Epigenetic Control of Gene Expression in the Normal and Malignant Human Prostate: A Rapid Response Which Promotes Therapeutic Resistance

Abstract
:1. Introduction: Prostate Cancer Is a Heterogeneous Disease Governed by Episodic Genomic Rearrangements
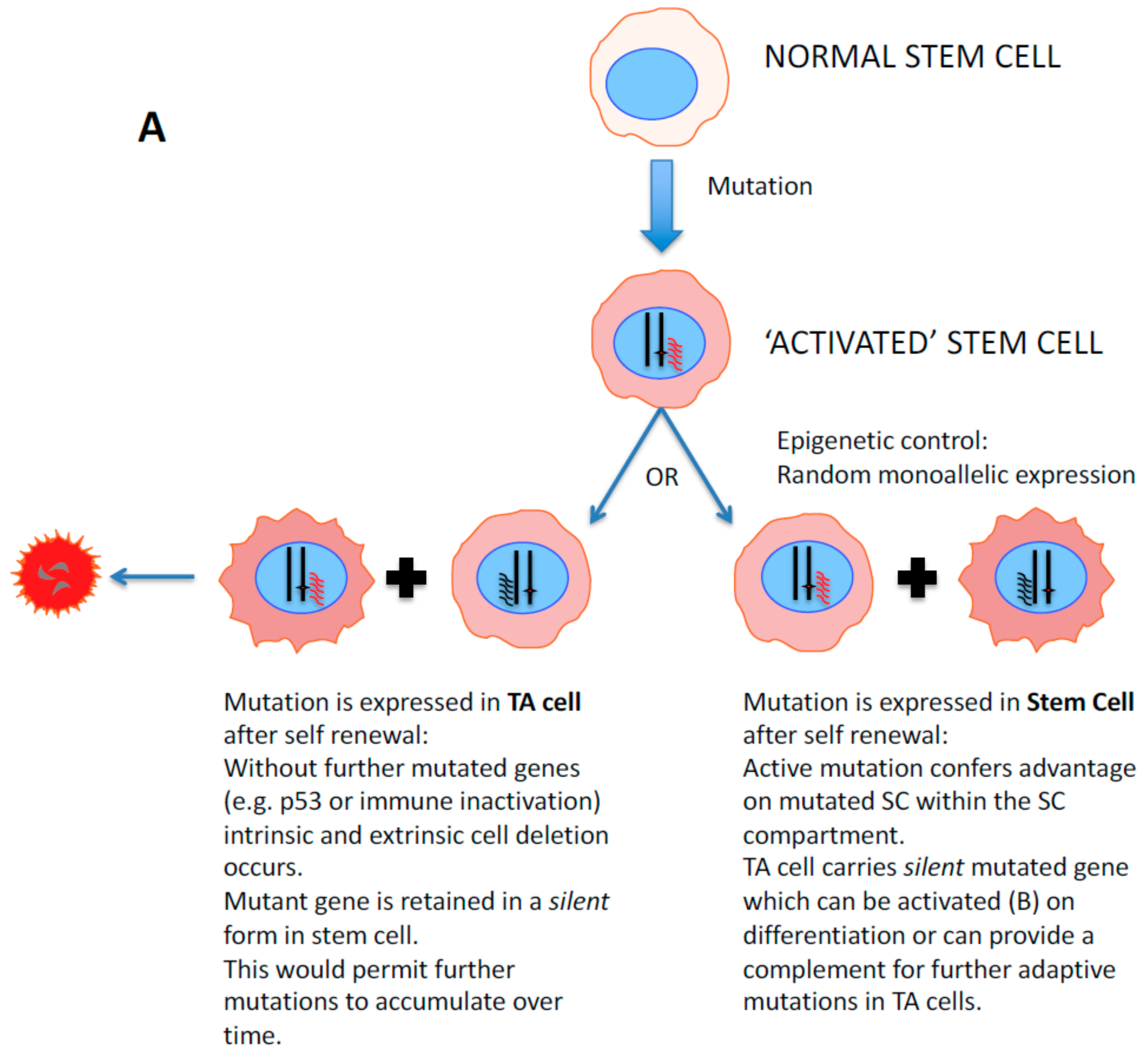
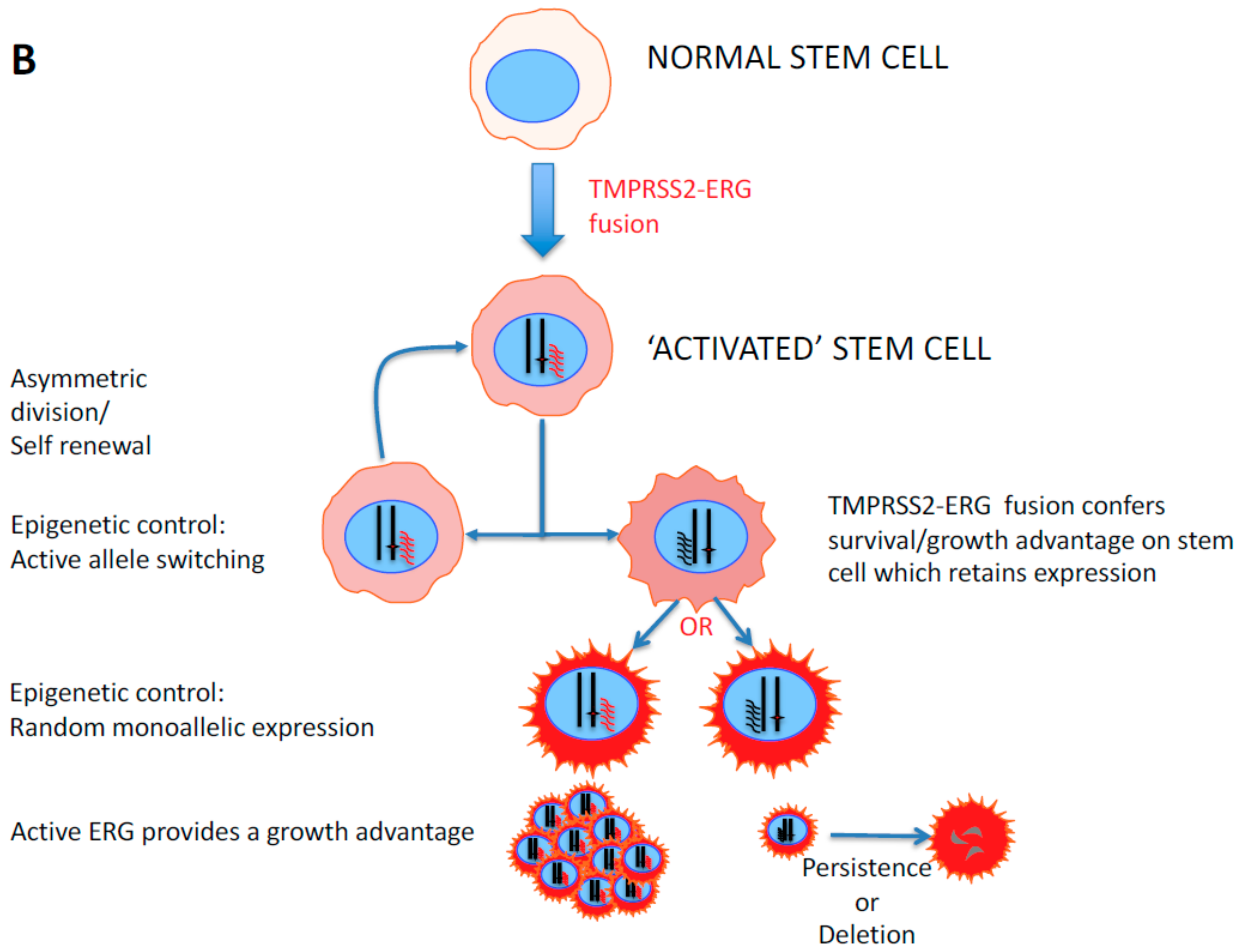
2. Stem Cell Versus Stochastic Mechanisms of Cancer Induction
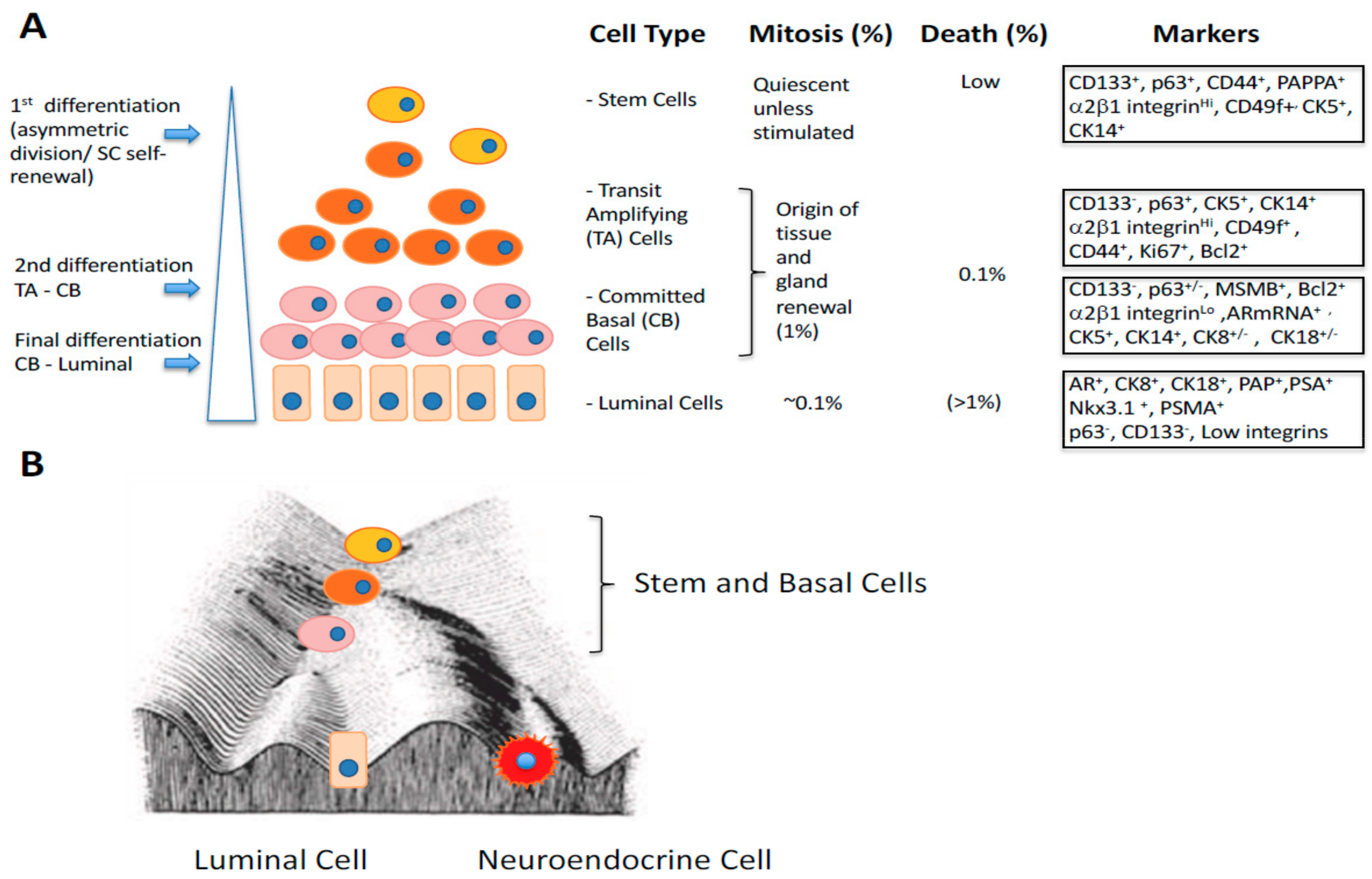
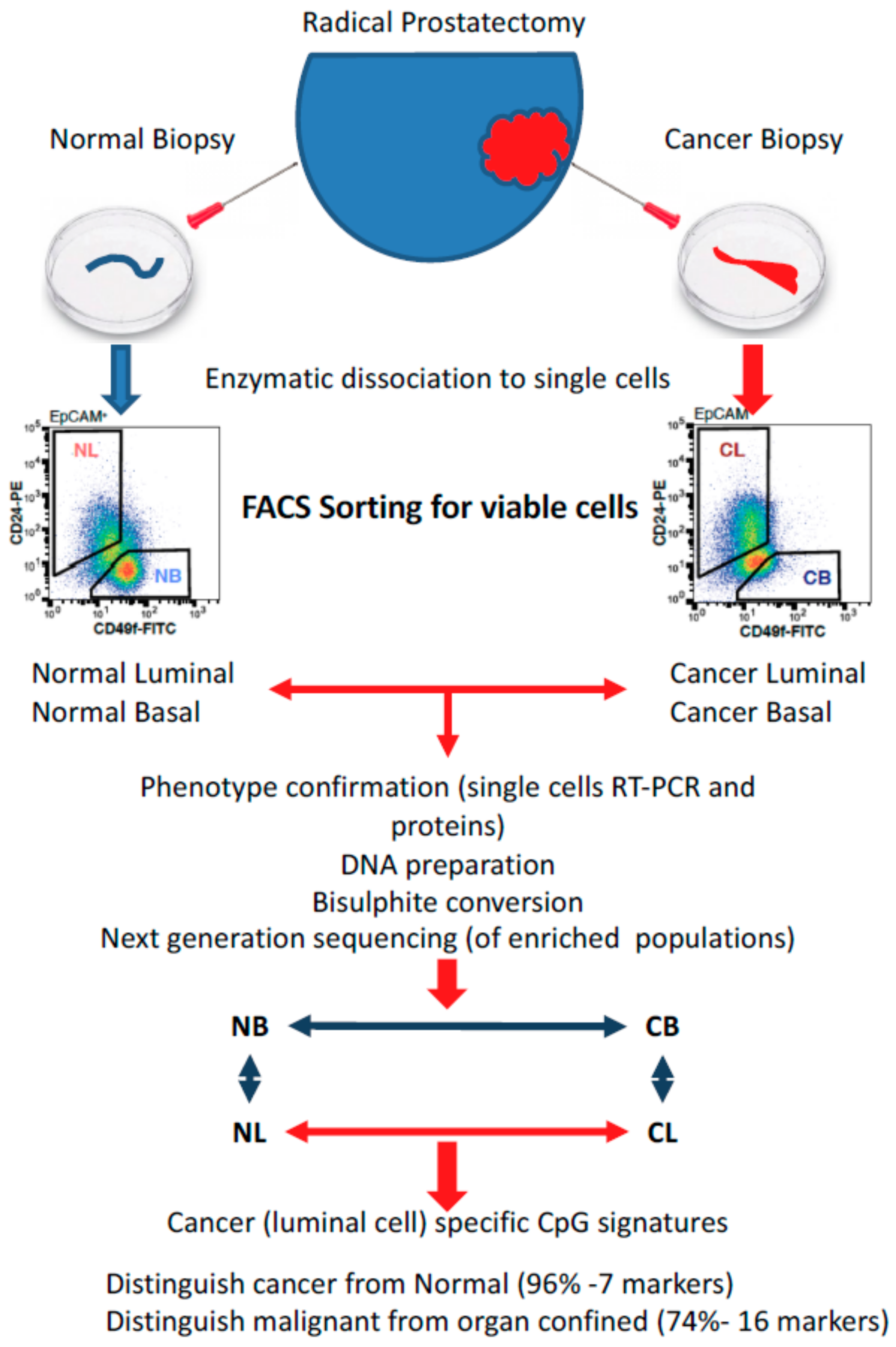
3. Gene Expression Changes During Epithelial Differentiation in Normal and Malignant Prostate
4. Epigenetics: A Mechanism for Phenotypic Flexibility in Development and Disease
5. Defining Epigenetics in Human Genetics: The Elegance and Simplicity of Waddington’s Concept
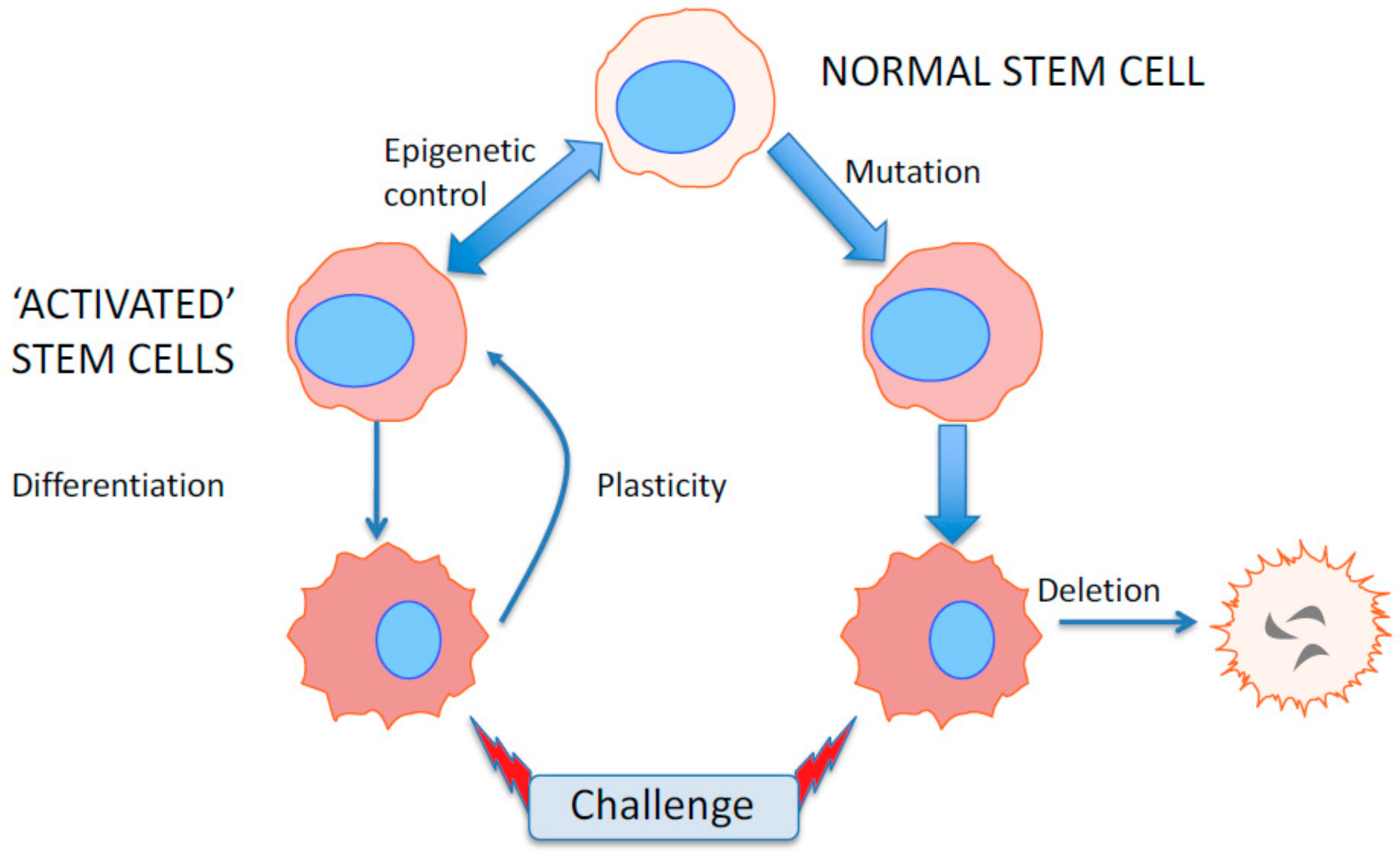
6. Epigenetics as a Flexible Response to Environmental and Microenvironmental Changes
7. The Epigenetic Landscape in Prostate Cancer
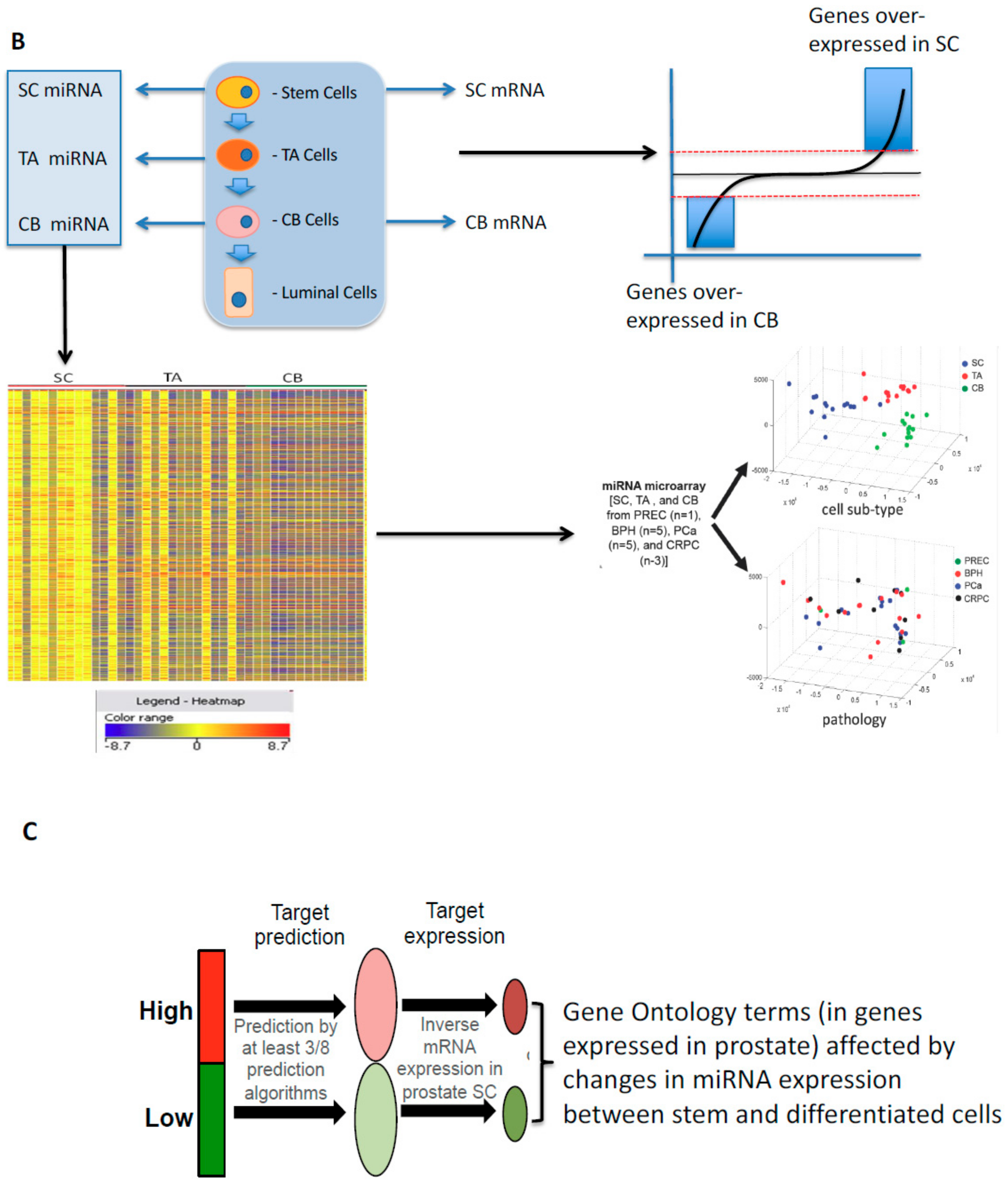
8. Small Non-Coding RNAs: The Rapid Reaction Force for Environmental Changes in Differentiation and Cancer Treatment Suppressor miRNAs and Onco-miRNAs: Designed or Selected for Cancers?
9. Developmental Changes in miRNAs in Prostate Epithelial Cells of Normal and Malignant Origins
10. Phenotypic Plasticity and a Stem-Like State as a Mechanism for Radio-Resistance
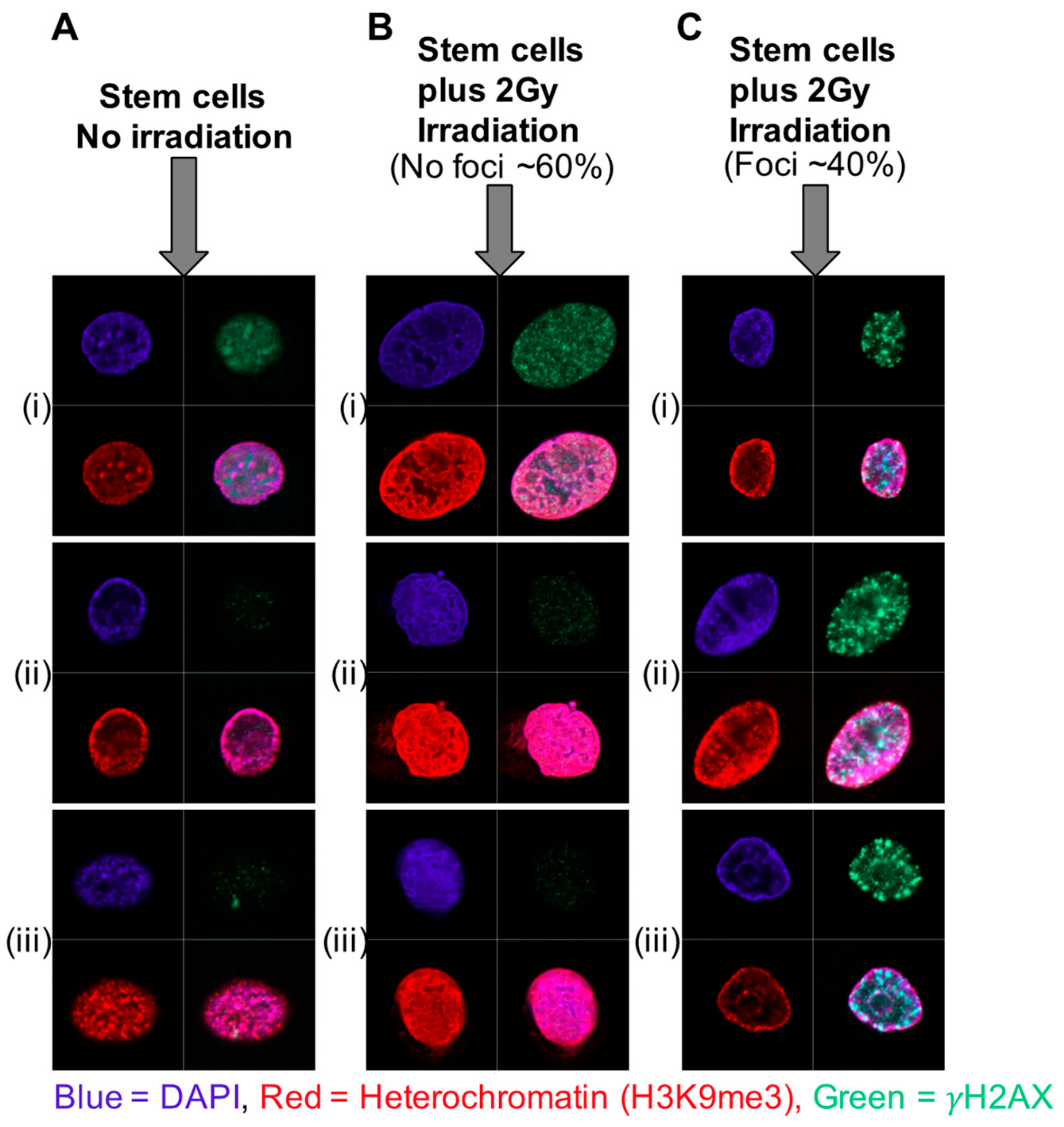
11. Increased Heterochromatin and Rapid Chromatin Condensation as a Mechanism for Radio-Resistance in Prostate Cancer Stem Cells—The Role of Histone Modifications
12. miRNA-Induced Changes in Chromatin Status as a Mechanism for Radio-Resistance in Prostate Cancer
13. The Paradoxical Role of Genomic Methylation in Prostate Epithelial Differentiation and Carcinogenesis
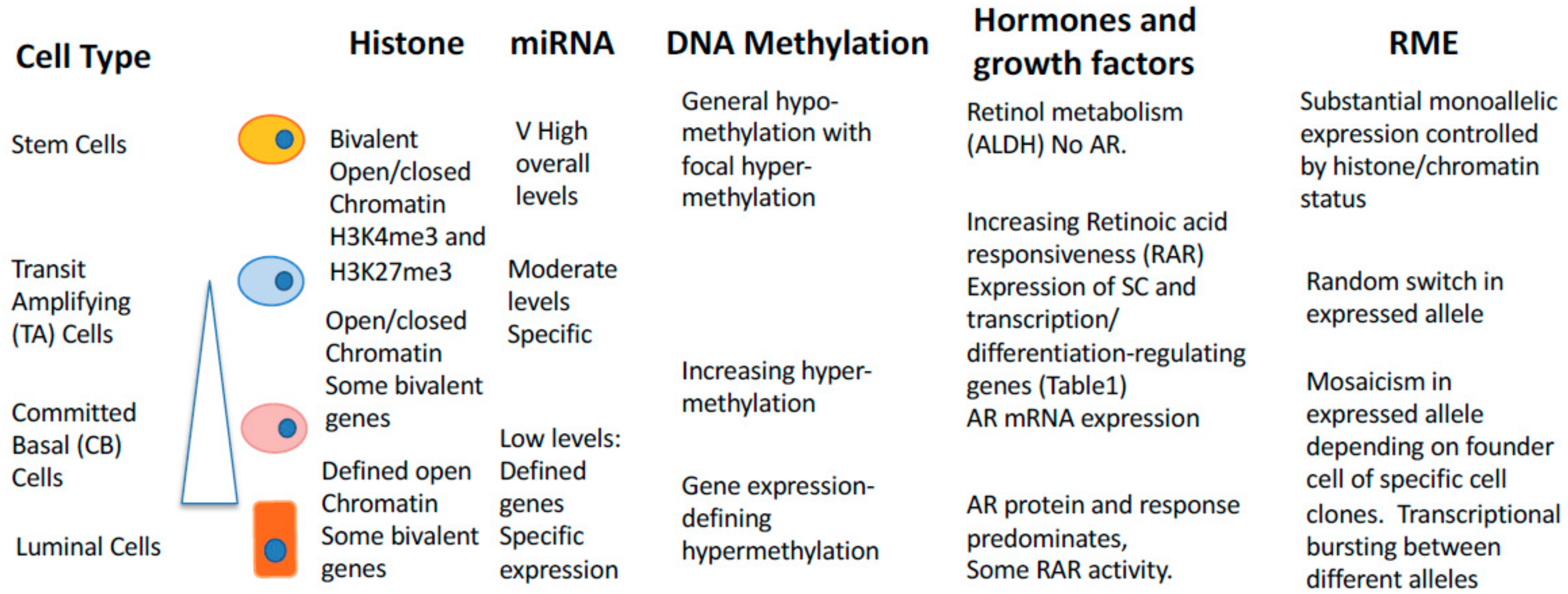
14. Epigenetic Control of Random Mono-Allelic Gene Expression in Development and Cancer
15. Random Monoallelic Gene Expression in Human Cancers
16. Conclusions: A Hypothesis for Epigenetic Control of Epithelial Cell Differentiation in Human Prostate
- Treatment with inhibitors of histone deacetylase, (at a concentration about 100-fold lower than that used in cytotoxic cancer treatments) resulted in a 40% increase in SC radio-sensitivity, whilst not affecting the more differentiated and radiosensitive cells. The in vitro data strongly promotes the use of HDAC inhibitors (at sub-toxic doses) as radio-sensitizers in prostate cancer treatments.
- Radiotherapy (and chemotherapy) patients are often treated with glucocorticoids to counteract the side-effects of treatment. In our studies of miR99a/100 in primary PCa, pre-treatment with dexamethasone stimulated miR99a/100, reducing SMARC expression and decreased radiotherapy responses, whereas combination treatment with the GCR inhibitor Mifepristone increased radio-sensitivity by stimulating the expression levels of miR-99a/100 and decreasing SMARC-induced chromatin condensation. Therefore, the clinical use of GCR inhibitors should clinically enhance radiotherapy and perhaps reduce tumor relapse.
- Overexpression of exogenous miR-548c-3p, which is highly expressed in PCa SC, made radiosensitive CB cells more resistant to irradiation, by induction of a more stem-like state. Thus, inhibition of the SC-preserving activity of high miR-548c-3p levels should also increase clinical radiotherapy efficacy.
17. Future Perspectives and Challenges
Funding
Acknowledgments
Conflicts of Interest
References
- Packer, J.R.; Maitland, N.J. The Molecular and Cellular Origin of Human Prostate Cancer. Biochim. Biophys. Acta 2016, 1863, 1238–1260. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Theodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel Plus Prednisone or Mitoxantrone Plus Prednisone for Advanced Prostate Cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef]
- Gleason, D.F. Classification of prostatic carcinomas. Cancer Chemother. Rep. 1966, 50, 125–128. [Google Scholar] [PubMed]
- MacIntosh, A.C.; Stower, M.; Reid, N.; Maitland, N.J. Precise microdissection of human prostate cancers reveals genotypic heterogeneity. Cancer Res. 1998, 58, 23–28. [Google Scholar] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Hayward, S.W.; Wang, Y.; Cao, M.; Hom, Y.K.; Zhang, B.; Grossfeld, G.D.; Sudilovsky, D.; Cunha, G.R. Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer Res. 2001, 61, 8135–8142. [Google Scholar] [PubMed]
- Hall, A.J.; Maitland, N.J.; Stower, M.; Lang, S.H. Primary prostate stromal cells modulate the morphology and migration of primary prostate epithelial cells in type 1 collagen gels. Cancer Res. 2002, 62, 58–62. [Google Scholar] [PubMed]
- Mo, F.; Lin, D.; Takhar, M.; Ramnarine, V.R.; Dong, X.; Bell, R.H.; Volik, S.V.; Wang, K.; Xue, H.; Wang, Y.; et al. Stromal Gene Expression Is Predictive for Metastatic Primary Prostate Cancer. Eur. Urol. 2018, 73, 524–532. [Google Scholar] [CrossRef]
- Dunne, P.D.; McArt, D.G.; Bradley, C.A.; O’Reilly, P.G.; Barrett, H.L.; Cummins, R.; O’Grady, T.; Arthur, K.; Loughrey, M.; Allen, W.L.; et al. Challenging the cancer molecular stratification dogma: Intratumoral heterogeneity undermines consensus molecular subtypes and potential diagnostic value in colorectal cancer. Clin. Cancer Res. 2016, 22, 4095–4104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The Genomic Complexity of Primary Human Prostate Cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.-M.; Robinson, R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The Mutational Landscape of Lethal Castration-Resistant Prostate Cancer. Nature 2013, 487, 239–243. [Google Scholar] [CrossRef]
- Sharifi, N. Mechanisms of Androgen Receptor Activation in Castration-Resistant Prostate Cancer. Endocrinology 2013, 154, 4010–4017. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of Resistance in Castration-Resistant Prostate Cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [CrossRef]
- Goldstein, A.; Toro, P.V.; Lee, J.; Silberstein, J.L.; Nakazawa, M.; Waters, I.; Cravero, K.; Chu, D.; Cochran, R.L.; Kim, M.; et al. Detection Fidelity of AR Mutations in Plasma Derived Cell-Free DNA. Oncotarget 2017, 8, 15651–15662. [Google Scholar] [CrossRef] [PubMed]
- Endrullat, C.; Glökler, J.; Franke, P.; Frohme, M. Standardization and Quality Management in Next-Generation Sequencing. Appl. Transl. Genomics 2016, 10, 2–9. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 2017, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, A.; M͡oz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; Manno, G.L.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Cell Types in the Mouse Cortex and Hippocampus Revealed by Single-Cell RNA-Seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Werner, B.; Barnes, C.P.; Graham, T.A.; Sottoriva, A. Identification of Neutral Tumor Evolution Across Cancer Types. Nat. Genet. 2016, 48, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated Evolution of Prostate Cancer Genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.-P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome Sequencing Identifies Recurrent SPOP, FOXA1 and MED12 Mutations in Prostate Cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Wang, C.; Deng, Y.; Yu, L.; Huang, H. Destruction of Full-LengthAndrogen Receptor by Wild-Type SPOP, but Not Prostate-Cancer-Associated Mutants. Cell Rep. 2014, 6, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.W.; Galsky, M.D.; Scher, H.I.; Magidson, J.; Wassmann, K.; Lee, G.-S.M.; Katz, L.; Subudhi, S.K.; Anand, A.; Fleisher, M.; et al. A Whole-Blood RNA Transcript-Based Prognostic Model in Men with Castration-Resistant Prostate Cancer: A Prospective Study. Lancet Oncol. 2012, 13, 1105–1113. [Google Scholar] [CrossRef]
- Olmos, D.; Brewer, D.; Clark, J.; Danila, D.C.; Parker, C.; Attard, G.; Fleisher, M.; Reid, A.H.; Castro, E.; Sandhu, S.K.; et al. Prognostic Value of Blood mRNA Expression Signatures in Castration-Resistant Prostate Cancer: A Prospective, Two-Stage Study. Lancet Oncol. 2012, 13, 1114–1124. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human Acute Myeloid Leukemia Is Organized as a Hierarchy That Originates From a Primitive Hematopoietic Cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Rane, J.K.; Droop, A.P.; Pellacani, D.; Polson, E.S.; Simms, M.S.; Collins, A.T.; Caves, L.S.D.; Maitland, N.J. Conserved Two-Step Regulatory Mechanism of Human Epithelial Differentiation. Stem Cell Rep. 2014, 2, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Rane, J.K.; Scaravilli, M.; Ylipää, A.; Pellacani, D.; Mann, V.M.; Simms, M.S.; Nykter, M.; Collins, A.T.; Visakorpi, T.; Maitland, N.J. MicroRNA Expression Profile of Primary Prostate Cancer Stem Cells as a Source of Biomarkers and Therapeutic Targets. Eur. Urol. 2015, 67, 7–10. [Google Scholar] [CrossRef]
- Rane, J.K.; Ylipaa, A.; Adamson, R.; Mann, V.M.; Simms, M.S.; Collins, A.T.; Visakorpi, T.; Nykter, M.; Maitland, N.J. Construction of Therapeutically Relevant Human Prostate Epithelial Fate Map by Utilising miRNA and mRNA Microarray Expression Data. Br. J. Cancer 2015, 113, 611–615. [Google Scholar] [CrossRef]
- Maitland, N.J.; Frame, F.M.; Polson, E.S.; Lewis, J.L.; Collins, A.T. Prostate Cancer Stem Cells: Do They Have a Basal or Luminal Phenotype? Hormones Cancer 2011, 2, 47–61. [Google Scholar] [CrossRef]
- Goldstein, A.S.; Huang, J.; Guo, C.; Garraway, I.P.; Witte, O.N. Identification of a Cell of Origin for Human Prostate Cancer. Science 2010, 329, 568–571. [Google Scholar] [CrossRef]
- Lawson, D.A.; Zong, Y.; Memarzadeh, S.; Xin, L.; Huang, J.; Witte, O.N. Basal Epithelial Stem Cells Are Efficient Targets for Prostate Cancer Initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 2610–2615. [Google Scholar] [CrossRef]
- Wang, X.I.; Kruithof-de Julio, M.; Economides, K.D.; Walker, D.; Yu, H.; Halili, M.V.; Hu, Y.; Price, S.M.; Abate-Shen, C.; Shen, M.M.; et al. A Luminal Epithelial Stem Cell That Is a Cell of Origin for Prostate Cancer. Nature 2009, 461, 495–500. [Google Scholar] [CrossRef]
- Tomasetti, C.; Vogelstein, B. Cancer Etiology. Variation in Cancer Risk Among Tissues Can Be Explained by the Number of Stem Cell Divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef]
- Calabrese, P.; Tavaré, S.; Shibata, D. Pretumor Progression: Clonal Evolution of Human Stem Cell Populations. Am. J. Pathol. 2004, 164, 1337–1346. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Schumacher, B. The P53 Network: Cellular and Systemic DNA Damage Responses in Aging and Cancer. Trends Genet. TIG 2012, 28, 128–136. [Google Scholar] [CrossRef]
- Victorelli, S.; Passos, J.F. Telomeres and Cell Senescence—Size Matters Not. EBioMedicine 2017, 21, 14–20. [Google Scholar] [CrossRef]
- Nonn, L.; Ananthanarayanan, V.; Gann, P.H. Evidence for Field Cancerization of the Prostate. Prostate 2009, 69, 1470–1479. [Google Scholar] [CrossRef] [Green Version]
- Mehrotra, J.; Varde, S.; Wang, H.; Chiu, H.; Vargo, J.; Gray, K.; Nagle, R.B.; Neri, J.R.; Mazumder, A. Quantitative, Spatial Resolution of the Epigenetic Field Effect in Prostate Cancer. Prostate 2008, 68, 152–160. [Google Scholar] [CrossRef]
- Slaughter, P.; Southwick, H.W.; Smejkal, W. Field Cancerization in Oral Stratified Squamous Epithelium; Clinical Implications of Multicentric Origin. Cancer 1953, 6, 963–968. [Google Scholar] [CrossRef]
- Gundem, G.; Van Loo, P.; Kremeyer, B.; Alexandrov, L.B.; Tubio, J.M.C.; Papaemmanuil, E.; Brewer, D.S.; Kallio, H.M.L.; Hognas, G.; Annala, M.; et al. The Evolutionary History of Lethal Metastatic Prostate Cancer. Nature 2015, 520, 353–357. [Google Scholar] [CrossRef] [Green Version]
- Birnie, R.; Bryce, S.D.; Roome, C.; Dussupt, V.; Droop, A.; Lang, S.H.; Berry, P.A.; Hyde, C.F.; Lewis, J.L.; Stower, M.J.; et al. Gene Expression Profiling of Human Prostate Cancer Stem Cells Reveals a Pro-Inflammatory Phenotype and the Importance of Extracellular Matrix Interactions. Genome Biol. 2008, 2008, R83. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective Identification of Tumorigenic Prostate Cancer Stem Cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [Green Version]
- Frame, F.M.; Pellacani, D.; Collins, A.T.; Maitland, N.J. Harvesting Human Prostate Tissue Material and Culturing Primary Prostate Epithelial Cells. Methods Mol. Biol. 2016, 1443, 181–201. [Google Scholar] [CrossRef]
- Vezina, C.M.; Allgeier, S.H.; Fritz, W.A.; Moore, R.W.; Strerath, M.; Bushman, W.; Peterson, R.E. Retinoic Acid Induces Prostatic Bud Formation. Dev. Dyn. 2008, 237, 1321–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldridge, E.E.; Walker, H.F.; Stower, M.J.; Simms, M.S.; Mann, V.M.; Collins, A.T.; Pellacani, D.; Maitland, N.J. Retinoic Acid Represses Invasion and Stem Cell Phenotype by Induction of the Metastasis Suppressors RARRES1 and LXN. Oncogenesis 2013, 2, e45. [Google Scholar] [CrossRef] [PubMed]
- Waltregny, D.; Leav, I.; Signoretti, S.; Soung, P.; Lin, D.; Merk, F.; Adams, J.Y.; Bhattacharya, N.; Cirenei, N.; Loda, M. Androgen-Driven Prostate Epithelial Cell Proliferation and Differentiation in Vivo Involve the Regulation of P27. Mol. Endocrinol. 2001, 15, 765–782. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.A.; Hodgson, M.C.; Loermans, S.D.; Hooper, J.; Endersby, R.; Bentel, J.M. Transcriptional Regulation of the Homeobox Gene NKX3.1 by All-Trans Retinoic Acid in Prostate Cancer Cells. J. Cell. Biochem. 2006, 99, 1409–1419. [Google Scholar] [CrossRef] [PubMed]
- Rane, J.K.; Pellacani, D.; Maitland, N.J. Advanced Prostate Cancer—A Case for Adjuvant Differentiation Therapy. Nat. Rev. Urol. 2012, 9, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Goossens, C.; Deboel, L.; Swinnen, J.V.; Roskams, T.; Manin, M.; Rombauts, W.; Verhoeven, G. Both Retinoids and Androgens Are Required to Maintain or Promote Functional Differentiation in Reaggregation Cultures of Human Prostate Epithelial Cells. Prostate 2002, 53, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Gonzalez, G.C.; Droop, A.P.; Rippon, H.J.; Tiemann, K.; Pellacani, D.; Georgopoulos, L.J.; Maitland, N.J. Retinoic Acid and Androgen Receptors Combine to Achieve Tissue Specific Control of Human Prostatic Transglutaminase Expression: A Novel Regulatory Network with Broader Significance. Nucleic Acids Res. 2012, 40, 4825–4840. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, S.; Zhang, Z.; He, J.; Xu, Y.; Liu, S. ROCK Has a Crucial Role in Regulating Prostate Tumor Growth Through Interaction with C-Myc. Oncogene 2014, 33, 5582–5591. [Google Scholar] [CrossRef] [PubMed]
- Claassen, D.A.; Desler, M.M.; Rizzino, A. ROCK Inhibition Enhances the Recovery and Growth of Cryopreserved Human Embryonic Stem Cells and Human Induced Pluripotent Stem Cells. Mol. Reprod. Dev. 2009, 76, 722–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent Fusion of TMPRSS2 and ETS Transcription Factor Genes in Prostate Cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Polson, E.S.; Lewis, J.; Celik, H.; Mann, V.M.; Stower, M.J.; Simms, M.S.; Rodrigues, G.; Collins, A.T.; Maitland, N.J. Monoallelic Expression of TMPRSS2/ERG in Prostate Cancer Stem Cells. Nat. Commun. 2013, 4, 1623. [Google Scholar] [CrossRef] [PubMed]
- Frame, F.M.; Maitland, N.J. Cancer Stem Cells Provide New Insights into the Therapeutic Responses of Human Prostate Cancer. In Stem Cells and Prostate Cancer; Cramer, S.D., Ed.; Springer: New York, NY, USA, 2013; pp. 51–75. [Google Scholar]
- Petrie, K.; Zelent, A.; Waxman, S. Differentiation Therapy of Acute Myeloid Leukemia: Past, Present and Future. Curr. Opin. Hematol. 2009, 16, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.-C.; Hsieh, H.-Y.; Chu, P.-C.; Chen, C.S. Therapeutic Opportunities of Targeting Histone Deacetylase Isoforms to Eradicate Cancer Stem Cells. Int. J. Mol. Sci. 2018, 19, 1939. [Google Scholar] [CrossRef] [PubMed]
- Salvador, M.A.; Wicinski, J.; Cabaud, O.; Toiron, Y.; Finetti, P.J.; Lelièvre, H.; Kraus-Berthier, L.; Depil, S.; Bertucci, F.; Collette, Y.; et al. The Histone Deacetylase Inhibitor Abexinostat Induces Cancer Stem Cells Differentiation in Breast Cancer with Low Xist Expression. Clin. Cancer Res. 2013, 19, 6520–6531. [Google Scholar] [CrossRef] [Green Version]
- Waddington, C.M. Organisers and Genes; Cambridge University Press: Cambridge, MA, USA, 1940. [Google Scholar]
- Papp, B.; Plath, K. Epigenetics of Reprogramming to Induced Pluripotency. Cell 2013, 152, 1324–1343. [Google Scholar] [CrossRef]
- Aranda-Anzaldo, A.; Dent, M.A.R. Landscaping the Epigenetic Landscape of Cancer. Prog. Biophys. Mol. Biol. 2018, 2018, 155–174. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic Modulators, Modifiers and Mediators in Cancer Aetiology and Progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic Reprogramming During Pancreatic Cancer Progression Links Anabolic Glucose Metabolism to Distant Metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Visakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinänen, R.; Palmberg, C.; Palotie, A.; Tammela, T.; Isola, J.; Kallioniemi, O.P. In Vivo Amplification of the Androgen Receptor Gene and Progression of Human Prostate Cancer. Nat. Genet. 1995, 9, 401–406. [Google Scholar] [CrossRef]
- Horns, R.C., Jr.; Dower, W.J.; Schimke, R.T. Gene Amplification in a Leukemic Patient Treated with Methotrexate. J. Clin. Oncol. 1984, 2, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The Epigenetic Progenitor Origin of Human Cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef]
- Przyborski, S.; Carr-Wilkinson, J.; Robson, C.N.; Heer, R. A Novel Model of Urinary Tract Differentiation, Tissue Regeneration, and Disease: Reprogramming Human Prostate and Bladder Cells into Induced Pluripotent Stem Cells. Eur. Urol. 2013, 64, 753–761. [Google Scholar] [CrossRef] [Green Version]
- Maitland, N.J. Stem Cells in the Normal and Malignant Prostate. In Prostate Cancer; Tindall, D.J., Ed.; Springer: New York, NY, USA, 2013; pp. 3–41. [Google Scholar]
- Neilson, J.R.; Zheng, G.X.Y.; Burge, C.B.; Sharp, P.A. Dynamic Regulation of miRNA Expression in Ordered Stages of Cellular Development. Genes Dev. 2007, 21, 578–589. [Google Scholar] [CrossRef]
- Mathieu, J.; Ruohola-Baker, H. Regulation of Stem Cell Populations by microRNAs. Transcript. Transl. Regul. Stem Cells 2013, 786, 329–351. [Google Scholar] [CrossRef] [Green Version]
- Catto, J.W.F.; Alcaraz, A.; Bjartell, A.S.; De Vere White, R.; Evans, C.P.; Fussel, S.; Hamdy, F.C.; Kallioniemi, O.; Mengual, L.; Schlomm, T.; et al. MicroRNA in Prostate, Bladder, and Kidney Cancer: A Systematic Review. Eur. Urol. 2011, 59, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Zoni, E.; Van Der Horst, G.; van de Merbel, A.F.; Chen, L.; Rane, J.K.; Pelger, R.C.M.; Collins, A.T.; Visakorpi, T.; Snaar-Jagalska, B.E.; Maitland, N.J.; et al. miR-25 Modulates Invasiveness and Dissemination of Human Prostate Cancer Cells via Regulation of Av- and A6-Integrin Expression. Cancer Res. 2015, 75, 2326–2336. [Google Scholar] [CrossRef]
- Liu, C.; Kelnar, K.; Vlassov, A.V.; Brown, D.; Wang, J.; Tang, D.G. Distinct microRNA Expression Profiles in Prostate Cancer Stem/Progenitor Cells and Tumor-Suppressive Functions of Let-7. Cancer Res. 2012, 72, 3393–3404. [Google Scholar] [CrossRef]
- Leonardo, T.R.; Schultheisz, H.L.; Loring, J.F. The Functions of microRNAs in Pluripotency and Reprogramming. Nat. Cell Biol. 2012, 14, 1114–1121. [Google Scholar] [CrossRef]
- Jalava, S.E.; Urbanucci, A.; Latonen, L.; Waltering, K.K. Androgen-Regulated miR-32 Targets BTG2 and Is Overexpressed in Castration-Resistant Prostate Cancer. Oncogene 2012, 31, 4460–4471. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Ranjbar, B.; Ganjalikhany, M.R.; Khan, F.M.; Schmitz, U.; Wolkenhauer, O.; Gupta, S.K. MicroRNA and Transcription Factor Gene Regulatory Network Analysis Reveals Key Regulatory Elements Associated with Prostate Cancer Progression. PLoS ONE 2016, 11, e0168760. [Google Scholar] [CrossRef] [PubMed]
- Formosa, A.; Markert, E.K.; Lena, A.M.; Italiano, D.O. MicroRNAs, miR-154, miR-299-5p, miR-376a, miR-376c, miR-377, miR-381, miR-487b, miR-485-3p, miR-495 and miR-654-3p, Mapped to the 14q32. 31 Locus regulate proliferation, apoptosis, migration and invasion in metastatic prostate cancer cells. Oncogene 2014, 33, 5173–5182. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.; Majid, S.; Shahryari, V.; Arora, S.; Yamamura, S.; Chang, I.; Zaman, M.S.; Deng, G.; Tanaka, Y.; Dahiya, R. miRNA-708 Control of CD44+ Prostate Cancer-Initiating Cells. Cancer Res. 2012, 72, 3618–3630. [Google Scholar] [CrossRef] [Green Version]
- Josson, S.; Lao, K.; Chung, L.W.; Johnstone, P.A.; Sung, S.-Y.; Sung, S. Radiation modulation of microRNA in prostate cancer cell lines. Prostate 2008, 68, 1599–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.M.; Cheong, H.S.; Lee, J.-K. Genome-Wide Detection of Allelic Gene Expression in Hepatocellular Carcinoma Cells Using a Human Exome SNP Chip. Gene 2014, 551, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Chivukula, R.R.; Shi, G.; Acharya, A.; Mills, E.W.; Zeitels, L.R.; Anandam, J.L.; Abdelnaby, A.A.; Balch, G.C.; Mansour, J.C.; Yopp, A.C.; et al. An Essential Mesenchymal Function for miR-143/145 in Intestinal Epithelial Regeneration. Cell 2014, 157, 1104–1116. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Guo, W.; Liu, T.; Wang, X.; Tu, X.; Xiong, D.; Chen, S.; Lai, Y.; Du, H.; Chen, G.; et al. Identification of miRs-143 and -145 That Is Associated with Bone Metastasis of Prostate Cancer and Involved in the Regulation of EMT. PloS ONE 2011, 6, e20341. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Hsu, A.K.; Sajdak, J.; Qin, J.; Pavlidis, P. Coexpression Analysis of Human Genes Across Many Microarray Data Sets. Genome Res. 2004, 14, 1085–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turdo, A.; Veschi, V.; Gaggianesi, M.; Chinnici, A.; Bianca, P.; Todaro, M.; Stassi, G. Meeting the Challenge of Targeting Cancer Stem Cells. Front. Cell Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef]
- Skvortsov, S.; Debbage, P.; Lukas, P.; Skvortsova, I. Crosstalk Between DNA Repair and Cancer Stem Cell (CSC) Associated Intracellular Pathways. Semin. Cancer Biol. 2015, 31, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Skvortsova, I.; Debbage, P.; Kumar, V.; Skvortsov, S. Radiation Resistance: Cancer Stem Cells (CSCs) and Their Enigmatic Pro-Survival Signaling. Semin. Cancer Biol. 2015, 35, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Lukas, C.; Bartek, J. More Than Just a Focus: The Chromatin Response to DNA Damage and Its Role in Genome Integrity Maintenance. Nat. Cell Biol. 2011, 13, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Takata, H.; Hanafusa, T.; Mori, T.; Shimura, M.; Iida, Y.; Ishikawa, K.; Yoshikawa, K.; Yoshikawa, Y.; Maeshima, K. Chromatin Compaction Protects Genomic DNA From Radiation Damage. Edited by Yamini Dalal. PLoS ONE 2013, 8, e75622. [Google Scholar] [CrossRef]
- Falk, M.; Lukášová, E.; Kozubek, S. Chromatin Structure Influences the Sensitivity of DNA to Γ-Radiation. Biochim. Biophys. Acta 2008, 1783, 2398–2414. [Google Scholar] [CrossRef] [PubMed]
- Srikantan, S.; Abdelmohsen, K.; Lee, E.K.; Tominaga, K.; Subaran, S.S.; Kuwano, Y.; Kulshrestha, R.; Panchakshari, R.; Kim, H.H.; Yang, X.; et al. Translational Control of TOP2A Influences Doxorubicin Efficacy. Mol. Cell. Biol. 2011, 31, 3790–3801. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.C.N.; Xie, W.; Yang, M.; Hsieh, C.-L.; Drouin, S.; Lee, G.-S.M.; Kantoff, P.W. Expression Differences of Circulating microRNAs in Metastatic Castration Resistant Prostate Cancer and Low-Risk, Localized Prostate Cancer. Prostate 2013, 73, 346–354. [Google Scholar] [CrossRef]
- Hall, A.W.; Battenhouse, A.M.; Shivram, H.; Morris, A.R.; Cowperthwaite, M.C.; Shpak, M.; Iyer, V.R. Bivalent Chromatin Domains in Glioblastoma Reveal a Subtype-Specific Signature of Glioma Stem Cells. Cancer Res. 2018, 78, 2463–2474. [Google Scholar] [CrossRef]
- Voigt, P.; Tee, W.W.; Reinberg, D. A Double Take on Bivalent Promoters. Genes Dev. 2013, 27, 1318–1338. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Wan, M.; Zhang, B.; Peng, Y.; Zhou, Y.; Pi, C.; Xu, X.; Ye, L.; Zhou, X.; Zheng, L. Bivalent Histone Modifications and Development. Curr. Stem Cell Res. Ther. 2018, 13, 83–90. [Google Scholar] [CrossRef]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–237. [Google Scholar] [CrossRef]
- Almassalha, L.M.; Bauer, G.M.; Wu, W.; Cherkezyan, L.; Zhang, D.; Kendra, A.; Gladstein, S.; Chandler, J.E.; VanDerway, D.; Seagle, B.L.; et al. Macrogenomic Engineering via Modulation of the Scaling of Chromatin Packing Density. Nat. Biomed. Eng. 2017, 1, 902–913. [Google Scholar] [CrossRef]
- Sato, K.; Imai, T.; Okayasu, R.; Shimokawa, T. Heterochromatin Domain Number Correlates with X-Ray and Carbon-Ion Radiation Resistance in Cancer Cells. Radiat. Res. 2014, 182, 408–419. [Google Scholar] [CrossRef]
- Frame, F.M.; Pellacani, D.; Collins, A.T.; Simms, M.S.; Mann, V.M.; Jones, D.D.; Meuth, M.; Bristow, R.G.; Maitland, N.J. HDAC Inhibitor Confers Radiosensitivity to Prostate Stem-Like Cells. Br. J. Cancer 2013, 109, 3023–3033. [Google Scholar] [CrossRef]
- Lafon-Hughes, L.; Di Tomaso, M.V.; Liddle, P.; Toledo, A.; Reyes-Ábalos, A.; Folle, G.A. Preferential Localization of γH2AX Foci in Euchromatin of Retina Rod Cells After DNA Damage Induction. Chromosome Res. 2013, 21, 789–803. [Google Scholar] [CrossRef]
- Frame, F.M.; Noble, A.R.; Klein, S.; Walker, H.F. Tumor Heterogeneity and Therapy Resistance-Implications for Future Treatments of Prostate Cancer. J. Cancer Metastasis 2017, 3, 302. [Google Scholar] [CrossRef]
- Horsman, M.R.; Overgaard, J. The Impact of Hypoxia and Its Modification of the Outcome of Radiotherapy. J. Radiat. 2016, 57, 90–98. [Google Scholar] [CrossRef]
- Johnson, A.B.; Barton, M.C. Hypoxia-Induced and Stress-Specific Changes in Chromatin Structure and Function. Mutat. Res. Fund. Mol. Mech. Mutagenesis 2007, 618, 149–162. [Google Scholar] [CrossRef]
- Prickaerts, P.; Adriaens, M.E.; van den Beucken, T.; Koch, E.; Dubois, L.; Dahlmans, V.E.H.; Gits, C.; Evelo, C.T.A.; Chan-Seng-Yue, M.; Wouters, B.G.; et al. Hypoxia Increases Genome-Wide Bivalent Epigenetic Marking by Specific Gain of H3K27me3. Epigenet. Chromatin 2016, 9, 1–19. [Google Scholar] [CrossRef]
- Galanis, E.; Anderson, S.K.; Miller, C.R.; Sarkaria, J.N.; Jaeckle, K.; Buckner, J.C.; Ligon, K.L.; Ballman, K.V.; Moore, D.F., Jr.; Nebozhyn, M.; et al. Phase I/II Trial of Vorinostat Combined with Temozolomide and Radiation Therapy for Newly Diagnosed Glioblastoma: Results of Alliance N0874/ABTC 02. Neuro-Oncology 2018, 20, 546–556. [Google Scholar] [CrossRef]
- Mueller, A.C.; Sun, D.; Dutta, A. The miR-99 Family Regulates the DNA Damage Response Through Its Target SNF2H. Oncogene 2013, 32, 1164–1172. [Google Scholar] [CrossRef]
- Rane, J.K.; Erb, H.H.H.; Nappo, G.; Mann, V.M.; Simms, S.; Collins, A.T.; Visakorpi, T.; Maitland, N.J. Inhibition of the Glucocorticoid Receptor Results in an Enhanced miR-99a/100-Mediated Radiation Response in Stem-Like Cells from Human Prostate Cancers. Oncotarget 2016, 7, 51965–51980. [Google Scholar] [CrossRef]
- Mulder, K.W.; Wang, X.; Escriu, C.; Ito, Y.; Schwarz, R.F.; Gillis, J.; Sirokmány, G.; Donati, G.; Uribe-Lewis, S.; Pavlidis, P.; et al. Diverse Epigenetic Strategies Interact to Control Epidermal Differentiation. Nat. Cell Biol. 2012, 14, 753–763. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Rodrigues, D.N.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [Green Version]
- Henneman, L.; Van Miltenburg, M.H.; Michalak, M.; Braumuller, T.M.; Jaspers, J.E.; Drenth, A.P.; de Korte-Grimmerink, R.; Gogola, E.; Szuhai, K.; Schlicker, A.; et al. Selective Resistance to the PARP Inhibitor Olaparib in a Mouse Model for BRCA1-Deficient Metaplastic Breast Cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 8409–8414. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.-T.; Yeh, Y.-M.; Chuang, C.-M.; Yang, S.Y.; Chang, J.-W.; Sun, S.-P.; Wang, Y.-S.; Chao, K.-C.; Wang, L.H. Glucocorticoids Mediate Induction of microRNA-708 to Suppress Ovarian Cancer Metastasis Through Targeting Rap1B. Nat. Commun. 2015, 6, 5917. [Google Scholar] [CrossRef]
- Taplin, M.-E.; Manola, J.; Oh, W.K.; Kantoff, P.W.; Bubley, G.J.; Smith, M.; Barb, D.; Mantzoros, C.; Gelmann, E.P.; Balk, S.P. A Phase II Study of Mifepristone (RU-486) in Castration-Resistant Prostate Cancer, with a Correlative Assessment of Androgen-Related Hormones. BJU Int. 2008, 101, 1084–1089. [Google Scholar] [CrossRef]
- Abeshouse, A.; Jaeil, A.; Rehan, A.; Adrian, A.; Samirkumar, A.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Kin Sung, K.W.; Rigoutsis, I.; Loring, J.; et al. Dynamic Changes in the Human Methylome During Differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef]
- Hawkins, R.D.; Hon, G.C.; Lee, L.K.; Ngo, Q.M.; Lister, R.; Pelizzola, M.; Edsall, L.E.; Kuan, S.; Luu, Y.; Klugman, S.; et al. Distinct Epigenomic Landscapes of Pluripotent and Lineage-Committed Human Cells. Stem Cell 2010, 6, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.H.; Morton, R.A.; Epstein, J.I.; Brooks, J.D.; Campbell, P.A.; Bova, G.S.; Hsieh, W.S.; Isaacs, W.B.; Nelson, W.G. Cytidine Methylation of Regulatory Sequences Near the Pi-Class Glutathione S Transferase Gene Accompanies Human Prostatic Carcinogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 11733–11737. [Google Scholar] [CrossRef]
- Nakayama, M.; Bennett, C.J.; Hicks, J.L.; Epstein, J.I.; Platz, E.A.; Nelson, W.G.; De Marzo, A.M. Hypermethylation of the Human Glutathione S-Transferase-Pi Gene (GSTP1) CpG Island Is Present in a Subset of Proliferative Inflammatory Atrophy Lesions but Not in Normal or Hyperplastic Epithelium of the Prostate: A Detailed Study Using Laser-Capture Microdissection. Am. J. Pathol. 2003, 163, 923–933. [Google Scholar]
- Luo, J.-H.; Ding, Y.; Chen, R.; Michalopoulos, G.; Nelson, J.; Tseng, G.; Yu, Y.P. Genome Genome-wide Methylation Analysis of Prostate Tissues Reveals Global Methylation Patterns of Prostate Cancer. Am. J. Pathol. 2013, 182, 2028–2036. [Google Scholar] [CrossRef]
- Aryee, M.J.; Liu, W.; Engelmann, J.C.; Nuhn, P.; Gurel, M.; Haffner, M.C.; Esopi, D.; Irizarry, R.A.; Getzenberg, R.H.; Nelson, W.G.; et al. DNA Methylation Alterations Exhibit Intraindividual Stability and Interindividual Heterogeneity in Prostate Cancer Metastases. Sci. Transl. Med. 2013, 5, 169ra10. [Google Scholar] [CrossRef]
- Angermueller, C.; Clark, S.J.; Lee, H.J.; Macaulay, I.C.; Teng, M.J.; Hu, T.X.; Krueger, F.; Smallwood, S.; Ponting, C.P.; Voet, T.; et al. Parallel Single-Cell Sequencing Links Transcriptional and Epigenetic Heterogeneity. Nat. Methods 2016, 13, 229–232. [Google Scholar] [CrossRef]
- Pellacani, D.; Kestoras, D.; Droop, A.P.; Frame, F.M.; Berry, P.A.; Lawrence, M.G.; Stower, M.J.; Simms, M.S.; Mann, V.M.; Collins, A.T.; et al. DNA Hypermethylation in Prostate Cancer Is a Consequence of Aberrant Epithelial Differentiation and Hyperproliferation. Cell Death Diff. 2014, 21, 761–773. [Google Scholar] [CrossRef]
- Wang, Q.; Williamson, M.; Bott, S.; Brookman-Amissah, N.; Freeman, A.; Nariculam, J.; Hubank, M.J.F.; Ahmed, A.; Masters, J.R. Hypomethylation of WNT5A, CRIP1 and S100P in Prostate Cancer. Oncogene 2007, 26, 6560–6565. [Google Scholar] [CrossRef]
- Yamamoto, H.; Oue, N.; Sato, A.; Hasegawa, Y.; Matsubara, A.; Yasui, W.; Kikuchi, A. Wnt5a Signaling Is Involved in the Aggressiveness of Prostate Cancer and Expression of Metalloproteinase. Oncogene 2010, 29, 2036–2046. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, D.T.; Zheng, Y.; Wittner, B.S.; Lee, R.J.; Zhu, H.; Broderick, K.T.; Desai, R.I.; Fox, D.B.; Brannigan, B.W.; Trautwein, J.; et al. RNA-Seq of Single Prostate CTCs Implicates Noncanonical Wnt Signaling in Antiandrogen Resistance. Science 2015, 349, 1351–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.P.; Ding, Y.; Chen, R.; Liao, S.G.; Ren, B.-G.; Michalopoulos, A.; Michalopoulos, G.; Nelson, J.; Tseng, G.C.; Luo, J.-H. Whole-Genome Methylation Sequencing Reveals Distinct Impact of Differential Methylations on Gene Transcription in Prostate Cancer. Am. J. Pathol. 2013, 183, 1960–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellacani, D.; Droop, A.P.; Frame, F.; Simms, M.S.; Mann, V.M.; Collins, A.T.; Eaves, C.J.; Maitland, N.J. Phenotype-Independent DNA Methylation Changes in Prostate Cancer. Br. J. Cancer 2018, 119, 1133–1143. [Google Scholar] [CrossRef]
- Kim, J.H.; Dhanasekaran, S.M.; Prensner, J.R.; Cao, X.; Robinson, D.; Kalyana-Sundaram, S.; Huang, C.; Shankar, S.; Jing, X.; Iyer, M.; et al. Deep Sequencing Reveals Distinct Patterns of DNA Methylation in Prostate Cancer. Genome Res. 2011, 21, 1028–1041. [Google Scholar] [CrossRef]
- Doi, A.; Park, I.-H.; Wen, B.; Murakami, P.; Aryee, M.J.; Irizarry, R.; Herb, B.; Ladd-Acosta, C.; Rho, J.; Loewer, S.; et al. Differential Methylation of Tissue and Cancerspecific CpG Island Shores Distinguishes Human Induced Pluripotent Stem Cells, Embryonic Stem Cells and Fibroblasts. Nat. Genet. 2009, 41, 1350–1353. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone Modifications at Human Enhancers Reflect Global Cell-Type-Specific Gene Expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The Accessible Chromatin Landscape of the Human Genome. Nature 2012, 489, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Romanoski, C.E.B.; Glass, C. The Selection and Function of Cell Type Specific Enhancers. Nat. Rev. Mol. Cell Biol. 2015, 16, 144–154. [Google Scholar] [CrossRef]
- Foster, C.S.; Falconer, A.; Dodson, A.R.; Norman, A.R.; Dennis, N.; Fletcher, A.; Southgate, C.; Dowe, A.; Dearnaley, D.; Jhavar, S.; et al. Transcription Factor E2F3 Overexpressed in Prostate Cancer Independently Predicts Clinical Outcome. Oncogene 2004, 23, 5871–5879. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Liu, C.; Zhou, B.; Bi, L.; Huang, H.; Lin, T.; Xu, K. Role of EZH2 in the Growth of Prostate Cancer Stem Cells Isolated From LNCaP Cells. Int. J. Mol. Sci. 2013, 14, 11981–11993. [Google Scholar] [CrossRef] [PubMed]
- Lyon, M.F. Gene Action in the X-Chromosome of the Mouse (Mus Musculus L.). Nature 1961, 190, 372–373. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.K.; Disteche, C.M. Silence of the Fathers: Early X Inactivation. BioEssays 2004, 26, 821–824. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Blackwood, J.K.; Williamson, S.C.; Greaves, L.C.; Wilson, L.; Rigas, A.C.; Sandher, R.; Pickard, R.S.; Robson, C.N.; Turnbull, D.M.; Taylor, R.W.; et al. In Situ Lineage Tracking of Human Prostatic Epithelial Stem Cell Fate Reveals a Common Clonal Origin for Basal and Luminal Cells. J. Pathol. 2011, 225, 181–188. [Google Scholar] [CrossRef]
- Eckersley-Maslin, M.A.; Thybert, D.; Bergmann, J.H.; Marioni, J.C.; Flicek, P.; Spector, D.L. Random Monoallelic Gene Expression Increases Upon Embryonic Stem Cell Differentiation. Dev. Cell 2014, 28, 351–365. [Google Scholar] [CrossRef]
- Deng, Q.; Ramsköld, D.; Reinius, B.; Sandberg, R. Single-Cell RNA-Seq Reveals Dynamic, Random Monoallelic Gene Expression in Mammalian Cells. Science 2014, 343, 193–196. [Google Scholar] [CrossRef] [Green Version]
- Dar, R.D.; Razooky, B.S.; Singh, A.; Trimeloni, T.V.; McCollum, J.M.; Cox, C.D.; Simpson, M.L.; Weinberger, L.S. Transcriptional burst frequency and burst size are equally modulated across the human genome. Proc. Natl. Acad. Sci. USA 2012, 109, 17454–17459. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.; Shalek, A.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Gendrel, A.; Attia, M.; Chen, C.-J.; Diabangouaya, P.; Servant, N.; Barillot, E.; Heard, E. Developmental Dynamics and Disease Potential of Random Monoallelic Gene Expression. Dev. Cell 2014, 28, 366–380. [Google Scholar] [CrossRef] [Green Version]
- Gimelbrant, A.; Hutchinson, J.N.; Thompson, B.R.; Chess, A. Widespread Monoallelic Expression on Human Autosomes. Science 2007, 318, 1136–1140. [Google Scholar] [CrossRef]
- Nag, A.; Vigneau, S.; Savova, V.; Zwemer, L.M. Chromatin Signature Identifies Monoallelic Gene Expression Across Mammalian Cell Types. G3 2015, 5, 1713–1720. [Google Scholar] [CrossRef] [Green Version]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Wang, J.; Valo, Z.; Bowers, C.W.; Smith, D.D.; Liu, Z.; Singer-Sam, J. Dual DNA Methylation Patterns in the CNS Reveal Developmentally Poised Chromatin and Monoallelic Expression of Critical Genes. PLoS ONE 2010, 5, e13843. [Google Scholar] [CrossRef]
- Lin, B.; Lee, H.; Yoon, J.G.; Madan, A.; Wayner, E.; Tonning, S.; Hothi, P.; Schroeder, B.; Ulasoc, I.; Foltz, G.; et al. Global Analysis of H3K4me3 and H3K27me3 Profiles in Glioblastoma Stem Cells and Identification of SLC17A7 as a Bivalent Tumor Suppressor Gene. Oncotarget 2015, 6, 5369–5381. [Google Scholar] [CrossRef]
- Trotman, L.C.; Niki, M.; Dotan, Z.A.; Koutcher, J.A.; Di Cristofano, A.; Xiao, A.; Khoo, A.S.; Roy-Burman, P.; Greenberg, N.M.; van Dyke, T.; et al. Pten Dose Dictates Cancer Progression in the Prostate. PLoS Biol. 2003, 1, e9. [Google Scholar] [CrossRef]
- Wang, S.; Garcia, A.J.; Wu, M.; Aawson, D.; Witte, O.N.; Wu, H. Pten Deletion Leads to the Expansion of a Prostatic Stem/Progenitor Cell Subpopulation and Tumor Initiation. Proc. Natl. Acad. Sci. USA 2006, 103, 1480–1485. [Google Scholar] [CrossRef]
- Whang, Y.E.; Wu, X.; Suzuki, H.; Reiter, R.E.; Tran, C.; Vessella, R.L.; Said, J.W.; Isaacs, W.B.; Sawyers, C.L. Inactivation of the Tumor Suppressor PTEN/MMAC1 in Advanced Human Prostate Cancer Through Loss of Expression. Proc. Natl. Acad. Sci. USA 1998, 95, 5246–5250. [Google Scholar] [CrossRef]
- Suzuki, A.; de la Pompa, J.L.; Stambolic, V.; Elia, A.J.; Sasaki, T.; del Barco Barrantes, I.; Ho, A.; Wakeham, A.; Itie, A.; Fukumoto, M.; et al. High Cancer Susceptibility and Embryonic Lethality Associated with Mutation of the PTEN Tumor Suppressor Gene in Mic. Curr. Biol. 1998, 8, 1169–1178. [Google Scholar] [CrossRef]
- Suzuki, H.; Freije, D.; Nusskern, D.R.; Okami, K.; Cairns, P.; Sidransky, D.; Isaacs, W.B.; Bova, B.S. Interfocal Heterogeneity of PTEN/MMAC1 Gene Alterations in Multiple Metastatic Prostate Cancer Tissues. Cancer Res. 1998, 58, 204–209. [Google Scholar]
- Raval, A.; Tanner, S.M.; Byrd, C.; Angerman, E.B.; Perko, J.D.; Chen, S.-S.; Hackanson, B.; Grever, M.R.; Lucas, D.M.; Matkovic, J.J.; et al. Downregulation of Death-Associated Protein Kinase 1 (DAPK1) in Chronic Lymphocytic Leukemia. Cell 2007, 129, 879–890. [Google Scholar] [CrossRef] [Green Version]
- Park, D.; Zhong, Y.; Lu, Y.; Rycaj, K.; Gong, S.; Chen, X.; Liu, X.; Chao, H.P.; Whitney, P.; Calhoun-Davis, T.; et al. Stem Cell and Neurogenic Gene-Expression Profiles Link Prostate Basal Cells to Aggressive Prostate Cancer. Nat. Commun. 2016, 7, 1–15. [Google Scholar] [CrossRef]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjöblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The Genomic Landscapes of Human Breast and Colorectal Cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational Heterogeneity in Cancer and the Search for New Cancer-Associated Genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Rashid, N.U.; Sperling, S.; Bolli, N.; Wedge, D.C.; Van Loo, P.; Tai, Y.T.; Shammas, M.A.; Fulciniti, M.; Samur, M.K.; Richardson, P.G.; et al. Differential and Limited Expression of Mutant Alleles in Multiple Myeloma. Blood 2014, 124, 3110–3117. [Google Scholar] [CrossRef]
- Efeyan, A.; Serrano, M. P53: Guardian of the Genome and Policeman of the Oncogenes. Cell Cycle 2007, 6, 1006–1010. [Google Scholar] [CrossRef]
- Finn, O.J. A Believer’s Overview of Cancer Immunosurveillance and Immunotherapy. J. Immunol. 2018, 200, 385–391. [Google Scholar] [CrossRef]
- Cerveira, N.; Ribeiro, F.R.; Peixoto, A.; Costa, V.; Henrique, R.; Jerónimo, C.; Teixeira, M.R. TMPRSS2-ERG Gene Fusion Causing ERG Overexpression Precedes Chromosome Copy Number Changes in Prostate Carcinomas and Paired HGPIN Lesions. Neoplasia 2006, 8, 826–832. [Google Scholar] [CrossRef]
- Taoudi, S.; Bee, T.; Hilton, A.; Knezevic, K.; Scott, J.; Willson, T.A.; Collin, C.; Thomas, T.; Voss, A.K.; Kile, B.T.; et al. ERG Dependence Distinguishes Developmental Control of Hematopoietic Stem Cell Maintenance from Hematopoietic Specification. Genes Dev. 2011, 25, 251–262. [Google Scholar] [CrossRef]
- Cooper, C.S.; Eeles, R.; Wedge, D.C.; Van Loo, P.; Gundem, G.; Alexandrov, L.B.; Kremeyer, B.; Butler, A.; Lynch, A.G.; Camacho, N.; et al. Analysis of the Genetic Phylogeny of Multifocal Prostate Cancer Identifies Multiple Independent Clonal Expansions in Neoplastic and Morphologically Normal Prostate Tissue. Nat. Genet. 2015, 47, 367–372. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
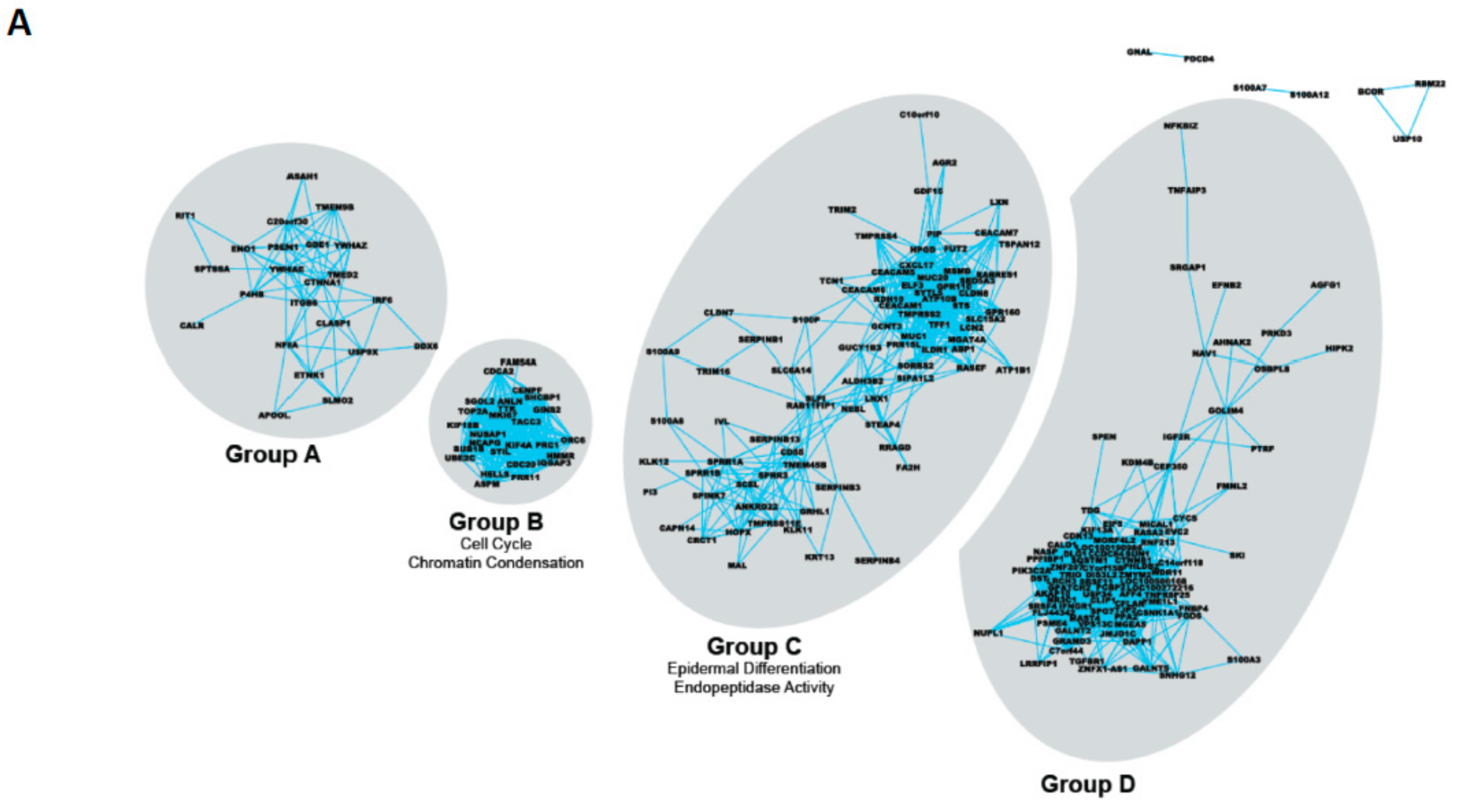
| Gene Group | Common Gene Ontology Terms | Selected Members |
|---|---|---|
| A | None | PSEN1 ITGB6 IRF6 |
| B | Cell cycle Chromatin condensation | CDCA2 TOP2A CDC20 |
| C | Epidermal differentiation Endopeptidase activity | TMPRSS2 S100P SPINK7 ELF3 LXN MSMB |
| D | (Lens) Epithelial development | CTNNB1 IGFR2 |
| TF Identity | Full Name | Principal Role |
|---|---|---|
| RXR | Retinoid X receptor: acts as a homodimer or as a heterodimer with other receptors (e.g., VDR). Binds co-repressors of transcription (as a repressor) until a conformation change occurs after ligand binding. | Reproduction, cellular differentiation, bone development, haematopoiesis and pattern formation during embryogenesis |
| VDR | Vitamin D Receptor: homodimer in the absence of ligand then heterodimerises with RXR to increase transcription of a number of genes. Interacts with SMAD3 and MED1, NCOA1, NCOA2, NCOA3 and NCOA6 coactivators. | Mineral metabolism (calcium homeostasis) although VDR regulates a variety of other pathways, such as those involved in the immune response and cancer. Keratinocyte, mammary and prostate differentiation. |
| GCR | Glucocorticoid Receptor (N3CR1): acts both as a transcription factor and modulator of other transcription factors by binding to glucocorticoid response elements (GRE), both in the cell nucleus and mitochondria. | Affects inflammatory responses, cellular proliferation and differentiation in target tissues. Also involved in chromatin remodeling and RNA stability/degradation. |
| TAZ | Transcriptional co-activator with PDZ-binding motif (or WWTR1): acts as a transcriptional co-activator, downstream of the Hippo pathway. Regulated by soluble extra-cellular factors, cell–cell adhesions and mechano-transduction. Interacts with and regulates multiple transcription factors, e.g., Runx2 PPAR TBX5, TBX5, TEADs, TTF-1 and PAX3. | Organ development, stem cell differentiation and development of human cancer. Mesenchymal stem cell differentiation, promoting cell proliferation and epithelial-mesenchymal transition (EMT). TAZ senses different cellular signals such as cell density and the extracellular matrix stiffness. Significantly overexpressed in breast cancer samples and papillary thyroid carcinoma tissues. |
| SRF | Serum Response Factor: member of the MADS box superfamily of transcription factors, and binds to the serum response element (SRE) in the promoter region of target genes. SRF regulates the activity of many immediate-early genes, e.g., c-fos. A downstream target of many pathways; for example, the mitogen-activated protein kinase pathway (MAPK). | Stimulates cell cycle regulation, apoptosis, cell growth, and cell differentiation. In embryonic development, Expression controls the formation of mesoderm and is crucial for the growth of skeletal muscle. Interaction of SRF with other proteins, such as steroid hormone receptors, may contribute to the regulation of muscle growth by steroids. |
| HSF1 | Heat shock transcription factor 1: an inactive monomer in a complex with Hsp40/Hsp70 and Hsp90. Target genes include major inducible heat shock proteins such as Hsp72 and noncoding RNA within Satellite III repeat regions. Upon stress, such as elevated temperature, HSF1 is released from the chaperone complex and trimerizes. HSF1 is then transported into the nucleus where it is hyperphosphorylated and binds to heat shock elements in DNA. | Master regulator of stress responses, mammalian development, insulin metabolism, cell division, transcriptional reprogramming/chromatin status. |
| ROCK2 | Rho-associated coiled coil-containing protein kinase 2: regulates smooth muscle contraction, actin cytoskeleton organization, stress fiber and focal adhesion formation, neurite retraction, cell adhesion and motility via phosphorylation of ADD1, BRCA2, CNN1, EZR, DPYSL2, EP300, MSN, MYL9/MLC2, NPM1, RDX, PPP1R12A and VIM. Phosphorylates SORL1 and IRF4. Acts as a negative regulator of VEGF-induced angiogenic endothelial cell activation and inhibits keratinocyte terminal differentiation. | Regulates cytoplasmic actin and cell polarity. Major regulator of epithelial terminal differentiation. |
| SC Signature | Specific PCa CSC Signature | Specific CRPC CSC Signature |
|---|---|---|
| Upregulated miRNA | ||
| miR-302 family | miR-33a * | let-7i * |
| miR-371 family | miR-181a-2 * | miR-136 |
| miR-484 | miR-323-3p | miR-143 |
| miR-548c-3p | miR-411 * | miR-214 * |
| miR-487b | miR-362-5p | |
| miR-532-3p | miR-516a-5p | |
| miR-1271 | miR-542-5p | |
| miR-545 | ||
| miR-1913 | ||
| Downregulated miRNA | ||
| let-7 family | miR-302c | miR-125b-2 * |
| miR-8 family | miR-519c-3p | miR-708 |
| miR-10 family | miR-574-5p | |
| miR-17-92 family | miR-1181 | |
| miR-99a/100 | ||
| miR-143 | ||
| miR-145 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frame, F.M.; Maitland, N.J. Epigenetic Control of Gene Expression in the Normal and Malignant Human Prostate: A Rapid Response Which Promotes Therapeutic Resistance. Int. J. Mol. Sci. 2019, 20, 2437. https://doi.org/10.3390/ijms20102437
Frame FM, Maitland NJ. Epigenetic Control of Gene Expression in the Normal and Malignant Human Prostate: A Rapid Response Which Promotes Therapeutic Resistance. International Journal of Molecular Sciences. 2019; 20(10):2437. https://doi.org/10.3390/ijms20102437
Chicago/Turabian StyleFrame, Fiona M., and Norman J. Maitland. 2019. "Epigenetic Control of Gene Expression in the Normal and Malignant Human Prostate: A Rapid Response Which Promotes Therapeutic Resistance" International Journal of Molecular Sciences 20, no. 10: 2437. https://doi.org/10.3390/ijms20102437
APA StyleFrame, F. M., & Maitland, N. J. (2019). Epigenetic Control of Gene Expression in the Normal and Malignant Human Prostate: A Rapid Response Which Promotes Therapeutic Resistance. International Journal of Molecular Sciences, 20(10), 2437. https://doi.org/10.3390/ijms20102437