The Effects of Systemic and Local Acidosis on Insulin Resistance and Signaling



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
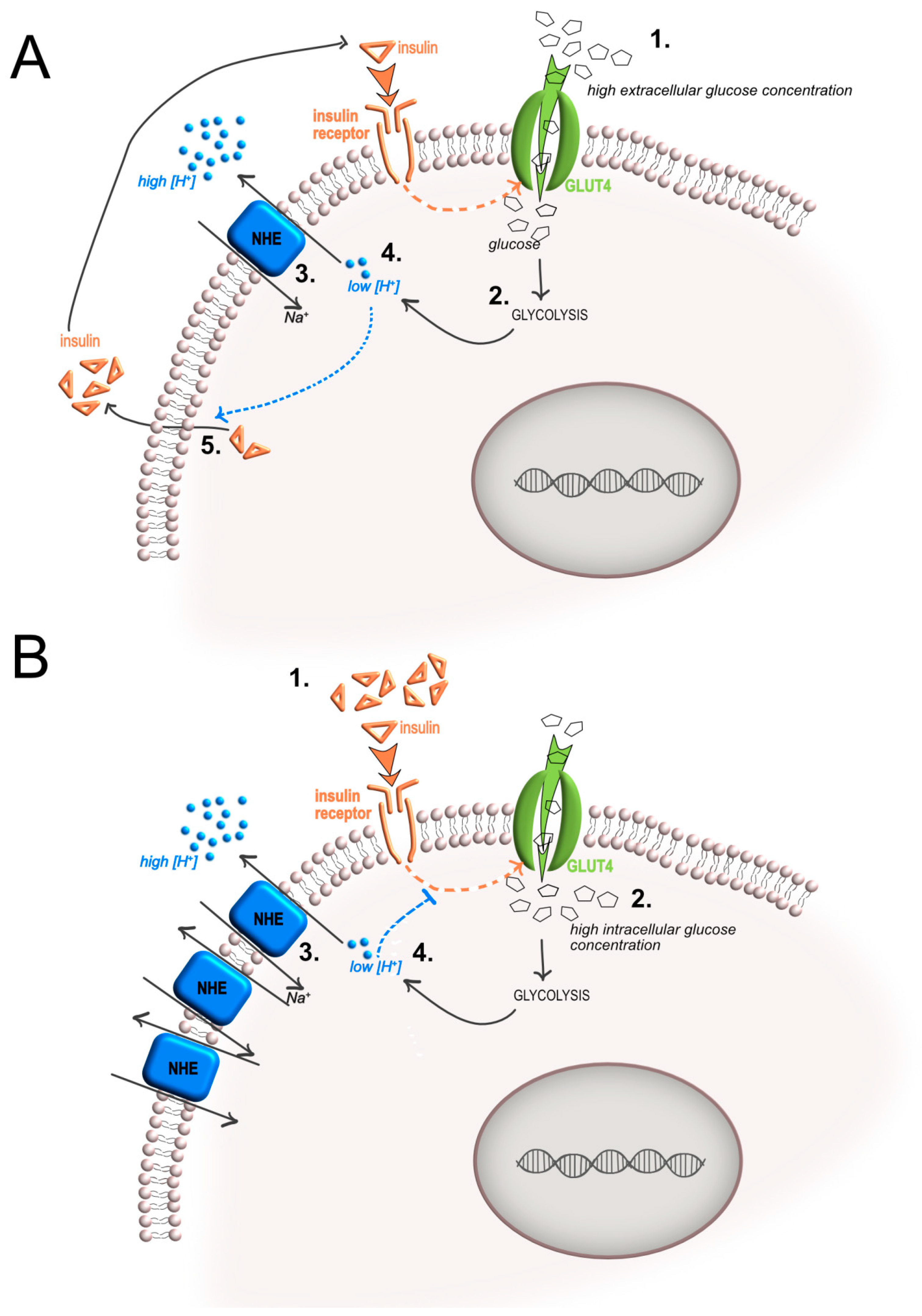
2. Insulin-Mediated Increase of Glycolysis and Local Acidosis
3. Acidosis-Mediated Effects on Insulin Activity and Resistance
3.1. Effects of Acidosis on Insulin Sensitivity and Release
3.2. Effects of Acidosis on Insulin Receptor Expression, Activation, and Signaling
3.3. Effect of Acidosis on the Expression of GLUT Transporters
4. Intracellular pH Regulation by Proton/Ion Transporters and Insulin Secretion
5. Insulin Receptors and Sensing Extracellular Acidosis
6. Insulin Receptor-Induced Inflammatory Pathway and Acidosis
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| acidoCEST MRI | acido chemical exchange saturation transfer magnetic resonance imaging |
| AIDS | acquired immunodeficiency syndrome |
| ASICS | acid-sensing ion channels |
| CA | carbonic anhydrases |
| [18F]FDG PET | 18F-fluoro-2-deoxy-d-glucose positron emission tomography |
| GLUT | glucose transporters |
| GPCR | metabotropic proton-sensing G protein-coupled receptors |
| GTPases | guanosintrifosfato hydrolases |
| HE | Na+/H+ exchanger |
| HIF | hypoxia-inducible factor |
| HK | hexokinase |
| IκBα | the nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha |
| IR | insulin receptor |
| IRS-1 | insulin receptor substrate 1 |
| LDH | lactic dehydrogenase |
| LDH-A | lactate dehydrogenase a |
| MCT | monocarboxylate transporter |
| MEK/ERK | mitogen-activated protein kinases/extracellular signal-regulated kinases |
| MSC | mesechymal stromal cells |
| NAD | nicotinammide adenina dinucleotide |
| NF-κB | Nuclear Factor kappaB |
| NHE | sodium–hydrogen exchanger |
| NSIS | nutrient-stimulated insulin secretion |
| OGR1 | ovarian cancer G protein-coupled receptor 1 |
| OxPhos | oxidative phosphorylation |
| PFK | phosphofructokinase |
| PGI | phosphoglucose isomerase |
| pHi | intracellular pH |
| PK | pyruvate kinase |
| PKM2 | pyruvate kinase isozymes m2 |
| SIRT-1 | Sirtuin 1 |
| TGF-β | transforming growth factor beta |
| TPI | triosephosphate isomerase |
| TRPV | transient receptor potential vanilloid |
| V-ATPase | vacuolar ATP-ase |
References
- Arnett, T. Regulation of bone cell function by acid-base balance. Proc. Nutr. Soc. 2003, 62, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Ratel, S.; Duche, P.; Hennegrave, A.; Van Praagh, E.; Bedu, M. Acid-base balance during repeated cycling sprints in boys and men. J. Appl. Physiol. (1985) 2002, 92, 479–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frassetto, L.A.; Morris, R.C., Jr.; Sebastian, A. Effect of age on blood acid-base composition in adult humans: Role of age-related renal functional decline. Am. J. Physiol. 1996, 271, F1114–F1122. [Google Scholar] [CrossRef] [PubMed]
- Kolosenko, I.; Avnet, S.; Baldini, N.; Viklund, J.; De Milito, A. Therapeutic implications of tumor interstitial acidification. Semin. Cancer Biol. 2017, 43, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Massa, A.; Perut, F.; Chano, T.; Woloszyk, A.; Mitsiadis, T.A.; Avnet, S.; Baldini, N. The effect of extracellular acidosis on the behaviour of mesenchymal stem cells in vitro. Eur. Cell. Mater. 2017, 33, 252–267. [Google Scholar] [CrossRef] [PubMed]
- Marunaka, Y.; Yoshimoto, K.; Aoi, W.; Hosogi, S.; Ikegaya, H. Low pH of interstitial fluid around hippocampus of the brain in diabetic OLETF rats. Mol. Cell. Ther. 2014, 2, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Chiang, S.H.; Saltiel, A.R. Insulin signaling and the regulation of glucose transport. Mol. Med. 2004, 10, 65–71. [Google Scholar] [PubMed]
- Goguen, J.M.; Halperin, M.L. Can insulin administration cause an acute metabolic acidosis in vivo? An experimental study in dogs. Diabetologia 1993, 36, 813–816. [Google Scholar] [CrossRef]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- Adeva-Andany, M.; Lopez-Ojen, M.; Funcasta-Calderon, R.; Ameneiros-Rodriguez, E.; Donapetry-Garcia, C.; Vila-Altesor, M.; Rodriguez-Seijas, J. Comprehensive review on lactate metabolism in human health. Mitochondrion 2014, 17, 76–100. [Google Scholar] [CrossRef]
- Seheult, J.; Fitzpatrick, G.; Boran, G. Lactic acidosis: An update. Clin. Chem. Lab. Med. 2017, 55, 322–333. [Google Scholar] [CrossRef]
- Robergs, R.A.; Ghiasvand, F.; Parker, D. Biochemistry of exercise-induced metabolic acidosis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R502–R516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Price, N.T. The proton-linked monocarboxylate transporter (MCT) family: Structure, function and regulation. Biochem. J. 1999, 343, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Spugnini, E.P.; Sonveaux, P.; Stock, C.; Perez-Sayans, M.; De Milito, A.; Avnet, S.; Garcia, A.G.; Harguindey, S.; Fais, S. Proton channels and exchangers in cancer. Biochim. Biophys. Acta 2015, 1848, 2715–2726. [Google Scholar] [CrossRef] [PubMed]
- Naylor, J.M.; Kronfeld, D.S.; Freeman, D.E.; Richardson, D. Hepatic and extrahepatic lactate metabolism in sheep: Effects of lactate loading and pH. Am. J. Physiol. 1984, 247, E747–E755. [Google Scholar] [CrossRef] [PubMed]
- Ewaschuk, J.B.; Naylor, J.M.; Zello, G.A. d-lactate in human and ruminant metabolism. J. Nutr. 2005, 135, 1619–1625. [Google Scholar] [CrossRef]
- Claus, P.; Gimenes, A.M.; Castro, J.R.; Mantovani, M.M.; Kanayama, K.K.; Simoes, D.M.N.; Schwartz, D.S. Blood lactate concentration in diabetic dogs. Can. Vet. J. 2017, 58, 817–822. [Google Scholar]
- Lu, J.; Zello, G.A.; Randell, E.; Adeli, K.; Krahn, J.; Meng, Q.H. Closing the anion gap: Contribution of d-lactate to diabetic ketoacidosis. Clin. Chim. Acta 2011, 412, 286–291. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Goldenberg, J.M.; Cardenas-Rodriguez, J.; Pagel, M.D. Preliminary Results that Assess Metformin Treatment in a Preclinical Model of Pancreatic Cancer Using Simultaneous [(18)F]FDG PET and acidoCEST MRI. Mol. Imaging Biol. 2018, 20, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Longo, D.L.; Bartoli, A.; Consolino, L.; Bardini, P.; Arena, F.; Schwaiger, M.; Aime, S. In Vivo Imaging of Tumor Metabolism and Acidosis by Combining PET and MRI-CEST pH Imaging. Cancer Res. 2016, 76, 6463–6470. [Google Scholar] [CrossRef] [PubMed]
- Yajima, M.; Ui, M. Carbohydrate metabolism and its response to catecholamines as modified in alkalotic rat. Am. J. Physiol. 1975, 228, 1046–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uajima, M.; Ui, M. Hydrocortisone restoration of the pH-dependent metabolic responses to catecholamines. Am. J. Physiol. 1975, 228, 1053–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, M.; Yamatani, K.; Fukase, N.; Daimon, M.; Ohnuma, H.; Ogawa, A.; Tominaga, M.; Sasaki, H. Effect of acidosis on insulin binding and glucose uptake in isolated rat adipocytes. Tohoku J. Exp. Med. 1993, 169, 205–213. [Google Scholar] [CrossRef]
- Cuthbert, C.; Alberti, K.G. Acidemia and insulin resistance in the diabetic ketoacidotic rat. Metabolism 1978, 27, 1903–1916. [Google Scholar] [CrossRef]
- Mak, R.H. Effect of metabolic acidosis on insulin action and secretion in uremia. Kidney Int. 1998, 54, 603–607. [Google Scholar] [CrossRef]
- Reaich, D.; Graham, K.A.; Channon, S.M.; Hetherington, C.; Scrimgeour, C.M.; Wilkinson, R.; Goodship, T.H. Insulin-mediated changes in PD and glucose uptake after correction of acidosis in humans with CRF. Am. J. Physiol. 1995, 268, E121–E126. [Google Scholar] [CrossRef]
- Chano, T.; Avnet, S.; Kusuzaki, K.; Bonuccelli, G.; Sonveaux, P.; Rotili, D.; Mai, A.; Baldini, N. Tumour-specific metabolic adaptation to acidosis is coupled to epigenetic stability in osteosarcoma cells. Am. J. Cancer Res. 2016, 6, 859–875. [Google Scholar]
- Lamonte, G.; Tang, X.; Chen, J.L.; Wu, J.; Ding, C.K.; Keenan, M.M.; Sangokoya, C.; Kung, H.N.; Ilkayeva, O.; Boros, L.G.; et al. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab. 2013, 1, 23. [Google Scholar] [CrossRef] [Green Version]
- Corbet, C.; Draoui, N.; Polet, F.; Pinto, A.; Drozak, X.; Riant, O.; Feron, O. The SIRT1/HIF2alpha axis drives reductive glutamine metabolism under chronic acidosis and alters tumor response to therapy. Cancer Res. 2014, 74, 5507–5519. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wu, H.; Dai, C.; Pan, Q.; Ding, Z.; Hu, D.; Ji, B.; Luo, Y.; Hu, X. Beyond Warburg effect-dual metabolic nature of cancer cells. Sci. Rep. 2014, 4, 4927. [Google Scholar] [CrossRef] [PubMed]
- Peppicelli, S.; Bianchini, F.; Toti, A.; Laurenzana, A.; Fibbi, G.; Calorini, L. Extracellular acidity strengthens mesenchymal stem cells to promote melanoma progression. Cell Cycle 2015, 14, 3088–3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemma, S.; Di Pompo, G.; Porporato, P.E.; Sboarina, M.; Russell, S.; Gillies, R.J.; Baldini, N.; Sonveaux, P.; Avnet, S. MDA-MB-231 breast cancer cells fuel osteoclast metabolism and activity: A new rationale for the pathogenesis of osteolytic bone metastases. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3254–3264. [Google Scholar] [CrossRef] [PubMed]
- Counillon, L.; Bouret, Y.; Marchiq, I.; Pouyssegur, J. Na(+)/H(+) antiporter (NHE1) and lactate/H(+) symporters (MCTs) in pH homeostasis and cancer metabolism. Biochim. Biophys. Acta 2016, 1863, 2465–2480. [Google Scholar] [CrossRef] [PubMed]
- Cabalgante, M.J.; Gadola, L.; Luzardo, L.; Marquez, M.; Boggia, J.; Boim, M.A. Calcium citrate improves the epithelial-to-mesenchymal transition induced by acidosis in proximal tubular cells. J. Bras. Nefrol. 2012, 34, 343–348. [Google Scholar] [CrossRef]
- Uribe-San Martin, R.; Herrera-Molina, R.; Olavarria, L.; Ramirez, G.; von Bernhardi, R. Reduction of beta-amyloid-induced neurotoxicity on hippocampal cell cultures by moderate acidosis is mediated by transforming growth factor beta. Neuroscience 2009, 158, 1338–1347. [Google Scholar] [CrossRef]
- Smart, N.G.; Apelqvist, A.A.; Gu, X.; Harmon, E.B.; Topper, J.N.; MacDonald, R.J.; Kim, S.K. Conditional expression of Smad7 in pancreatic beta cells disrupts TGF-beta signaling and induces reversible diabetes mellitus. PLoS Biol. 2006, 4, e39. [Google Scholar] [CrossRef]
- Lin, H.M.; Lee, J.H.; Yadav, H.; Kamaraju, A.K.; Liu, E.; Zhigang, D.; Vieira, A.; Kim, S.J.; Collins, H.; Matschinsky, F.; et al. Transforming growth factor-beta/Smad3 signaling regulates insulin gene transcription and pancreatic islet beta-cell function. J. Biol. Chem. 2009, 284, 12246–12257. [Google Scholar] [CrossRef]
- Dhawan, S.; Dirice, E.; Kulkarni, R.N.; Bhushan, A. Inhibition of TGF-beta Signaling Promotes Human Pancreatic beta-Cell Replication. Diabetes 2016, 65, 1208–1218. [Google Scholar] [CrossRef]
- Hidaka, H.; Howard, B.V.; Ishibashi, F.; Kosmakos, F.C.; Craig, J.W.; Bennett, P.H.; Larner, J. Effect of pH and 3-hydroxybutyrate on insulin binding and action in cultured human fibroblasts. Diabetes 1981, 30, 402–406. [Google Scholar] [CrossRef]
- Hayata, H.; Miyazaki, H.; Niisato, N.; Yokoyama, N.; Marunaka, Y. Lowered extracellular pH is involved in the pathogenesis of skeletal muscle insulin resistance. Biochem. Biophys. Res. Commun. 2014, 445, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, J.; Cuthbert, C.; Hammond, V.A.; Alberti, K.G. The effects of metabolic acidosis in vivo on insulin binding to isolated rat adipocytes. Metabolism 1982, 31, 553–557. [Google Scholar] [CrossRef]
- Hara, H.; Hidaka, H.; Kosmakos, F.C.; Mott, D.M.; Vasquez, B.; Howard, B.V.; Bennett, P.H. Characterization of the human insulin receptor solubilized from cultured fibroblast and erythrocyte cell membrane preparations. J. Clin. Endocrinol. Metab. 1981, 52, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Van Putten, J.P.; Wieringa, T.; Krans, H.M. Low pH and ketoacids induce insulin receptor binding and postbinding alterations in cultured 3T3 adipocytes. Diabetes 1985, 34, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, H.; Yamauchi, K.; Ohara, N.; Miyamoto, T.; Ichikawa, K.; Hashizume, K. Effects of beta-hydroxy butyric acid on insulin binding to its receptor and on autophosphorylation of the receptor. Endocrinol. Jpn. 1990, 37, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Haft, C.R.; De La Luz Sierra, M.; Hamer, I.; Carpentier, J.L.; Taylor, S.I. Analysis of the juxtamembrane dileucine motif in the insulin receptor. Endocrinology 1998, 139, 1618–1629. [Google Scholar] [CrossRef]
- Kasuga, M.; Kahn, C.R.; Hedo, J.A.; Van Obberghen, E.; Yamada, K.M. Insulin-induced receptor loss in cultured human lymphocytes is due to accelerated receptor degradation. Proc. Natl. Acad. Sci. USA 1981, 78, 6917–6921. [Google Scholar] [CrossRef]
- Bertacca, A.; Ciccarone, A.; Cecchetti, P.; Vianello, B.; Laurenza, I.; Del Prato, S.; Benzi, L. High insulin levels impair intracellular receptor trafficking in human cultured myoblasts. Diabetes Res. Clin. Pract. 2007, 78, 316–323. [Google Scholar] [CrossRef]
- Salerno, M.; Avnet, S.; Bonuccelli, G.; Hosogi, S.; Granchi, D.; Baldini, N. Impairment of lysosomal activity as a therapeutic modality targeting cancer stem cells of embryonal rhabdomyosarcoma cell line RD. PLoS ONE 2014, 9, e110340. [Google Scholar] [CrossRef]
- Avnet, S.; Lemma, S.; Cortini, M.; Pellegrini, P.; Perut, F.; Zini, N.; Kusuzaki, K.; Chano, T.; Grisendi, G.; Dominici, M.; et al. Altered pH gradient at the plasma membrane of osteosarcoma cells is a key mechanism of drug resistance. Oncotarget 2016, 7, 63408–63423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glunde, K.; Guggino, S.E.; Solaiyappan, M.; Pathak, A.P.; Ichikawa, Y.; Bhujwalla, Z.M. Extracellular acidification alters lysosomal trafficking in human breast cancer cells. Neoplasia 2003, 5, 533–545. [Google Scholar] [CrossRef]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Taha, C.; Mitsumoto, Y.; Liu, Z.; Skolnik, E.Y.; Klip, A. The insulin-dependent biosynthesis of GLUT1 and GLUT3 glucose transporters in L6 muscle cells is mediated by distinct pathways. Roles of p21ras and pp70 S6 kinase. J. Biol. Chem. 1995, 270, 24678–24681. [Google Scholar] [CrossRef] [PubMed]
- Cormont, M.; Le Marchand-Brustel, Y. The role of small G-proteins in the regulation of glucose transport (review). Mol. Membr. Biol. 2001, 18, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Kaddai, V.; Gonzalez, T.; Keslair, F.; Gremeaux, T.; Bonnafous, S.; Gugenheim, J.; Tran, A.; Gual, P.; Le Marchand-Brustel, Y.; Cormont, M. Rab4b is a small GTPase involved in the control of the glucose transporter GLUT4 localization in adipocyte. PLoS ONE 2009, 4, e5257. [Google Scholar] [CrossRef]
- Kessler, A.; Tomas, E.; Immler, D.; Meyer, H.E.; Zorzano, A.; Eckel, J. Rab11 is associated with GLUT4-containing vesicles and redistributes in response to insulin. Diabetologia 2000, 43, 1518–1527. [Google Scholar] [CrossRef] [Green Version]
- Uhlig, M.; Passlack, W.; Eckel, J. Functional role of Rab11 in GLUT4 trafficking in cardiomyocytes. Mol. Cell. Endocrinol. 2005, 235, 1–9. [Google Scholar] [CrossRef]
- Oehlke, O.; Martin, H.W.; Osterberg, N.; Roussa, E. Rab11b and its effector Rip11 regulate the acidosis-induced traffic of V-ATPase in salivary ducts. J. Cell. Physiol. 2011, 226, 638–651. [Google Scholar] [CrossRef]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef]
- Iwanaga, T.; Kishimoto, A. Cellular distributions of monocarboxylate transporters: A review. Biomed. Res. 2015, 36, 279–301. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.S.; Morris, M.E. Monocarboxylate Transporters: Therapeutic Targets and Prognostic Factors in Disease. Clin. Pharmacol. Ther. 2016, 100, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damaghi, M.; Wojtkowiak, J.W.; Gillies, R.J. pH sensing and regulation in cancer. Front. Physiol. 2013, 4, 370. [Google Scholar] [CrossRef] [PubMed]
- Gunawardana, S.C.; Rocheleau, J.V.; Head, W.S.; Piston, D.W. Nutrient-stimulated insulin secretion in mouse islets is critically dependent on intracellular pH. BMC Endocr. Disord. 2004, 4, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juntti-Berggren, L.; Arkhammar, P.; Nilsson, T.; Rorsman, P.; Berggren, P.O. Glucose-induced increase in cytoplasmic pH in pancreatic beta-cells is mediated by Na+/H+ exchange, an effect not dependent on protein kinase C. J. Biol. Chem. 1991, 266, 23537–23541. [Google Scholar] [PubMed]
- Best, L.; Bone, E.A.; Meats, J.E.; Tomlinson, S. Is intracellular pH a coupling factor in nutrient-stimulated pancreatic islets? J. Mol. Endocrinol. 1988, 1, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Nabe, K.; Fujimoto, S.; Shimodahira, M.; Kominato, R.; Nishi, Y.; Funakoshi, S.; Mukai, E.; Yamada, Y.; Seino, Y.; Inagaki, N. Diphenylhydantoin suppresses glucose-induced insulin release by decreasing cytoplasmic H+ concentration in pancreatic islets. Endocrinology 2006, 147, 2717–2727. [Google Scholar] [CrossRef]
- Klip, A.; Ramlal, T.; Cragoe, E.J., Jr. Insulin-induced cytoplasmic alkalinization and glucose transport in muscle cells. Am. J. Physiol. 1986, 250, C720–C728. [Google Scholar] [CrossRef]
- Deisl, C.; Albano, G.; Fuster, D.G. Role of Na/H exchange in insulin secretion by islet cells. Curr. Opin. Nephrol. Hypertens. 2014, 23, 406–410. [Google Scholar] [CrossRef] [Green Version]
- Ohishi, M. Hypertension with diabetes mellitus: Physiology and pathology. Hypertens. Res. 2018, 41, 389–393. [Google Scholar] [CrossRef]
- Romero, J.R.; Rivera, A.; Conlin, P.R. Red blood cell Na+/H+ exchange activity is insulin resistant in hypertensive patients. Clin. Exp. Hypertens. 2002, 24, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Parui, R.; Gambhir, K.K.; Mehrotra, P.P. Changes in carbonic anhydrase may be the initial step of altered metabolism in hypertension. Biochem. Int. 1991, 23, 779–789. [Google Scholar] [PubMed]
- Holzer, P. Acid-sensitive ion channels and receptors. Handb. Exp. Pharmacol. 2009, 194, 283–332. [Google Scholar]
- Zheng, J. Molecular mechanism of TRP channels. Compr. Physiol. 2013, 3, 221–242. [Google Scholar] [PubMed]
- Cheng, Y.R.; Jiang, B.Y.; Chen, C.C. Acid-sensing ion channels: Dual function proteins for chemo-sensing and mechano-sensing. J. Biomed. Sci. 2018, 25, 46. [Google Scholar] [CrossRef] [PubMed]
- Avnet, S.; Di Pompo, G.; Chano, T.; Errani, C.; Ibrahim-Hashim, A.; Gillies, R.J.; Donati, D.M.; Baldini, N. Cancer-associated mesenchymal stroma fosters the stemness of osteosarcoma cells in response to intratumoral acidosis via NF-kappaB activation. Int. J. Cancer 2017, 140, 1331–1345. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.J.; Yang, W.S.; Lin, Y.W.; Wang, H.C.; Chen, C.C. Increase of insulin sensitivity and reversal of age-dependent glucose intolerance with inhibition of ASIC3. Biochem. Biophys. Res. Commun. 2008, 371, 729–734. [Google Scholar] [CrossRef]
- Uchida, K.; Tominaga, M. The role of thermosensitive TRP (transient receptor potential) channels in insulin secretion. Endocr. J. 2011, 58, 1021–1028. [Google Scholar] [CrossRef]
- Mogi, C.; Nakakura, T.; Okajima, F. Role of extracellular proton-sensing OGR1 in regulation of insulin secretion and pancreatic beta-cell functions. Endocr. J. 2014, 61, 101–110. [Google Scholar] [CrossRef]
- Punnia-Moorthy, A. Evaluation of pH changes in inflammation of the subcutaneous air pouch lining in the rat, induced by carrageenan, dextran and Staphylococcus aureus. J. Oral. Pathol. 1987, 16, 36–44. [Google Scholar] [CrossRef]
- Di Pompo, G.; Lemma, S.; Canti, L.; Rucci, N.; Ponzetti, M.; Errani, C.; Donati, D.M.; Russell, S.; Gillies, R.; Chano, T.; et al. Intratumoral acidosis fosters cancer-induced bone pain through the activation of the mesenchymal tumor-associated stroma in bone metastasis from breast carcinoma. Oncotarget 2017, 8, 54478–54496. [Google Scholar] [CrossRef] [PubMed]
- Nagasaka, A.; Mogi, C.; Ono, H.; Nishi, T.; Horii, Y.; Ohba, Y.; Sato, K.; Nakaya, M.; Okajima, F.; Kurose, H. The proton-sensing G protein-coupled receptor T-cell death-associated gene 8 (TDAG8) shows cardioprotective effects against myocardial infarction. Sci. Rep. 2017, 7, 7812. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yu, R.; Xiong, Y.; Du, F.; Zhu, S. A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids Health Dis. 2017, 16, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wu, W.; Li, D.; Guo, Y.; Ding, H. Overactivation of NF-kappaB impairs insulin sensitivity and mediates palmitate-induced insulin resistance in C2C12 skeletal muscle cells. Endocrine 2010, 37, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Ma, Y.; Yan, J.; Liu, J.; Li, L. Geniposide promotes autophagy to inhibit insulin resistance in HepG2 cells via P62/NFkappaB/GLUT4. Mol. Med. Rep. 2017, 16, 7237–7244. [Google Scholar] [CrossRef] [PubMed]
- Sauter, N.S.; Schulthess, F.T.; Galasso, R.; Castellani, L.W.; Maedler, K. The antiinflammatory cytokine interleukin-1 receptor antagonist protects from high-fat diet-induced hyperglycemia. Endocrinology 2008, 149, 2208–2218. [Google Scholar] [CrossRef] [PubMed]
- Feve, B.; Bastard, J.P. The role of interleukins in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2009, 5, 305–311. [Google Scholar] [CrossRef]
- Robey, I.F.; Baggett, B.K.; Kirkpatrick, N.D.; Roe, D.J.; Dosescu, J.; Sloane, B.F.; Hashim, A.I.; Morse, D.L.; Raghunand, N.; Gatenby, R.A.; et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009, 69, 2260–2268. [Google Scholar] [CrossRef]
- Della Guardia, L.; Thomas, M.A.; Cena, H. Insulin Sensitivity and Glucose Homeostasis Can Be Influenced by Metabolic Acid Load. Nutrients 2018, 10, 618. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldini, N.; Avnet, S. The Effects of Systemic and Local Acidosis on Insulin Resistance and Signaling. Int. J. Mol. Sci. 2019, 20, 126. https://doi.org/10.3390/ijms20010126
Baldini N, Avnet S. The Effects of Systemic and Local Acidosis on Insulin Resistance and Signaling. International Journal of Molecular Sciences. 2019; 20(1):126. https://doi.org/10.3390/ijms20010126
Chicago/Turabian StyleBaldini, Nicola, and Sofia Avnet. 2019. "The Effects of Systemic and Local Acidosis on Insulin Resistance and Signaling" International Journal of Molecular Sciences 20, no. 1: 126. https://doi.org/10.3390/ijms20010126
APA StyleBaldini, N., & Avnet, S. (2019). The Effects of Systemic and Local Acidosis on Insulin Resistance and Signaling. International Journal of Molecular Sciences, 20(1), 126. https://doi.org/10.3390/ijms20010126