maljukova, A.; Cepeda, D.; Fiegl, H.; Dafou, D.; Marth, C.; et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007, 67, 9006–9012. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.B.; Van Buskirk, H.A.; Dugan, K.A.; Copeland, T.D.; Cole, M.D. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell 1998, 94, 363–374. [Google Scholar] [CrossRef]
- Itzen, F.; Greifenberg, A.K.; Bosken, C.A.; Geyer, M. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014, 42, 7577–7590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Herbst, A.; Tworkowski, K.A.; Salghetti, S.E.; Tansey, W.P. Skp2 regulates Myc protein stability and activity. Mol. Cell 2003, 11, 1177–1188. [Google Scholar] [CrossRef]
- Von der Lehr, N.; Johansson, S.; Wu, S.; Bahram, F.; Castell, A.; Cetinkaya, C.; Hydbring, P.; Weidung, I.; Nakayama, K.; Nakayama, K.I.; et al. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol. Cell 2003, 11, 1189–1200. [Google Scholar] [CrossRef]
- Farrell, A.S.; Sears, R.C. MYC degradation. Cold Spring Harbor Perspect. Med. 2014, 4. [Google Scholar] [CrossRef]
- Garcia-Sanz, P.; Quintanilla, A.; Lafita, M.C.; Moreno-Bueno, G.; Garcia-Gutierrez, L.; Tabor, V.; Varela, I.; Shiio, Y.; Larsson, L.G.; Portillo, F.; et al. Sin3b interacts with Myc and decreases Myc levels. J. Biol. Chem. 2014, 289, 22221–22236. [Google Scholar] [CrossRef]
- Kurland, J.F.; Tansey, W.P. Myc-mediated transcriptional repression by recruitment of histone deacetylase. Cancer Res. 2008, 68, 3624–3629. [Google Scholar] [CrossRef]
- Thomas, L.R.; Wang, Q.; Grieb, B.C.; Phan, J.; Foshage, A.M.; Sun, Q.; Olejniczak, E.T.; Clark, T.; Dey, S.; Lorey, S.; et al. Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol. Cell 2015, 58, 440–452. [Google Scholar] [CrossRef]
- Hydbring, P.; Castell, A.; Larsson, L.G. MYC Modulation around the CDK2/p27/SKP2 Axis. Genes 2017, 8, 174. [Google Scholar] [CrossRef]
- Conacci-Sorrell, M.; Ngouenet, C.; Eisenman, R.N. Myc-nick: A cytoplasmic cleavage product of Myc that promotes alpha-tubulin acetylation and cell differentiation. Cell 2010, 142, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Conacci-Sorrell, M.; Ngouenet, C.; Anderson, S.; Brabletz, T.; Eisenman, R.N. Stress-induced cleavage of Myc promotes cancer cell survival. Genes Dev. 2014, 28, 689–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, S.; Poudel, K.R.; Roh-Johnson, M.; Brabletz, T.; Yu, M.; Borenstein-Auerbach, N.; Grady, W.N.; Bai, J.; Moens, C.B.; Eisenman, R.N.; et al. MYC-nick promotes cell migration by inducing fascin expression and Cdc42 activation. Proc. Natl. Acad. Sci. USA 2016, 113, E5481–E5490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V.; Lee, W.M. Identification of the human c-myc protein nuclear translocation signal. Mol. Cell. Biol. 1988, 8, 4048–4054. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef] [PubMed]
- Baudino, T.A.; Cleveland, J.L. The Max Network Gone Mad. Mol. Cell. Biol. 2001, 21, 691. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Spears, E.; Boone, D.N.; Li, Z.; Gregory, M.A.; Hann, S.R. Domain-specific c-Myc ubiquitylation controls c-Myc transcriptional and apoptotic activity. Proc. Natl. Acad. Sci. USA 2013, 110, 978–983. [Google Scholar] [CrossRef]
- Luscher, B. Function and regulation of the transcription factors of the Myc/Max/Mad network. Gene 2001, 277, 1–14. [Google Scholar] [CrossRef]
- Carroll, P.A.; Freie, B.W.; Mathsyaraja, H.; Eisenman, R.N. The MYC transcription factor network: Balancing metabolism, proliferation and oncogenesis. Front. Med. 2018, 12, 412–425. [Google Scholar] [CrossRef]
- Eilers, A.L.; Billin, A.N.; Liu, J.; Ayer, D.E. A 13-amino acid amphipathic alpha-helix is required for the functional interaction between the transcriptional repressor Mad1 and mSin3A. J. Biol. Chem. 1999, 274, 32750–32756. [Google Scholar] [CrossRef]
- Sun, Q.Y.; Ding, L.W.; Tan, K.T.; Chien, W.; Mayakonda, A.; Lin, D.C.; Loh, X.Y.; Xiao, J.F.; Meggendorfer, M.; Alpermann, T.; et al. Ordering of mutations in acute myeloid leukemia with partial tandem duplication of MLL (MLL-PTD). Leukemia 2017, 31, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.A.; Maclean, K.H.; Keller, U.B.; Pendeville, H.; Baudino, T.A.; Cleveland, J.L. Mnt loss triggers Myc transcription targets, proliferation, apoptosis, and transformation. Mol. Cell. Biol. 2004, 24, 1560–1569. [Google Scholar] [CrossRef] [PubMed]
- Hurlin, P.J.; Zhou, Z.Q.; Toyo-oka, K.; Ota, S.; Walker, W.L.; Hirotsune, S.; Wynshaw-Boris, A. Deletion of Mnt leads to disrupted cell cycle control and tumorigenesis. EMBO J. 2003, 22, 4584–4596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popov, N.; Wahlstrom, T.; Hurlin, P.J.; Henriksson, M. Mnt transcriptional repressor is functionally regulated during cell cycle progression. Oncogene 2005, 24, 8326–8337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretzner, L.; Blackwood, E.M.; Eisenman, R.N. Myc and Max proteins possess distinct transcriptional activities. Nature 1992, 359, 426. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Grove, L.; Prochownik, E.V. Lack of transcriptional repression by max homodimers. Oncogene 1998, 16, 2629. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, T.; Dunn, B.K.; Bar-Ner, M.; Cultraro, C.M.; Segal, S. Transfected wild-type and mutant max regulate cell growth and differentiation of murine erythroleukemia cells. Oncogene 1993, 8, 1263–1268. [Google Scholar]
- Lindeman, G.J.; Harris, A.W.; Bath, M.L.; Eisenman, R.N.; Adams, J.M. Overexpressed max is not oncogenic and attenuates myc-induced lymphoproliferation and lymphomagenesis in transgenic mice. Oncogene 1995, 10, 1013–1017. [Google Scholar]
- Yuza, Y.; Kawakami, M.; Takagi, K.; Yamazaki, Y.; Urashima, M. Max protein expression is associated with survival of children with lymphoblastic lymphoma. Pediatrics Int. 1999, 41, 637–640. [Google Scholar] [CrossRef]
- Makela, T.P.; Koskinen, P.J.; Vastrik, I.; Alitalo, K. Alternative forms of Max as enhancers or suppressors of Myc-ras cotransformation. Science 1992, 256, 373–377. [Google Scholar] [CrossRef]
- O’Shea, J.M.; Ayer, D.E. Coordination of nutrient availability and utilization by MAX- and MLX-centered transcription networks. Cold Spring Harbor Perspect. Med. 2013, 3, a014258. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, A.V.; Hu, C.D.; Kerppola, T.K. Visualization of Myc/Max/Mad family dimers and the competition for dimerization in living cells. Mol. Cell. Biol. 2004, 24, 4294–4308. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; Burley, S.K. Structural Aspects of Interactions Within the Myc/Max/Mad Network. In The Myc/Max/Mad Transcription Factor Network; Eisenman, R.N., Ed.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 123–143. [Google Scholar]
- Ferre-D’Amare, A.R.; Prendergast, G.C.; Ziff, E.B.; Burley, S.K. Recognition by Max of its cognate DNA through a dimeric b/HLH/Z domain. Nature 1993, 363, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; Burley, S.K. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell 2003, 112, 193–205. [Google Scholar] [CrossRef]
- O’Shea, E.K.; Klemm, J.D.; Kim, P.S.; Alber, T. X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science 1991, 254, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.J.; Halazonetis, T.D. Both the helix-loop-helix and the leucine zipper motifs of c-Myc contribute to its dimerization specificity with Max. Oncogene 1993, 8, 125–132. [Google Scholar] [PubMed]
- Fisher, F.; Goding, C.R. Single amino acid substitutions alter helix-loop-helix protein specificity for bases flanking the core CANNTG motif. EMBO J. 1992, 11, 4103–4109. [Google Scholar] [CrossRef]
- Dang, C.V.; Dolde, C.; Gillison, M.L.; Kato, G.J. Discrimination between related DNA sites by a single amino acid residue of Myc-related basic-helix-loop-helix proteins. Proc. Natl. Acad. Sci. USA 1992, 89, 599–602. [Google Scholar] [CrossRef]
- Jung, L.A.; Gebhardt, A.; Koelmel, W.; Ade, C.P.; Walz, S.; Kuper, J.; von Eyss, B.; Letschert, S.; Redel, C.; d’Artista, L.; et al. OmoMYC blunts promoter invasion by oncogenic MYC to inhibit gene expression characteristic of MYC-dependent tumors. Oncogene 2017, 36, 1911–1924. [Google Scholar] [CrossRef]
- Soucek, L.; Helmer-Citterich, M.; Sacco, A.; Jucker, R.; Cesareni, G.; Nasi, S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 1998, 17, 2463–2472. [Google Scholar] [CrossRef] [Green Version]
- Savino, M.; Annibali, D.; Carucci, N.; Favuzzi, E.; Cole, M.D.; Evan, G.I.; Soucek, L.; Nasi, S. The action mechanism of the Myc inhibitor termed Omomyc may give clues on how to target Myc for cancer therapy. PLoS ONE 2011, 6, e22284. [Google Scholar] [CrossRef]
- Soucek, L.; Whitfield, J.R.; Sodir, N.M.; Masso-Valles, D.; Serrano, E.; Karnezis, A.N.; Swigart, L.B.; Evan, G.I. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013, 27, 504–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Follis, A.V.; Hammoudeh, D.I.; Wang, H.; Prochownik, E.V.; Metallo, S.J. Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chem. Biol. 2008, 15, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Andrieu, G.; Belkina, A.C.; Denis, G.V. Clinical trials for BET inhibitors run ahead of the science. Drug Discov. Today Technol. 2016, 19, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Liu, C.; Liu, B.; Chen, J.; Wu, X.; Gong, W. JQ1: A novel potential therapeutic target. Die Pharmazie 2018, 73, 491–493. [Google Scholar] [PubMed]
- Clinical Trials Using BET Bromodomain Inhibitor ZEN-3694—National Cancer Institute. Available online: https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/bet-bromodomain-inhibitor-zen-3694 (accessed on 1 November 2018).
- Berthon, C.; Raffoux, E.; Thomas, X.; Vey, N.; Gomez-Roca, C.; Yee, K.; Taussig, D.C.; Rezai, K.; Roumier, C.; Herait, P.; et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: A dose-escalation, phase 1 study. Lancet Haematol. 2016, 3, e186–e195. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Dowlati, A.; LoRusso, P.M.; Eder, J.P.; Anderson, A.; Do, K.T.; Kagey, M.H.; Sirard, C.; Bradner, J.E.; Landau, S.B. Abstract A49: Clinically efficacy of the BET bromodomain inhibitor TEN-010 in an open-label substudy with patients with documented NUT-midline carcinoma (NMC). Mol. Cancer Ther. 2015, 14, A49. [Google Scholar] [CrossRef]
- Poole, C.; Zheng, W.; Lee, H.; Young, D.; Lodh, A.; Chadli, A.; van Riggelen, J. Targeting the MYC Oncogene in Burkitt Lymphoma through HSP90 Inhibition. Cancers 2018, 10, 448. [Google Scholar] [CrossRef]
- Lu, J.J.; Meng, L.H.; Shankavaram, U.T.; Zhu, C.H.; Tong, L.J.; Chen, G.; Lin, L.P.; Weinstein, J.N.; Ding, J. Dihydroartemisinin accelerates c-MYC oncoprotein degradation and induces apoptosis in c-MYC-overexpressing tumor cells. Biochem. Pharmacol. 2010, 80, 22–30. [Google Scholar] [CrossRef]
- Bayliss, R.; Burgess, S.G.; Leen, E.; Richards, M.W. A moving target: Structure and disorder in pursuit of Myc inhibitors. Biochem. Soc. Trans. 2017, 45, 709–717. [Google Scholar] [CrossRef]
- Gustafson, W.C.; Meyerowitz, J.G.; Nekritz, E.A.; Chen, J.; Benes, C.; Charron, E.; Simonds, E.F.; Seeger, R.; Matthay, K.K.; Hertz, N.T.; et al. Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell 2014, 26, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, M.; Poon, E.; Berry, T.; Carstensen, A.; Deubzer, H.E.; Rycak, L.; Jamin, Y.; Thway, K.; Robinson, S.P.; Roels, F.; et al. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell 2013, 24, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harbor Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sharma, L.; Lu, J.; Finch, P.; Fletcher, S.; Prochownik, E.V. Structurally diverse c-Myc inhibitors share a common mechanism of action involving ATP depletion. Oncotarget 2015, 6, 15857–15870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, T.; Cohen, S.B.; Desharnais, J.; Sonderegger, C.; Maslyar, D.J.; Goldberg, J.; Boger, D.L.; Vogt, P.K. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc. Natl. Acad. Sci. USA 2002, 99, 3830–3835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Stover, J.S.; Whitby, L.R.; Vogt, P.K.; Boger, D.L. Small molecule inhibitors of Myc/Max dimerization and Myc-induced cell transformation. Biorgan. Med. Chem. Lett. 2009, 19, 6038–6041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Giap, C.; Lazo, J.S.; Prochownik, E.V. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 2003, 22, 6151–6159. [Google Scholar] [CrossRef]
- Wang, H.; Chauhan, J.; Hu, A.; Pendleton, K.; Yap, J.L.; Sabato, P.E.; Jones, J.W.; Perri, M.; Yu, J.; Cione, E.; et al. Disruption of Myc-Max heterodimerization with improved cell-penetrating analogs of the small molecule 10074-G5. Oncotarget 2013, 4, 936–947. [Google Scholar] [CrossRef]
- Guo, J.; Parise, R.A.; Joseph, E.; Egorin, M.J.; Lazo, J.S.; Prochownik, E.V.; Eiseman, J.L. Efficacy, pharmacokinetics, tisssue distribution, and metabolism of the Myc-Max disruptor, 10058-F4 [Z,E]-5-[4-ethylbenzylidine]-2-thioxothiazolidin-4-one, in mice. Cancer Chemother. Pharmacol. 2009, 63, 615–625. [Google Scholar] [CrossRef]
- Clausen, D.M.; Guo, J.; Parise, R.A.; Beumer, J.H.; Egorin, M.J.; Lazo, J.S.; Prochownik, E.V.; Eiseman, J.L. In vitro cytotoxicity and in vivo efficacy, pharmacokinetics, and metabolism of 10074-G5, a novel small-molecule inhibitor of c-Myc/Max dimerization. J. Pharmacol. Exp. Ther. 2010, 335, 715–727. [Google Scholar] [CrossRef]
- Fletcher, S.; Prochownik, E.V. Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta 2015, 1849, 525–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Hammoudeh, D.I.; Follis, A.V.; Reese, B.E.; Lazo, J.S.; Metallo, S.J.; Prochownik, E.V. Improved low molecular weight Myc-Max inhibitors. Mol. Cancer Ther. 2007, 6, 2399–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, J.L.; Wang, H.; Hu, A.; Chauhan, J.; Jung, K.Y.; Gharavi, R.B.; Prochownik, E.V.; Fletcher, S. Pharmacophore identification of c-Myc inhibitor 10074-G5. Biorgan. Med. Chem. Lett. 2013, 23, 370–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, J.; Wang, H.; Yap, J.L.; Sabato, P.E.; Hu, A.; Prochownik, E.V.; Fletcher, S. Discovery of methyl 4′-methyl-5-(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)-[1,1′-biphenyl]-3-carboxylate, an improved small-molecule inhibitor of c-Myc-max dimerization. ChemMedChem 2014, 9, 2274–2285. [Google Scholar] [CrossRef] [PubMed]
- Wanner, J.; Romashko, D.; Werner, D.S.; May, E.W.; Peng, Y.; Schulz, R.; Foreman, K.W.; Russo, S.; Arnold, L.D.; Pingle, M.; et al. Reversible linkage of two distinct small molecule inhibitors of Myc generates a dimeric inhibitor with improved potency that is active in myc over-expressing cancer cell lines. PLoS ONE 2015, 10, e0121793. [Google Scholar] [CrossRef] [PubMed]
- Soodgupta, D.; Pan, D.; Cui, G.; Senpan, A.; Yang, X.; Lu, L.; Weilbaecher, K.N.; Prochownik, E.V.; Lanza, G.M.; Tomasson, M.H. Small Molecule MYC Inhibitor Conjugated to Integrin-Targeted Nanoparticles Extends Survival in a Mouse Model of Disseminated Multiple Myeloma. Mol. Cancer Ther. 2015, 14, 1286–1294. [Google Scholar] [CrossRef] [Green Version]
- Kiessling, A.; Wiesinger, R.; Sperl, B.; Berg, T. Selective inhibition of c-Myc/Max dimerization by a pyrazolo[1,5-a]pyrimidine. ChemMedChem 2007, 2, 627–630. [Google Scholar] [CrossRef]
- Kiessling, A.; Sperl, B.; Hollis, A.; Eick, D.; Berg, T. Selective inhibition of c-Myc/Max dimerization and DNA binding by small molecules. Chem. Biol. 2006, 13, 745–751. [Google Scholar] [CrossRef]
- Stellas, D.; Szabolcs, M.; Koul, S.; Li, Z.; Polyzos, A.; Anagnostopoulos, C.; Cournia, Z.; Tamvakopoulos, C.; Klinakis, A.; Efstratiadis, A. Therapeutic effects of an anti-Myc drug on mouse pancreatic cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Hart, J.R.; Garner, A.L.; Yu, J.; Ito, Y.; Sun, M.; Ueno, L.; Rhee, J.K.; Baksh, M.M.; Stefan, E.; Hartl, M.; et al. Inhibitor of MYC identified in a Krohnke pyridine library. Proc. Natl. Acad. Sci. USA 2014, 111, 12556–12561. [Google Scholar] [CrossRef] [Green Version]
- Fujimori, T.; Wirsching, P.; Janda, K.D. Preparation of a Kröhnke Pyridine Combinatorial Library Suitable for Solution-Phase Biological Screening. J. Comb. Chem. 2003, 5, 625–631. [Google Scholar] [CrossRef]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Zeller, K.I.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. An integrated database of genes responsive to the Myc oncogenic transcription factor: Identification of direct genomic targets. Genome Biol. 2003, 4, R69. [Google Scholar] [CrossRef] [PubMed]
- Schuhmacher, M.; Kohlhuber, F.; Holzel, M.; Kaiser, C.; Burtscher, H.; Jarsch, M.; Bornkamm, G.W.; Laux, G.; Polack, A.; Weidle, U.H.; et al. The transcriptional program of a human B cell line in response to Myc. Nucleic Acids Res. 2001, 29, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.H.; Mahankali, M.; Lee, S.J.; Hull, M.; Petrassi, H.M.; Chatterjee, A.K.; Schultz, P.G.; Jones, K.A.; Shen, W. Targeted Disruption of Myc-Max Oncoprotein Complex by a Small Molecule. ACS Chem. Biol. 2017, 12, 2715–2719. [Google Scholar] [CrossRef] [PubMed]
- Castell, A.; Yan, Q.; Fawkner, K.; Hydbring, P.; Zhang, F.; Verschut, V.; Franco, M.; Zakaria, S.M.; Bazzar, W.; Goodwin, J.; et al. A selective high affinity MYC-binding compound inhibits MYC:MAX interaction and MYC-dependent tumor cell proliferation. Sci. Rep. 2018, 8, 10064. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Teriete, P.; Hu, A.; Raveendra-Panickar, D.; Pendelton, K.; Lazo, J.S.; Eiseman, J.; Holien, T.; Misund, K.; Oliynyk, G.; et al. Direct inhibition of c-Myc-Max heterodimers by celastrol and celastrol-inspired triterpenoids. Oncotarget 2015, 6, 32380–32395. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.Y.; Wang, H.; Teriete, P.; Yap, J.L.; Chen, L.; Lanning, M.E.; Hu, A.; Lambert, L.J.; Holien, T.; Sundan, A.; et al. Perturbation of the c-Myc-Max protein-protein interaction via synthetic alpha-helix mimetics. J. Med. Chem. 2015, 58, 3002–3024. [Google Scholar] [CrossRef]
- Mo, H.; Henriksson, M. Identification of small molecules that induce apoptosis in a Myc-dependent manner and inhibit Myc-driven transformation. Proc. Natl. Acad. Sci. USA 2006, 103, 6344–6349. [Google Scholar] [CrossRef] [Green Version]
- Mo, H.; Vita, M.; Crespin, M.; Henriksson, M. Myc overexpression enhances apoptosis induced by small molecules. Cell Cycle 2006, 5, 2191–2194. [Google Scholar] [CrossRef]
- Jeong, K.C.; Ahn, K.O.; Yang, C.H. Small-molecule inhibitors of c-Myc transcriptional factor suppress proliferation and induce apoptosis of promyelocytic leukemia cell via cell cycle arrest. Mol. Biosyst. 2010, 6, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.C.; Kim, K.T.; Seo, H.H.; Shin, S.P.; Ahn, K.O.; Ji, M.J.; Park, W.S.; Kim, I.H.; Lee, S.J.; Seo, H.K. Intravesical instillation of c-MYC inhibitor KSI-3716 suppresses orthotopic bladder tumor growth. J. Urol. 2014, 191, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.K.; Ahn, K.O.; Jung, N.R.; Shin, J.S.; Park, W.S.; Lee, K.H.; Lee, S.J.; Jeong, K.C. Antitumor activity of the c-Myc inhibitor KSI-3716 in gemcitabine-resistant bladder cancer. Oncotarget 2014, 5, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, O.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble Docking in Drug Discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, D.A.C.D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham III, T.E.; DeBolt, S.; Fergusin, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Yao, R.; Sun, X.; Xie, Y.; Sun, X.; Yao, Y.; Li, H.; Li, Z.; Gao, J.; Xu, K. Identification of a novel c-Myc inhibitor with anti-tumor effects on multiple myeloma cells. Biosci. Rep. 2018, 38, BSR20181027. [Google Scholar] [CrossRef]
- Holien, T.; Vatsveen, T.K.; Hella, H.; Waage, A.; Sundan, A. Addiction to c-MYC in multiple myeloma. Blood 2012, 120, 2450–2453. [Google Scholar] [CrossRef] [Green Version]
- Szabo, A.G.; Gang, A.O.; Pedersen, M.O.; Poulsen, T.S.; Klausen, T.W.; Norgaard, P. Overexpression of c-myc is associated with adverse clinical features and worse overall survival in multiple myeloma. Leukemia Lymphoma 2016, 57, 2526–2534. [Google Scholar] [CrossRef]
- Spitzer, R.; Jain, A.N. Surflex-Dock: Docking benchmarks and real-world application. J. Comput.-Aided Mol. Des. 2012, 26, 687–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sybyl-X2.1. Available online: http://www.certara.com (accessed on 1 November 2018).
- Franke, N.E.; Niewerth, D.; Assaraf, Y.G.; van Meerloo, J.; Vojtekova, K.; van Zantwijk, C.H.; Zweegman, S.; Chan, E.T.; Kirk, C.J.; Geerke, D.P.; et al. Impaired bortezomib binding to mutant beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 2012, 26, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Carabet, L.A.; Lallous, N.; Leblanc, E.; Ban, F.; Morin, H.; Lawn, S.; Ghaidi, F.; Lee, J.; Mills, I.G.; Gleave, M.E.; et al. Computer-aided drug discovery of Myc-Max inhibitors as potential therapeutics for prostate cancer. Eur. J. Med. Chem. 2018, 160, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Bieberich, C.J.; Dang, C.V.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. MYC and Prostate Cancer. Genes Cancer 2010, 1, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Boutros, P.C.; Fraser, M.; Harding, N.J.; de Borja, R.; Trudel, D.; Lalonde, E.; Meng, A.; Hennings-Yeomans, P.H.; McPherson, A.; Sabelnykova, V.Y.; et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat. Genet. 2015, 47, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Dhanasekaran, S.M.; Mehra, R.; Tomlins, S.A.; Gu, W.; Yu, J.; Kumar-Sinha, C.; Cao, X.; Dash, A.; Wang, L.; et al. Integrative analysis of genomic aberrations associated with prostate cancer progression. Cancer Res. 2007, 67, 8229–8239. [Google Scholar] [CrossRef] [PubMed]
- Barfeld, S.J.; Urbanucci, A.; Itkonen, H.M.; Fazli, L.; Hicks, J.L.; Thiede, B.; Rennie, P.S.; Yegnasubramanian, S.; DeMarzo, A.M.; Mills, I.G. c-Myc Antagonises the Transcriptional Activity of the Androgen Receptor in Prostate Cancer Affecting Key Gene Networks. EBioMedicine 2017, 18, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Bernard, D.; Pourtier-Manzanedo, A.; Gil, J.; Beach, D.H. Myc confers androgen-independent prostate cancer cell growth. J. Clin. Investig. 2003, 112, 1724–1731. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef]
- Akamatsu, S.; Inoue, T.; Ogawa, O.; Gleave, M.E. Clinical and molecular features of treatment-related neuroendocrine prostate cancer. Int. J. Urol. Offi. J. Jpn. Urol. Assoc. 2018, 25, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Maestro, 2018-4; Schrödinger LLC: New York, NY, USA, 2018.
- Nadiminty, N.; Tummala, R.; Liu, C.; Lou, W.; Evans, C.P.; Gao, A.C. NF-kappaB2/p52:c-Myc:hnRNPA1 Pathway Regulates Expression of Androgen Receptor Splice Variants and Enzalutamide Sensitivity in Prostate Cancer. Mol. Cancer Ther. 2015, 14, 1884–1895. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Bower, K.E.; Beuscher, A.E.T.; Zhou, B.; Bobkov, A.A.; Olson, A.J.; Vogt, P.K. Stabilizers of the Max homodimer identified in virtual ligand screening inhibit Myc function. Mol. Pharmacol. 2009, 76, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.G.D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Berjanskii, M.V.; Neal, S.; Wishart, D.S. PREDITOR: A web server for predicting protein torsion angle restraints. Nucleic Acids Res. 2006, 34, W63–W69. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Muller, I.; Larsson, K.; Frenzel, A.; Oliynyk, G.; Zirath, H.; Prochownik, E.V.; Westwood, N.J.; Henriksson, M.A. Targeting of the MYCN protein with small molecule c-MYC inhibitors. PLoS ONE 2014, 9, e97285. [Google Scholar] [CrossRef]
- Hammoudeh, D.I.; Follis, A.V.; Prochownik, E.V.; Metallo, S.J. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J. Am. Chem. Soc. 2009, 131, 7390–7401. [Google Scholar] [CrossRef] [PubMed]
- Fisette, O.; Lague, P.; Gagne, S.; Morin, S. Synergistic applications of MD and NMR for the study of biological systems. J. Biomed. Biotechnol. 2012, 2012, 254208. [Google Scholar] [CrossRef] [PubMed]
- Ambadipudi, S.; Zweckstetter, M. Targeting intrinsically disordered proteins in rational drug discovery. Expert Opin. Drug Discov. 2016, 11, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.; Cuchillo, R. The impact of small molecule binding on the energy landscape of the intrinsically disordered protein C-myc. PLoS ONE 2012, 7, e41070. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Pei, J.; Lai, L. Binding site detection and druggability prediction of protein targets for structure-based drug design. Curr. Pharm. Des. 2013, 19, 2326–2333. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group ULC: Montreal, QC, Canada, 2018.
- Soga, S.; Shirai, H.; Kobori, M.; Hirayama, N. Use of amino acid composition to predict ligand-binding sites. J. Chem. Inf. Model. 2007, 47, 400–406. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Fiser, A.; Sali, A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar]
- Gonzalez, V.; Hurley, L.H. The c-MYC NHE III(1): Function and regulation. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 111–129. [Google Scholar] [CrossRef]
- Brooks, T.A.; Hurley, L.H. Targeting MYC Expression through G-Quadruplexes. Genes Cancer 2010, 1, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Hurley, L.H. Structure of the biologically relevant G-quadruplex in the c-MYC promoter. Nucleosides Nucleotides Nucleic Acids 2006, 25, 951–968. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S. Quadruplex nucleic acids as targets for anticancer therapeutics. Nat. Rev. Chem. 2017, 1, 0041. [Google Scholar] [CrossRef]
- Ambrus, A.; Chen, D.; Dai, J.; Jones, R.A.; Yang, D. Solution structure of the biologically relevant G-quadruplex element in the human c-MYC promoter. Implications for G-quadruplex stabilization. Biochemistry 2005, 44, 2048–2058. [Google Scholar] [CrossRef]
- Mathad, R.I.; Hatzakis, E.; Dai, J.; Yang, D. c-MYC promoter G-quadruplex formed at the 5′-end of NHE III1 element: Insights into biological relevance and parallel-stranded G-quadruplex stability. Nucleic Acids Res. 2011, 39, 9023–9033. [Google Scholar] [CrossRef] [PubMed]
- Brooks, T.A.; Hurley, L.H. The role of supercoiling in transcriptional control of MYC and its importance in molecular therapeutics. Nat. Rev. Cancer 2009, 9, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Paulo, A.; Castillo, C.C.; Neidle, S. Targeting Promoter Quadruplex Nucleic Acids for Cancer Therapy; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Duarte, A.R.; Cadoni, E.; Ressurreicao, A.S.; Moreira, R.; Paulo, A. Design of Modular G-quadruplex Ligands. ChemMedChem 2018, 13, 869–893. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Wu, Y.L.; Tanaka, Y.; Zhang, W. Small molecules targeting c-Myc oncogene: Promising anti-cancer therapeutics. Int. J. Biol. Sci. 2014, 10, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Dexheimer, T.S.; Carey, S.S.; Zuohe, S.; Gokhale, V.M.; Hu, X.; Murata, L.B.; Maes, E.M.; Weichsel, A.; Sun, D.; Meuillet, E.J.; et al. NM23-H2 may play an indirect role in transcriptional activation of c-myc gene expression but does not cleave the nuclease hypersensitive element III(1). Mol. Cancer Ther. 2009, 8, 1363–1377. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.K.; Kumar, P.; Halder, K.; Verma, A.; Kar, A.; Parent, J.L.; Basundra, R.; Kumar, A.; Chowdhury, S. Metastases suppressor NM23-H2 interaction with G-quadruplex DNA within c-MYC promoter nuclease hypersensitive element induces c-MYC expression. Nucleic Acids Res. 2009, 37, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Seenisamy, J.; Rezler, E.M.; Powell, T.J.; Tye, D.; Gokhale, V.; Joshi, C.S.; Siddiqui-Jain, A.; Hurley, L.H. The dynamic character of the G-quadruplex element in the c-MYC promoter and modification by TMPyP4. J. Am. Chem. Soc. 2004, 126, 8702–8709. [Google Scholar] [CrossRef]
- Seenisamy, J.; Bashyam, S.; Gokhale, V.; Vankayalapati, H.; Sun, D.; Siddiqui-Jain, A.; Streiner, N.; Shin-Ya, K.; White, E.; Wilson, W.D.; et al. Design and synthesis of an expanded porphyrin that has selectivity for the c-MYC G-quadruplex structure. J. Am. Chem. Soc. 2005, 127, 2944–2959. [Google Scholar] [CrossRef] [PubMed]
- Ou, T.M.; Lin, J.; Lu, Y.J.; Hou, J.Q.; Tan, J.H.; Chen, S.H.; Li, Z.; Li, Y.P.; Li, D.; Gu, L.Q.; et al. Inhibition of cell proliferation by quindoline derivative (SYUIQ-05) through its preferential interaction with c-myc promoter G-quadruplex. J. Med. Chem. 2011, 54, 5671–5679. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, K.; Xuan, S.; Toh, Z.; Zhang, D.; Shao, F. A Pt(II)-Dip complex stabilizes parallel c-myc G-quadruplex. Chem. Commun. 2013, 49, 4758–4760. [Google Scholar] [CrossRef]
- Wu, P.; Ma, D.L.; Leung, C.H.; Yan, S.C.; Zhu, N.; Abagyan, R.; Che, C.M. Stabilization of G-quadruplex DNA with platinum(II) Schiff base complexes: Luminescent probe and down-regulation of c-myc oncogene expression. Chemistry 2009, 15, 13008–13021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, F.; Li, H.; Liu, C.; Xia, J.; Ma, L.; Chu, W.; Zhang, Z.; Chen, C.; Li, S.; et al. Recent progress and future potential for metal complexes as anticancer drugs targeting G-quadruplex DNA. Curr. Med. Chem. 2012, 19, 2957–2975. [Google Scholar] [CrossRef]
- Quarfloxin in Patients With Low to Intermediate Grade Neuroendocrine Carcinoma—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00780663 (accessed on 6 December 2018).
- Duan, W.; Rangan, A.; Vankayalapati, H.; Kim, M.Y.; Zeng, Q.; Sun, D.; Han, H.; Fedoroff, O.Y.; Nishioka, D.; Rha, S.Y.; et al. Design and synthesis of fluoroquinophenoxazines that interact with human telomeric G-quadruplexes and their biological effects. Mol. Cancer Ther. 2001, 1, 103–120. [Google Scholar]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O’Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A.; et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Santos, N.D.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef] [Green Version]
- A Phase I Study of CX5461—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02719977 (accessed on 6 December 2018).
- Felsenstein, K.M.; Saunders, L.B.; Simmons, J.K.; Leon, E.; Calabrese, D.R.; Zhang, S.; Michalowski, A.; Gareiss, P.; Mock, B.A.; Schneekloth, J.S., Jr. Small Molecule Microarrays Enable the Identification of a Selective, Quadruplex-Binding Inhibitor of MYC Expression. ACS Chem. Biol. 2016, 11, 139–148. [Google Scholar] [CrossRef]
- Calabrese, D.R.; Chen, X.; Leon, E.C.; Gaikwad, S.M.; Phyo, Z.; Hewitt, W.M.; Alden, S.; Hilimire, T.A.; He, F.; Michalowski, A.M.; et al. Chemical and structural studies provide a mechanistic basis for recognition of the MYC G-quadruplex. Nat. Commun. 2018, 9, 4229. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Carver, M.; Hurley, L.H.; Yang, D. Solution structure of a 2:1 quindoline-c-MYC G-quadruplex: Insights into G-quadruplex-interactive small molecule drug design. J. Am. Chem. Soc. 2011, 133, 17673–17680. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.H.; Wang, Y.Q.; Yu, Z.Y.; Hu, L.N.; Ou, T.M.; Chen, S.B.; Huang, Z.S.; Tan, J.H. Discovery of a New Four-Leaf Clover-Like Ligand as a Potent c-MYC Transcription Inhibitor Specifically Targeting the Promoter G-Quadruplex. J. Med. Chem. 2018, 61, 2447–2459. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.B.; Tan, J.H.; Ou, T.M.; Huang, S.L.; An, L.K.; Luo, H.B.; Li, D.; Gu, L.Q.; Huang, Z.S. Pharmacophore-based discovery of triaryl-substituted imidazole as new telomeric G-quadruplex ligand. Biorgan. Med. Chem. Lett. 2011, 21, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.W.; Reddy, K.R.; Knolker, H.J. Occurrence, biogenesis, and synthesis of biologically active carbazole alkaloids. Chem. Rev. 2012, 112, 3193–3328. [Google Scholar] [CrossRef] [PubMed]
- Stump, S.; Mou, T.C.; Sprang, S.R.; Natale, N.R.; Beall, H.D. Crystal structure of the major quadruplex formed in the promoter region of the human c-MYC oncogene. PLoS ONE 2018, 13, e0205584. [Google Scholar] [CrossRef] [PubMed]
- Weaver, M.J.; Kearns, A.K.; Stump, S.; Li, C.; Gajewski, M.P.; Rider, K.C.; Backos, D.S.; Reigan, P.R.; Beall, H.D.; Natale, N.R. AIMing towards improved antitumor efficacy. Biorgan. Med. Chem. Lett. 2015, 25, 1765–1770. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Li, C.; Mosher, M.D.; Rider, K.C.; Zhou, P.; Crawford, R.L.; Fusco, W.; Paszczynski, A.; Natale, N.R. Design, synthesis and biological evaluation of a novel class of anticancer agents: Anthracenylisoxazole lexitropsin conjugates. Biorgan. Med. Chem. 2009, 17, 1671–1680. [Google Scholar] [CrossRef] [Green Version]
- Monsen, R.C.; Trent, J.O. G-quadruplex virtual drug screening: A review. Biochimie 2018, 152, 134–148. [Google Scholar] [CrossRef]
- Ma, D.L.; Chan, D.S.; Fu, W.C.; He, H.Z.; Yang, H.; Yan, S.C.; Leung, C.H. Discovery of a natural product-like c-myc G-quadruplex DNA groove-binder by molecular docking. PLoS ONE 2012, 7, e43278. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Phan, A.T.; Kuryavyi, V.; Gaw, H.Y.; Patel, D.J. Small-molecule interaction with a five-guanine-tract G-quadruplex structure from the human MYC promoter. Nat. Chem. Biol. 2005, 1, 167–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.J.; Park, H.J. In silico identification of novel ligands for G-quadruplex in the c-MYC promoter. J. Comput.-Aided Mol. Des. 2015, 29, 339–348. [Google Scholar] [CrossRef]
- Jain, A.N. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.Q.; Chen, S.B.; Zan, L.P.; Ou, T.M.; Tan, J.H.; Luyt, L.G.; Huang, Z.S. Identification of a selective G-quadruplex DNA binder using a multistep virtual screening approach. Chem. Commun. 2015, 51, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Rocca, R.; Costa, G.; Artese, A.; Parrotta, L.; Ortuso, F.; Maccioni, E.; Pinato, O.; Greco, M.L.; Sissi, C.; Alcaro, S.; et al. Hit Identification of a Novel Dual Binder for h-telo/c-myc G-Quadruplex by a Combination of Pharmacophore Structure-Based Virtual Screening and Docking Refinement. ChemMedChem 2016, 11, 1721–1733. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Bhat, J.; Mondal, S.; Sengupta, P.; Chatterjee, S. In Silico Screening and Binding Characterization of Small Molecules toward a G-Quadruplex Structure Formed in the Promoter Region of c-MYC Oncogene. ACS Omega 2017, 2, 4382–4397. [Google Scholar] [CrossRef]
- Shan, C.; Lin, J.; Hou, J.Q.; Liu, H.Y.; Chen, S.B.; Chen, A.C.; Ou, T.M.; Tan, J.H.; Li, D.; Gu, L.Q.; et al. Chemical intervention of the NM23-H2 transcriptional programme on c-MYC via a novel small molecule. Nucleic Acids Res. 2015, 43, 6677–6691. [Google Scholar] [CrossRef]
- Huth, J.R.; Yu, L.; Collins, I.; Mack, J.; Mendoza, R.; Isaac, B.; Braddock, D.T.; Muchmore, S.W.; Comess, K.M.; Fesik, S.W.; et al. NMR-driven discovery of benzoylanthranilic acid inhibitors of far upstream element binding protein binding to the human oncogene c-myc promoter. J. Med. Chem. 2004, 47, 4851–4857. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- QED—Computational Resources for Drug Discovery. Available online: crdd.osdd.net/oscadd/qed/ (accessed on 1 November 2018).
- Sterling, T.; Irwin, J.J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.-P.; Chari, A.; Ciferri, C.; Liu, W.-T.; Rémigy, H.-W.; Stark, H.; Wiesmann, C. Cryo-EM in drug discovery: Achievements, limitations and prospects. Nat. Rev. Drug Discov. 2018, 17, 471. [Google Scholar] [CrossRef] [PubMed]














© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carabet, L.A.; Rennie, P.S.; Cherkasov, A. Therapeutic Inhibition of Myc in Cancer. Structural Bases and Computer-Aided Drug Discovery Approaches. Int. J. Mol. Sci. 2019, 20, 120. https://doi.org/10.3390/ijms20010120
Carabet LA, Rennie PS, Cherkasov A. Therapeutic Inhibition of Myc in Cancer. Structural Bases and Computer-Aided Drug Discovery Approaches. International Journal of Molecular Sciences. 2019; 20(1):120. https://doi.org/10.3390/ijms20010120
Chicago/Turabian StyleCarabet, Lavinia A., Paul S. Rennie, and Artem Cherkasov. 2019. "Therapeutic Inhibition of Myc in Cancer. Structural Bases and Computer-Aided Drug Discovery Approaches" International Journal of Molecular Sciences 20, no. 1: 120. https://doi.org/10.3390/ijms20010120
APA StyleCarabet, L. A., Rennie, P. S., & Cherkasov, A. (2019). Therapeutic Inhibition of Myc in Cancer. Structural Bases and Computer-Aided Drug Discovery Approaches. International Journal of Molecular Sciences, 20(1), 120. https://doi.org/10.3390/ijms20010120