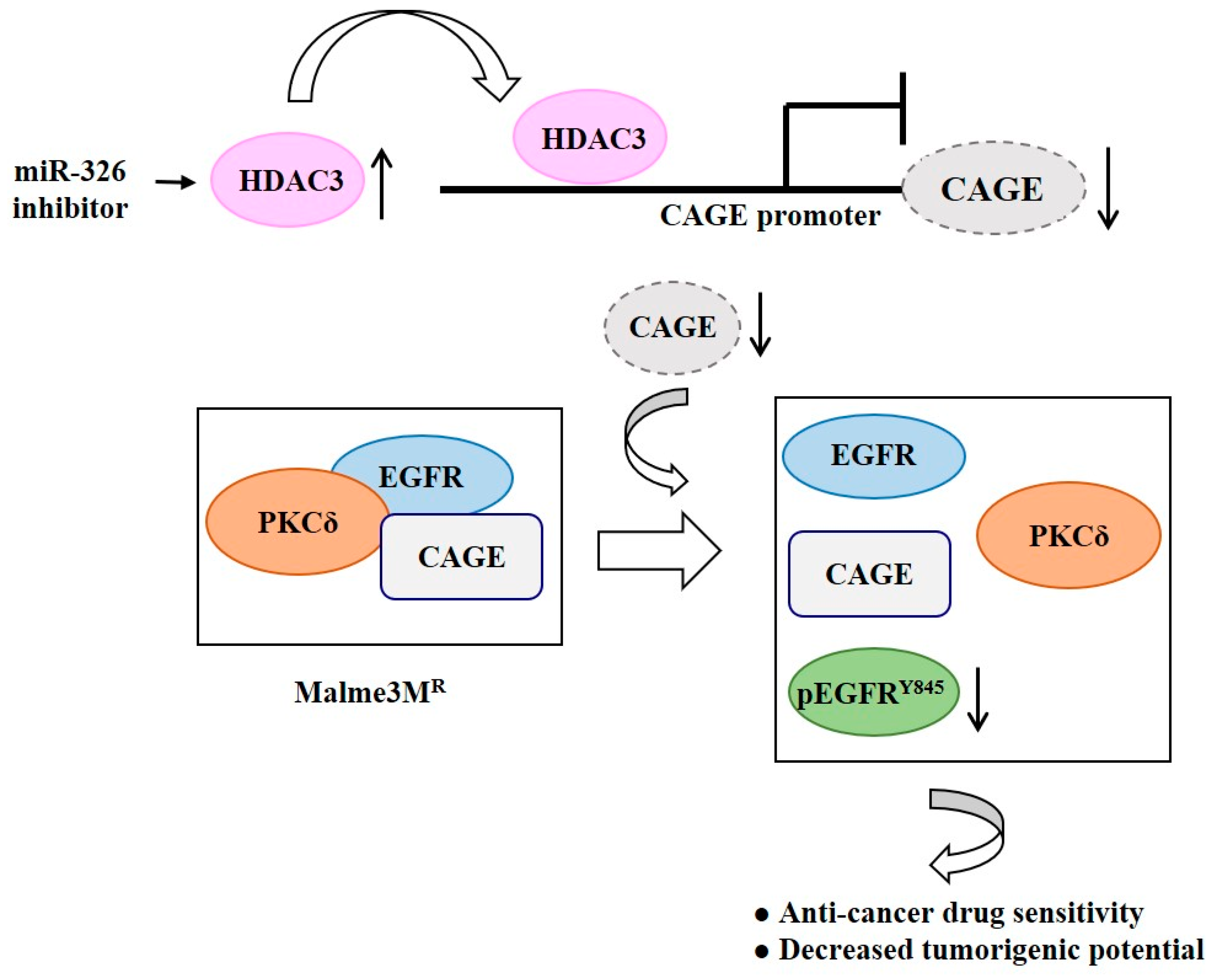
Role of HDAC3-miRNA-CAGE Network in Anti-Cancer Drug-Resistance


Abstract
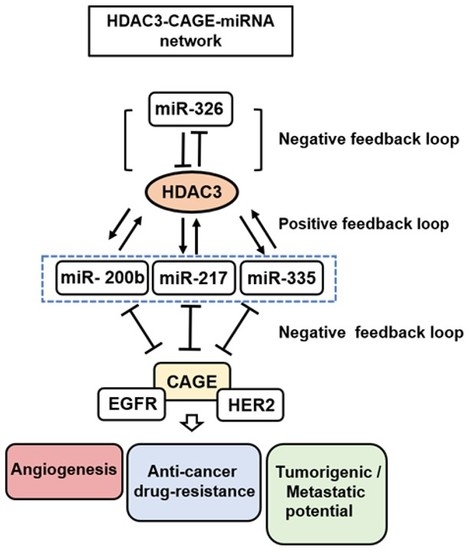
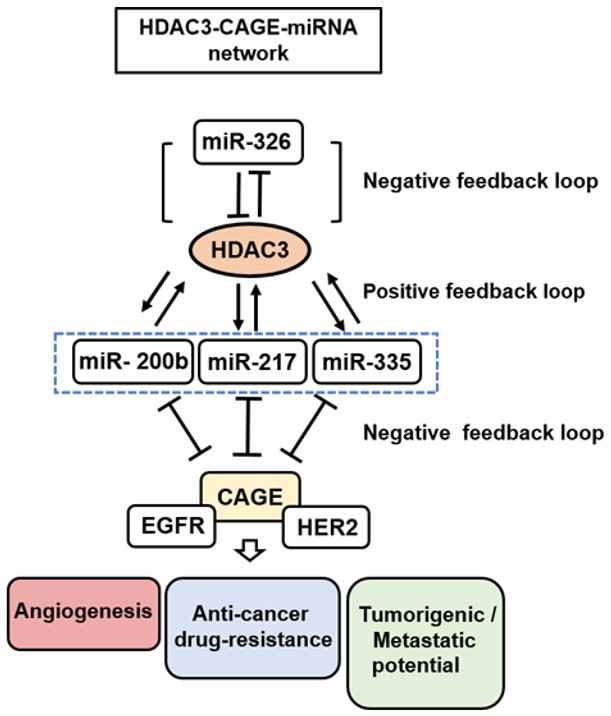
:
{kind=link}
{kind=link}
{kind=link}
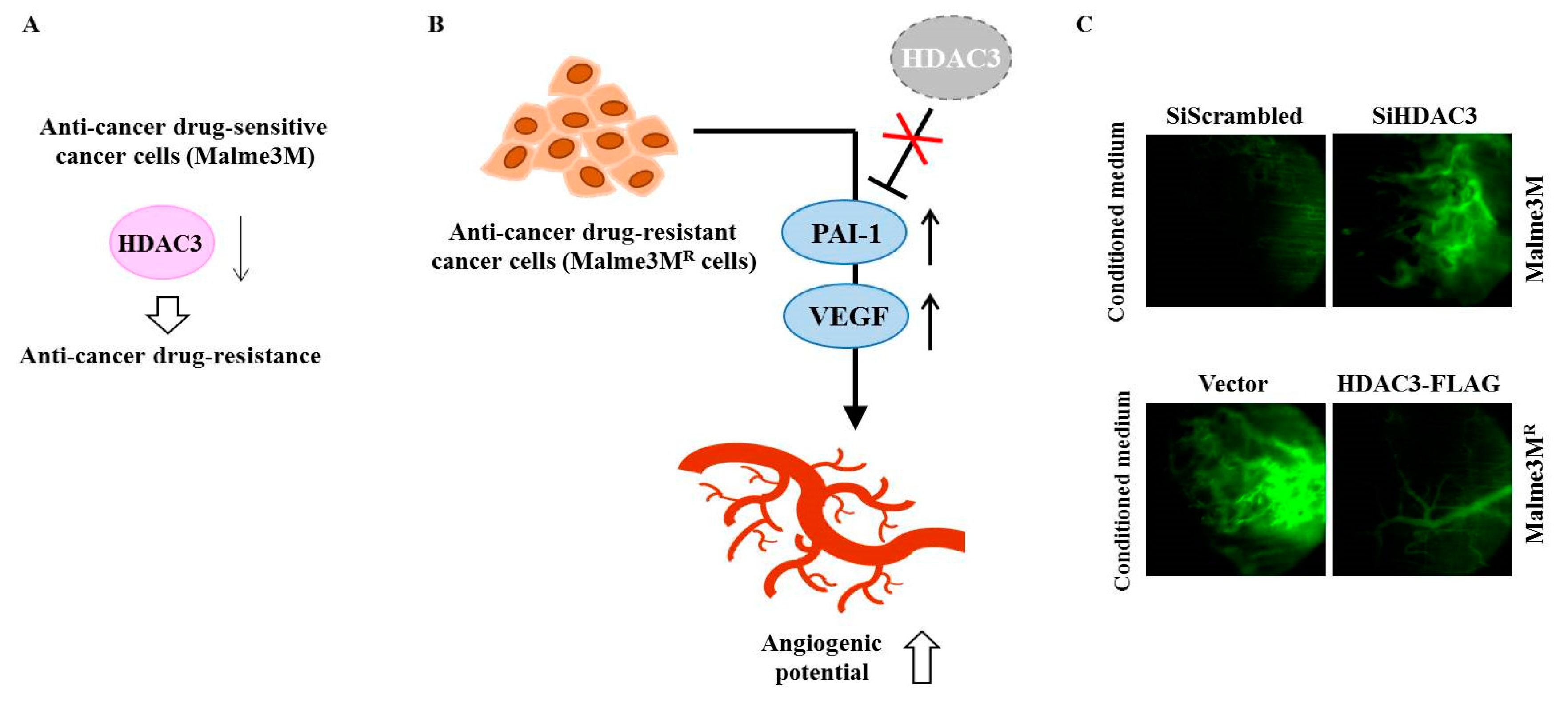{kind=link}
{kind=link}
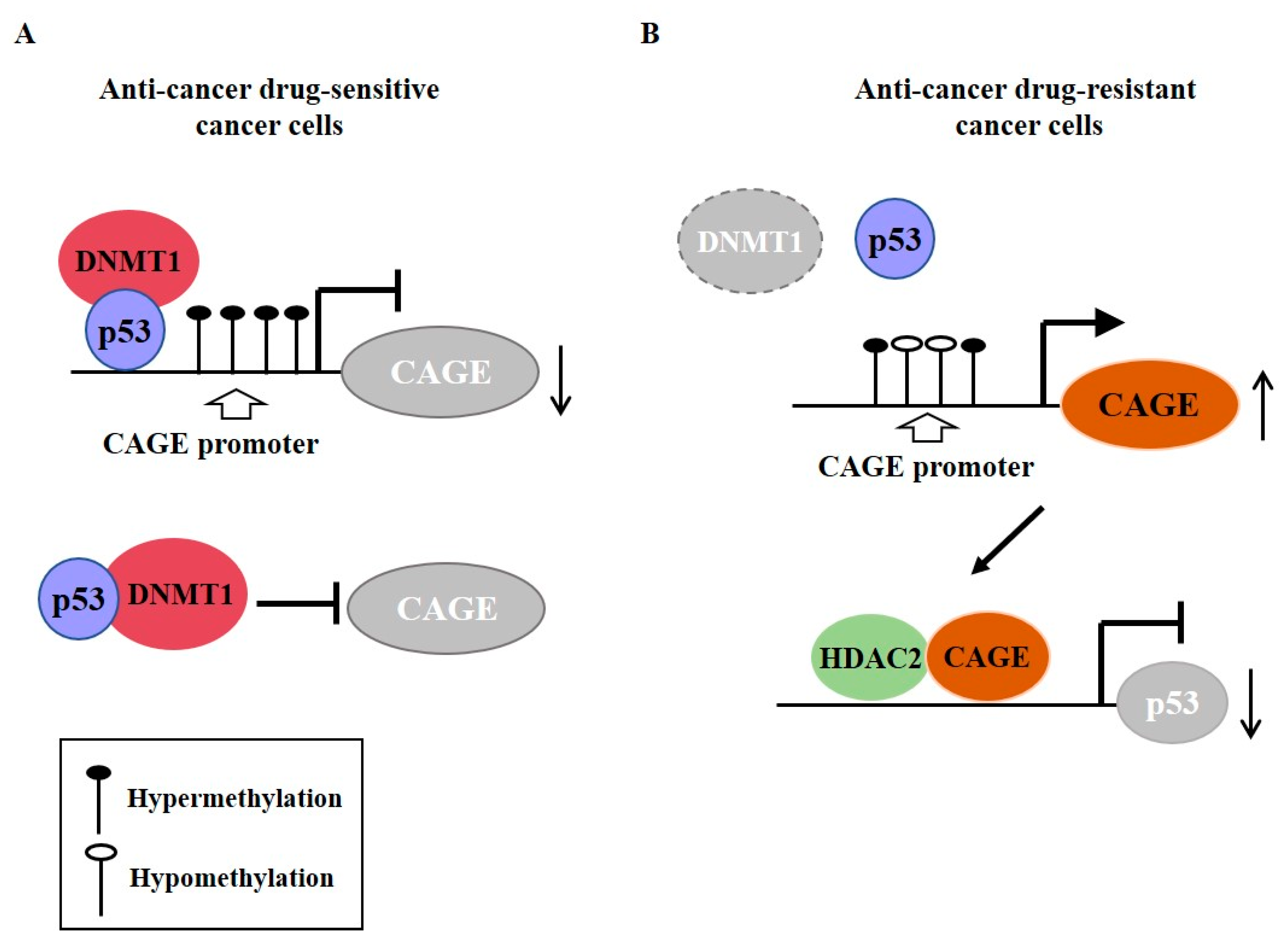{kind=link}
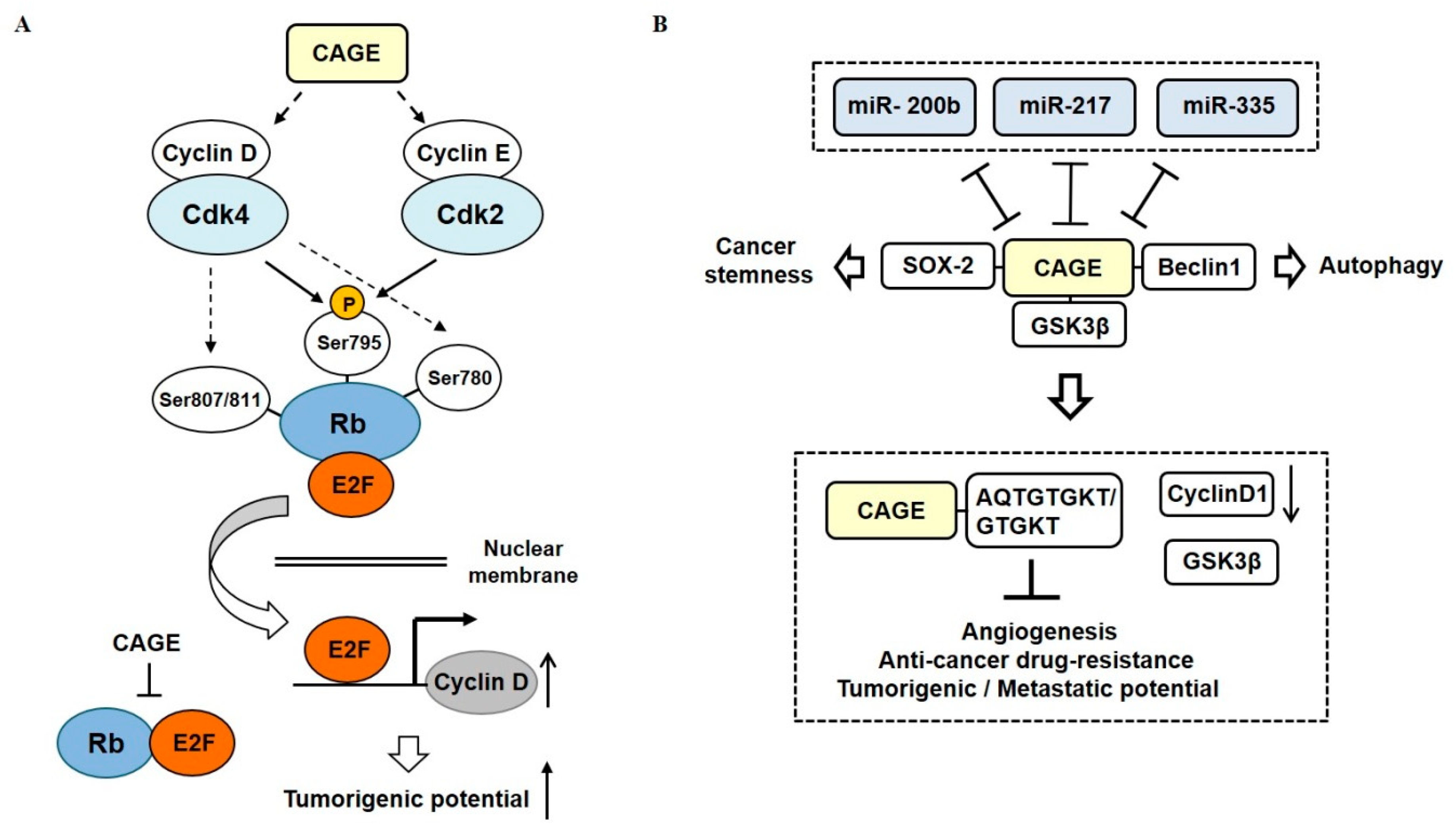{kind=link}
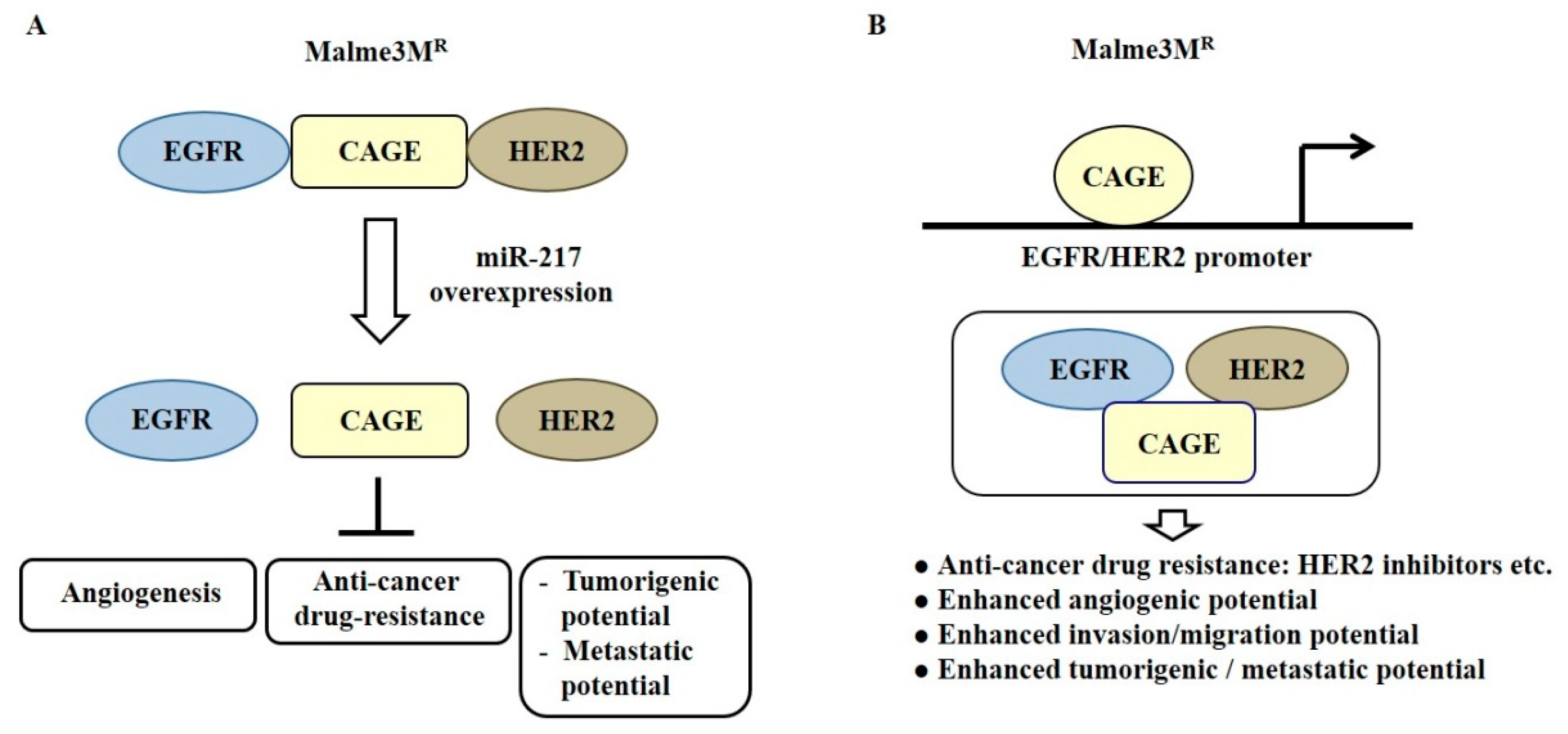{kind=link}
{kind=link}
1. HDAC3 as a Target for Development of Anti-Cancer Drugs
2. Anti-Cancer Therapy Targeting CSCS in Relation to HDACs
3. Role of HDAC3 in Anti-Cancer Drug Resistance
4. HDAC3-miRNA Network in Angiogenesis and Anti-Cancer Drug Resistance
5. Role of CAGE-miRNA Network in Anti-Cancer Drug Resistance
6. HDAC3 Functions Upstream of CAGE and Targets CAGE
7. Discussion and Conclusions
Funding
Conflicts of Interest
References
- Mahlknecht, U.; Emiliani, S.; Najfeld, V.; Young, S.; Verdin, E. Genomic organization and chromosomal localization of the human histone deacetylase 3 gene. Genomics 1999, 56, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.; Wang, J.; Nawaz, Z.; Liu, J.M.; Qin, J.; Wong, J. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 2000, 19, 4342–4350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Kalkum, M.; Chait, B.T.; Roeder, R.G. The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol. Cell 2002, 9, 611–623. [Google Scholar] [CrossRef]
- Chen, L.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Mahlknecht, U.; Will, J.; Varin, A.; Hoelzer, D.; Herbein, G. Histone deacetylase 3, a class I histone deacetylase, suppresses MAPK11-mediated activating transcription factor-2 activation and represses TNF gene expression. J. Immunol. 2004, 173, 3979–3990. [Google Scholar] [CrossRef] [PubMed]
- Pathania, R.; Ramachandran, S.; Mariappan, G.; Thakur, P.; Shi, H.; Choi, J.H.; Manicassamy, S.; Kolhe, R.; Prasad, P.D.; Sharma, S.; et al. Combined Inhibition of DNMT and HDAC Blocks the Tumorigenicity of Cancer Stem-like Cells and Attenuates Mammary Tumor Growth. Cancer Res. 2016, 76, 3224–3235. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Choi, K.C.; Choi, H.K.; Kang, H.B.; Kim, M.J.; Lee, Y.H.; Lee, O.H.; Lee, J.; Kim, Y.J.; Jun, W.; et al. HDAC3 selectively represses CREB3-mediated transcription and migration of metastatic breast cancer cells. Cell. Mol. Life Sci. 2010, 67, 3499–3510. [Google Scholar] [CrossRef]
- Bardai, F.H.; D’Mello, S.R. Selective toxicity by HDAC3 in neurons: Regulation by Akt and GSK3beta. J. Neurosci. 2011, 31, 1746–1751. [Google Scholar] [CrossRef]
- Lundh, M.; Christensen, D.P.; Damgaard Nielsen, M.; Richardson, S.J.; Dahllof, M.S.; Skovgaard, T.; Berthelsen, J.; Dinarello, C.A.; Stevenazzi, A.; Mascagni, P.; et al. Histone deacetylases 1 and 3 but not 2 mediate cytokine-induced beta cell apoptosis in INS-1 cells and dispersed primary islets from rats and are differentially regulated in the islets of type 1 diabetic children. Diabetologia 2012, 55, 2421–2431. [Google Scholar] [CrossRef] [Green Version]
- Bendinelli, P.; Matteucci, E.; Maroni, P.; Desiderio, M.A. NF-kappaB activation, dependent on acetylation/deacetylation, contributes to HIF-1 activity and migration of bone metastatic breast carcinoma cells. Mol. Cancer Res. 2009, 7, 1328–1341. [Google Scholar] [CrossRef]
- Miao, L.J.; Huang, F.X.; Sun, Z.T.; Zhang, R.X.; Huang, S.F.; Wang, J. Stat3 inhibits Beclin 1 expression through recruitment of HDAC3 in nonsmall cell lung cancer cells. Tumor Biol. 2014, 35, 7097–7103. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.S.; Alam, H.; Li, N.; Wagner, K.W.; Chung, J.; Ahn, Y.W.; Lee, M.G. Transcriptional repression of histone deacetylase 3 by the histone demethylase KDM2A is coupled to tumorigenicity of lung cancer cells. J. Biol. Chem. 2014, 289, 7483–7496. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Park, H.; Kim, Y.; Kim, H.; Jeoung, D. HDAC3 acts as a negative regulator of angiogenesis. BMB Rep. 2014, 47, 227–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaskara, S.; Knutson, S.K.; Jiang, G.; Chandrasekharan, M.B.; Wilson, A.J.; Zheng, S.; Yenamandra, A.; Locke, K.; Yuan, J.L.; Bonine-Summers, A.R.; et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 2010, 18, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.W.; Hiebert, S.W. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell. 2008, 30, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Byun, D.S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, L.; Wu, Y.; Zhang, R.; Zhang, M.; Liao, D.; Wang, G.; Qin, G.; Xu, R.H.; Kang, T. CBX4 Suppresses Metastasis via Recruitment of HDAC3 to the Runx2 Promoter in Colorectal Carcinoma. Cancer Res. 2016, 76, 7277–7289. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.H.; Ko, H.; Jeon, H.; Sung, G.J.; Park, S.Y.; Jun, W.J.; Lee, Y.H.; Lee, J.; Lee, S.W.; Yoon, H.G.; et al. Delphinidin induces apoptosis via cleaved HDAC3-mediated p53 acetylation and oligomerization in prostate cancer cells. Oncotarget 2016, 7, 56767–56780. [Google Scholar] [CrossRef] [Green Version]
- Hanigan, T.W.; Aboukhatwa, S.M.; Taha, T.Y.; Frasor, J.; Petukhov, P.A. Divergent JNK Phosphorylation of HDAC3 in Triple-Negative Breast Cancer Cells Determines HDAC Inhibitor Binding and Selectivity. Cell Chem. Biol. 2017, 24, 1356–1367.e8. [Google Scholar] [CrossRef]
- Yang, M.; Dang, X.; Tan, Y.; Wang, M.; Li, X.; Li, G. I-7ab inhibited the growth of TNBC cells via targeting HDAC3 and promoting the acetylation of p53. Biomed. Pharmacother. 2018, 99, 220–226. [Google Scholar] [CrossRef]
- Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Ebert, M.P.; Pross, M.; Dietel, M.; Denkert, C.; Rocken, C. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: A retrospective analysis. Lancet Oncol. 2008, 9, 139–148. [Google Scholar] [CrossRef]
- Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.R.; Niesporek, S.; Denkert, C.; Dietel, M.; et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, L.; Pan, M.; Sun, J.; Lu, H.; Shen, Q.; Zhang, S.; Jiang, T.; Liu, L.; Jin, W.; Chen, Y.; et al. Histone deacetylase 3 inhibits expression of PUMA in gastric cancer cells. J. Mol. Med. 2013, 91, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Inks, E.S.; Li, X.; Hou, J.; Chou, C.J.; Zhang, J.; Jiang, Y.; Zhang, Y.; Xu, W. Discovery of the first N-hydroxycinnamamide-based histone deacetylase 1/3 dual inhibitors with potent oral antitumor activity. J. Med. Chem. 2014, 57, 3324–3341. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Botrugno, O.A.; Santoro, F.; Minucci, S. Histone deacetylase inhibitors as a new weapon in the arsenal of differentiation therapies of cancer. Cancer Lett. 2009, 280, 134–144. [Google Scholar] [CrossRef]
- Dvorakova, M.; Vanek, T. Histone deacetylase inhibitors for the treatment of cancer stem cells. MedChemComm 2016, 7, 2217–2231. [Google Scholar] [CrossRef]
- Liu, N.; Li, S.; Wu, N.; Cho, K.S. Acetylation and deacetylation in cancer stem-like cells. Oncotarget 2017, 8, 89315–89325. [Google Scholar] [CrossRef]
- Guzman, M.L.; Yang, N.; Sharma, K.K.; Balys, M.; Corbett, C.A.; Jordan, C.T.; Becker, M.W.; Steidl, U.; Abdel-Wahab, O.; Levine, R.L.; et al. Selective activity of the histone deacetylase inhibitor AR-42 against leukemia stem cells: A novel potential strategy in acute myelogenous leukemia. Mol. Cancer Ther. 2014, 13, 1979–1990. [Google Scholar] [CrossRef]
- Nalls, D.; Tang, S.N.; Rodova, M.; Srivastava, R.K.; Shankar, S. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS ONE 2011, 6, e24099. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Yadav, A.; Lang, J.C.; Teknos, T.N.; Kumar, P. Suberoylanilide hydroxamic acid (SAHA) reverses chemoresistance in head and neck cancer cells by targeting cancer stem cells via the downregulation of nanog. Genes Cancer 2015, 6, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Triner, D.; Shah, Y.M. Hypoxia-inducible factors: A central link between inflammation and cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Malek, S.N.; Zheng, P.; Liu, Y. Targeting HIF1alpha eliminates cancer stem cells in hematological malignancies. Cell Stem Cell 2011, 8, 399–411. [Google Scholar] [CrossRef]
- Kim, S.H.; Jeong, J.W.; Park, J.A.; Lee, J.W.; Seo, J.H.; Jung, B.K.; Bae, M.K.; Kim, K.W. Regulation of the HIF-1alpha stability by histone deacetylases. Oncol. Rep. 2007, 17, 647–651. [Google Scholar] [PubMed]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Geng, H.; Liu, Q.; Xue, C.; David, L.L.; Beer, T.M.; Thomas, G.V.; Dai, M.S.; Qian, D.Z. HIF1alpha protein stability is increased by acetylation at lysine 709. J. Biol. Chem. 2012, 287, 35496–35505. [Google Scholar] [CrossRef]
- Qian, D.Z.; Kachhap, S.K.; Collis, S.J.; Verheul, H.M.; Carducci, M.A.; Atadja, P.; Pili, R. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006, 66, 8814–8821. [Google Scholar] [CrossRef]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef]
- Lu, X.F.; Cao, X.Y.; Zhu, Y.J.; Wu, Z.R.; Zhuang, X.; Shao, M.Y.; Xu, Q.; Zhou, Y.J.; Ji, H.J.; Lu, Q.R.; et al. Histone deacetylase 3 promotes liver regeneration and liver cancer cells proliferation through signal transducer and activator of transcription 3 signaling pathway. Cell Death Dis. 2018, 9, 398. [Google Scholar] [CrossRef] [Green Version]
- Rinkenbaugh, A.L.; Cogswell, P.C.; Calamini, B.; Dunn, D.E.; Persson, A.I.; Weiss, W.A.; Lo, D.C.; Baldwin, A.S. IKK/NF-kappaB signaling contributes to glioblastoma stem cell maintenance. Oncotarget 2016, 7, 69173–69187. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Li, Q.Z.; Chen, L.; Chen, B.D.; Wang, B.; Zhang, X.J.; Li, W.P. Histone Deacetylase Inhibitor RGFP109 Overcomes Temozolomide Resistance by Blocking NF-kappaB-Dependent Transcription in Glioblastoma Cell Lines. Neurochem. Res. 2016, 41, 3192–3205. [Google Scholar] [CrossRef]
- Kiernan, R.; Bres, V.; Ng, R.W.; Coudart, M.P.; El Messaoudi, S.; Sardet, C.; Jin, D.Y.; Emiliani, S.; Benkirane, M. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J. Biol. Chem. 2003, 278, 2758–2766. [Google Scholar] [CrossRef]
- Salvador, M.A.; Wicinski, J.; Cabaud, O.; Toiron, Y.; Finetti, P.; Josselin, E.; Lelievre, H.; Kraus-Berthier, L.; Depil, S.; Bertucci, F.; et al. The histone deacetylase inhibitor abexinostat induces cancer stem cells differentiation in breast cancer with low Xist expression. Clin. Cancer Res. 2013, 19, 6520–6531. [Google Scholar] [CrossRef] [PubMed]
- Di Pompo, G.; Salerno, M.; Rotili, D.; Valente, S.; Zwergel, C.; Avnet, S.; Lattanzi, G.; Baldini, N.; Mai, A. Novel histone deacetylase inhibitors induce growth arrest, apoptosis, and differentiation in sarcoma cancer stem cells. J. Med. Chem. 2015, 58, 4073–4079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yin, Y.; Dorfman, R.G.; Zou, T.; Pan, Y.; Li, Y.; Wang, Y.; Zhou, Q.; Zhou, L.; Kong, B.; et al. Down-regulation of HDAC3 inhibits growth of cholangiocarcinoma by inducing apoptosis. Oncotarget 2017, 8, 99402–99413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, M.R.; Smigiel, J.M.; Junk, D.J.; Jackson, M.W. Cancer Stem Cell Plasticity Drives Therapeutic Resistance. Cancers 2016, 8. [Google Scholar] [CrossRef]
- El-Khoury, V.; Breuzard, G.; Fourre, N.; Dufer, J. The histone deacetylase inhibitor trichostatin A downregulates human MDR1 (ABCB1) gene expression by a transcription-dependent mechanism in a drug-resistant small cell lung carcinoma cell line model. Br. J. Cancer 2007, 97, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Jiang, Z.; Yin, P.; Li, Q.; Liu, J. Role for Class I histone deacetylases in multidrug resistance. Exp. Cell Res. 2012, 318, 177–186. [Google Scholar] [CrossRef]
- Kavallaris, M.; Kuo, D.Y.; Burkhart, C.A.; Regl, D.L.; Norris, M.D.; Haber, M.; Horwitz, S.B. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J. Clin. Investig. 1997, 100, 1282–1293. [Google Scholar] [CrossRef]
- Levallet, G.; Bergot, E.; Antoine, M.; Creveuil, C.; Santos, A.O.; Beau-Faller, M.; de Fraipont, F.; Brambilla, E.; Levallet, J.; Morin, F.; et al. High TUBB3 expression, an independent prognostic marker in patients with early non-small cell lung cancer treated by preoperative chemotherapy, is regulated by K-Ras signaling pathway. Mol. Cancer Ther. 2012, 11, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Verdier-Pinard, P.; Wang, F.; Martello, L.; Burd, B.; Orr, G.A.; Horwitz, S.B. Analysis of tubulin isotypes and mutations from taxol-resistant cells by combined isoelectrofocusing and mass spectrometry. Biochemistry 2003, 42, 5349–5357. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Sato, N.; Yanai, K.; Akiyoshi, T.; Nagai, S.; Wada, J.; Koga, K.; Mibu, R.; Nakamura, M.; Katano, M. Enhancement of paclitaxel-induced apoptosis by inhibition of mitogen-activated protein kinase pathway in colon cancer cells. Anticancer Res. 2009, 29, 261–270. [Google Scholar] [PubMed]
- Guan, J.; Yuan, Z.; He, J.; Wu, Z.; Liu, B.; Lin, X.; Mo, L.; Mo, H. Overexpression of caveolin-1 reduces Taxol resistance in human osteosarcoma cells by attenuating PI3K-Akt-JNK dependent autophagy. Exp. Ther. Med. 2016, 12, 2815–2822. [Google Scholar] [CrossRef] [PubMed]
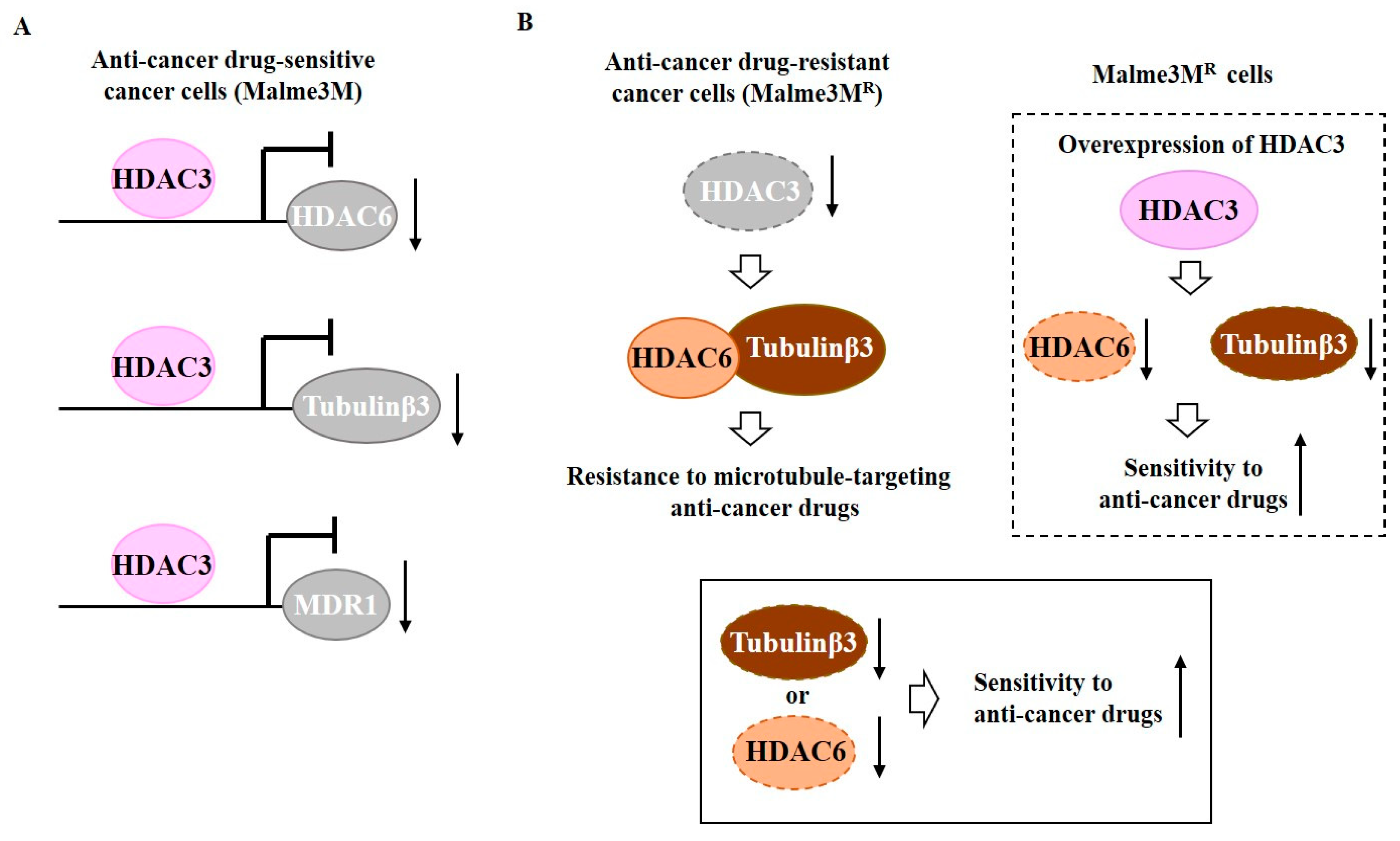
- Kim, Y.; Kim, H.; Jeoung, D. Tubulin Beta3 Serves as a Target of HDAC3 and Mediates Resistance to Microtubule-Targeting Drugs. Mol. Cells 2015, 38, 705–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.S.; Sceneay, J.; House, C.M.; Halse, H.M.; Liu, M.C.; George, J.; Hunnam, T.C.; Parker, B.S.; Haviv, I.; Ronai, Z.; et al. Vascular normalization by loss of Siah2 results in increased chemotherapeutic efficacy. Cancer Res. 2012, 72, 1694–1704. [Google Scholar] [CrossRef]
- Christian, P.A.; Fiandalo, M.V.; Schwarze, S.R. Possible role of death receptor-mediated apoptosis by the E3 ubiquitin ligases Siah2 and POSH. Mol. Cancer 2011, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.; Stebbins, J.L.; Dewing, A.; Qi, J.; Pellecchia, M.; Ronai, Z.A. Inhibition of Siah2 ubiquitin ligase by vitamin K3 (menadione) attenuates hypoxia and MAPK signaling and blocks melanoma tumorigenesis. Pigment Cell Melanoma Res. 2009, 22, 799–808. [Google Scholar] [CrossRef]
- Ma, B.; Chen, Y.; Chen, L.; Cheng, H.; Mu, C.; Li, J.; Gao, R.; Zhou, C.; Cao, L.; Liu, J.; et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nat. Cell Biol. 2015, 17, 95–103. [Google Scholar] [CrossRef]
- Chan, P.; Moller, A.; Liu, M.C.; Sceneay, J.E.; Wong, C.S.; Waddell, N.; Huang, K.T.; Dobrovic, A.; Millar, E.K.; O’Toole, S.A.; et al. The expression of the ubiquitin ligase SIAH2 (seven in absentia homolog 2) is mediated through gene copy number in breast cancer and is associated with a basal-like phenotype and p53 expression. Breast Cancer Res. 2011, 13, R19. [Google Scholar] [CrossRef] [PubMed]
- Grishina, I.; Debus, K.; Garcia-Limones, C.; Schneider, C.; Shresta, A.; Garcia, C.; Calzado, M.A.; Schmitz, M.L. SIAH-mediated ubiquitination and degradation of acetyl-transferases regulate the p53 response and protein acetylation. Biochim. Biophys. Acta 2012, 1823, 2287–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.L.; Ueki, N.; Hayman, M.J. The Ski protein negatively regulates Siah2-mediated HDAC3 degradation. Biochem. Biophys. Res. Commun. 2010, 399, 623–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
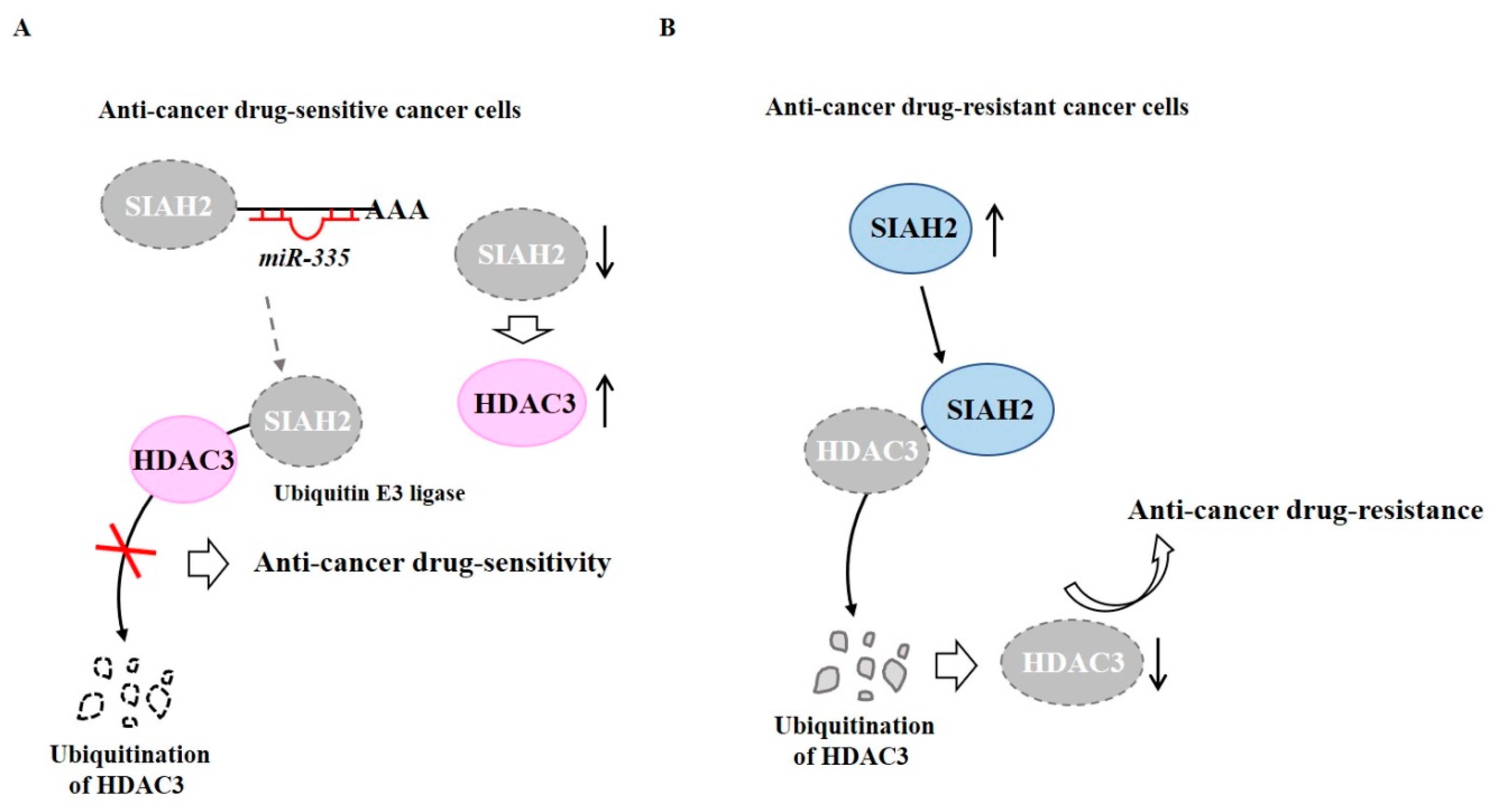
- Kim, Y.; Kim, H.; Park, D.; Jeoung, D. miR-335 Targets SIAH2 and Confers Sensitivity to Anti-Cancer Drugs by Increasing the Expression of HDAC3. Mol. Cells 2015, 38, 562–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Yang, X.; Xue, Y.W.; Zhang, X.F.; Wang, Y.; Liu, W.J.; Wu, X.J. Promoter methylation of glutathione S-transferase pi1 and multidrug resistance gene 1 in bronchioloalveolar carcinoma and its correlation with DNA methyltransferase 1 expression. Cancer 2009, 115, 3222–3232. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.; Polgar, O.; Huff, L.M.; Morisaki, K.; Bates, S.E. Histone modifications at the ABCG2 promoter following treatment with histone deacetylase inhibitor mirror those in multidrug-resistant cells. Mol. Cancer Res. 2008, 6, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, H.; Park, H.; Park, D.; Lee, H.; Lee, Y.S.; Choe, J.; Kim, Y.M.; Jeoung, D. miR-326-histone deacetylase-3 feedback loop regulates the invasion and tumorigenic and angiogenic response to anti-cancer drugs. J. Biol. Chem. 2014, 289, 28019–28039. [Google Scholar] [CrossRef]
- Park, H.; Kim, Y.; Park, D.; Jeoung, D. Nuclear localization signal domain of HDAC3 is necessary and sufficient for the expression regulation of MDR1. BMB Rep. 2014, 47, 342–347. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, Y.; Goh, H.; Jeoung, D. Histone Deacetylase-3/CAGE Axis Targets EGFR Signaling and Regulates the Response to Anti-Cancer Drugs. Mol. Cells 2016, 39, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Kefas, B.; Comeau, L.; Floyd, D.H.; Seleverstov, O.; Godlewski, J.; Schmittgen, T.; Jiang, J.; diPierro, C.G.; Li, Y.; Chiocca, E.A.; et al. The neuronal microRNA miR-326 acts in a feedback loop with notch and has therapeutic potential against brain tumors. J. Neurosci. 2009, 29, 15161–15168. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Wu, H.; Xia, J.; Li, Y.; Zhang, Y.; Huang, K.; Wagar, N.; Yoon, Y.; Cho, H.T.; Scala, S.; et al. Involvement of miR-326 in chemotherapy resistance of breast cancer through modulating expression of multidrug resistance-associated protein 1. Biochem. Pharmacol. 2010, 79, 817–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, B.; Lim, Y.; Lee, D.Y.; Park, S.Y.; Lee, H.; Kim, W.H.; Yang, H.; Bang, Y.J.; Jeoung, D.I. Identification and characterization of a novel cancer/testis antigen gene CAGE. Biochem. Biophys. Res. Commun. 2002, 292, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Iwata, T.; Fujita, T.; Hirao, N.; Matsuzaki, Y.; Okada, T.; Mochimaru, H.; Susumu, N.; Matsumoto, E.; Sugano, K.; Yamashita, N.; et al. Frequent immune responses to a cancer/testis antigen, CAGE, in patients with microsatellite instability-positive endometrial cancer. Clin. Cancer Res. 2005, 11, 3949–3957. [Google Scholar] [CrossRef] [PubMed]
- Liggins, A.P.; Lim, S.H.; Soilleux, E.J.; Pulford, K.; Banham, A.H. A panel of cancer-testis genes exhibiting broad-spectrum expression in haematological malignancies. Cancer Immun. 2010, 10, 8. [Google Scholar] [PubMed]
- Cho, B.; Lee, H.; Jeong, S.; Bang, Y.J.; Lee, H.J.; Hwang, K.S.; Kim, H.Y.; Lee, Y.S.; Kang, G.H.; Jeoung, D.I. Promoter hypomethylation of a novel cancer/testis antigen gene CAGE is correlated with its aberrant expression and is seen in premalignant stage of gastric carcinoma. Biochem. Biophys. Res. Commun. 2003, 307, 52–63. [Google Scholar] [CrossRef]
- Kim, Y.; Park, H.; Park, D.; Lee, Y.S.; Choe, J.; Hahn, J.H.; Lee, H.; Kim, Y.M.; Jeoung, D. Cancer/testis antigen CAGE exerts negative regulation on p53 expression through HDAC2 and confers resistance to anti-cancer drugs. J. Biol. Chem. 2010, 285, 25957–25968. [Google Scholar] [CrossRef] [PubMed]
- Por, E.; Byun, H.J.; Lee, E.J.; Lim, J.H.; Jung, S.Y.; Park, I.; Kim, Y.M.; Jeoung, D.I.; Lee, H. The cancer/testis antigen CAGE with oncogenic potential stimulates cell proliferation by up-regulating cyclins D1 and E in an AP-1- and E2F-dependent manner. J. Biol. Chem. 2010, 285, 14475–14485. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, H.; Park, D.; Lee, H.; Lee, Y.S.; Choe, J.; Kim, Y.M.; Jeon, D.; Jeoung, D. The pentapeptide Gly-Thr-Gly-Lys-Thr confers sensitivity to anti-cancer drugs by inhibition of CAGE binding to GSK3beta and decreasing the expression of cyclinD1. Oncotarget 2017, 8, 13632–13651. [Google Scholar] [CrossRef]
- Kim, Y.; Yeon, M.; Jeoung, D. DDX53 Regulates Cancer Stem Cell-Like Properties by Binding to SOX-2. Mol. Cells 2017, 40, 322–330. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Park, D.; Kim, H.; Choi, M.; Lee, H.; Lee, Y.S.; Choe, J.; Kim, Y.M.; Jeoung, D. miR-200b and cancer/testis antigen CAGE form a feedback loop to regulate the invasion and tumorigenic and angiogenic responses of a cancer cell line to microtubule-targeting drugs. J. Biol. Chem. 2013, 288, 36502–36518. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, H.; Park, D.; Han, M.; Lee, H.; Lee, Y.S.; Choe, J.; Kim, Y.M.; Jeoung, D. miR-217 and CAGE form feedback loop and regulates the response to anti-cancer drugs through EGFR and HER2. Oncotarget 2016, 7, 10297–10321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, R.E.; Huang, Q.; Thai, K.; Advani, S.L.; Lee, K.; Yuen, D.A.; Connelly, K.A.; Advani, A. Histone deacetylase inhibition attenuates diabetes-associated kidney growth: Potential role for epigenetic modification of the epidermal growth factor receptor. Kidney Int. 2011, 79, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.W.; Wu, M.S.; Huang, W.C.; Chen, C.C. HDAC inhibition decreases the expression of EGFR in colorectal cancer cells. PLoS ONE 2011, 6, e18087. [Google Scholar] [CrossRef] [PubMed]
- Robertson, E.D.; Weir, L.; Romanowska, M.; Leigh, I.M.; Panteleyev, A.A. ARNT controls the expression of epidermal differentiation genes through HDAC- and EGFR-dependent pathways. J. Cell Sci 2012, 125, 3320–3332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Shimizu, E.; Zhang, X.; Partridge, N.C.; Qin, L. EGFR signaling suppresses osteoblast differentiation and inhibits expression of master osteoblastic transcription factors Runx2 and Osterix. J. Cell. Biochem. 2011, 112, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Tiwari, A.K.; Shukla, S.; Robey, R.W.; Kim, I.W.; Parmar, S.; Bates, S.E.; Si, Q.S.; Goldblatt, C.S.; Abraham, I.; et al. Inhibiting the function of ABCB1 and ABCG2 by the EGFR tyrosine kinase inhibitor AG1478. Biochem. Pharmacol. 2009, 77, 781–793. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef]
- Walther, A.; Johnstone, E.; Swanton, C.; Midgley, R.; Tomlinson, I.; Kerr, D. Genetic prognostic and predictive markers in colorectal cancer. Nat. Rev. Cancer 2009, 9, 489–499. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Gao, X.; Han, W.; Zheng, Y.; Xu, H.; Zhang, C.; He, Q.; Zhang, L.; Li, Z.; et al. c-Met targeting enhances the effect of irradiation and chemical agents against malignant colon cells harboring a KRAS mutation. PLoS ONE 2014, 9, e113186. [Google Scholar] [CrossRef]
- Steinway, S.N.; Dang, H.; You, H.; Rountree, C.B.; Ding, W. The EGFR/ErbB3 Pathway Acts as a Compensatory Survival Mechanism upon c-Met Inhibition in Human c-Met+ Hepatocellular Carcinoma. PLoS ONE 2015, 10, e0128159. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, Y.; Kim, Y.; Jung, H.S.; Jeoung, D. Role of HDAC3-miRNA-CAGE Network in Anti-Cancer Drug-Resistance. Int. J. Mol. Sci. 2019, 20, 51. https://doi.org/10.3390/ijms20010051
Kwon Y, Kim Y, Jung HS, Jeoung D. Role of HDAC3-miRNA-CAGE Network in Anti-Cancer Drug-Resistance. International Journal of Molecular Sciences. 2019; 20(1):51. https://doi.org/10.3390/ijms20010051
Chicago/Turabian StyleKwon, Yoojung, Youngmi Kim, Hyun Suk Jung, and Dooil Jeoung. 2019. "Role of HDAC3-miRNA-CAGE Network in Anti-Cancer Drug-Resistance" International Journal of Molecular Sciences 20, no. 1: 51. https://doi.org/10.3390/ijms20010051
APA StyleKwon, Y., Kim, Y., Jung, H. S., & Jeoung, D. (2019). Role of HDAC3-miRNA-CAGE Network in Anti-Cancer Drug-Resistance. International Journal of Molecular Sciences, 20(1), 51. https://doi.org/10.3390/ijms20010051