Involvement of Salicylic Acid in Anthracnose Infection in Tea Plants Revealed by Transcriptome Profiling

 ,
,  and
and
Abstract
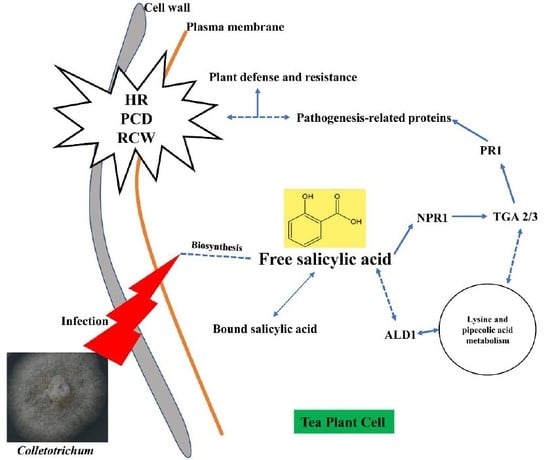
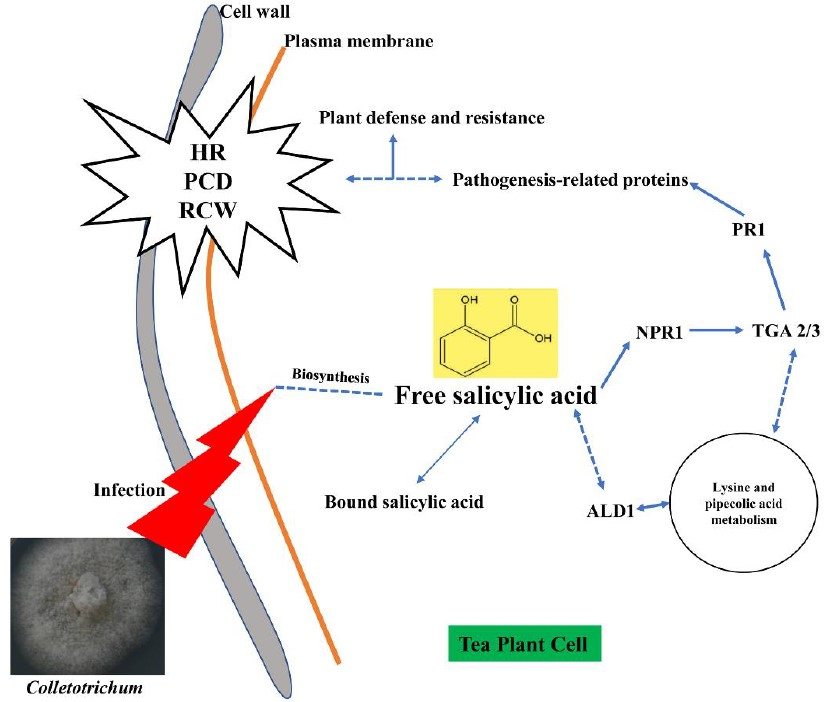
:
1. Introduction
2. Results
2.1. Symptoms of Infected Tea Plants
2.2. Transcriptome Profiling
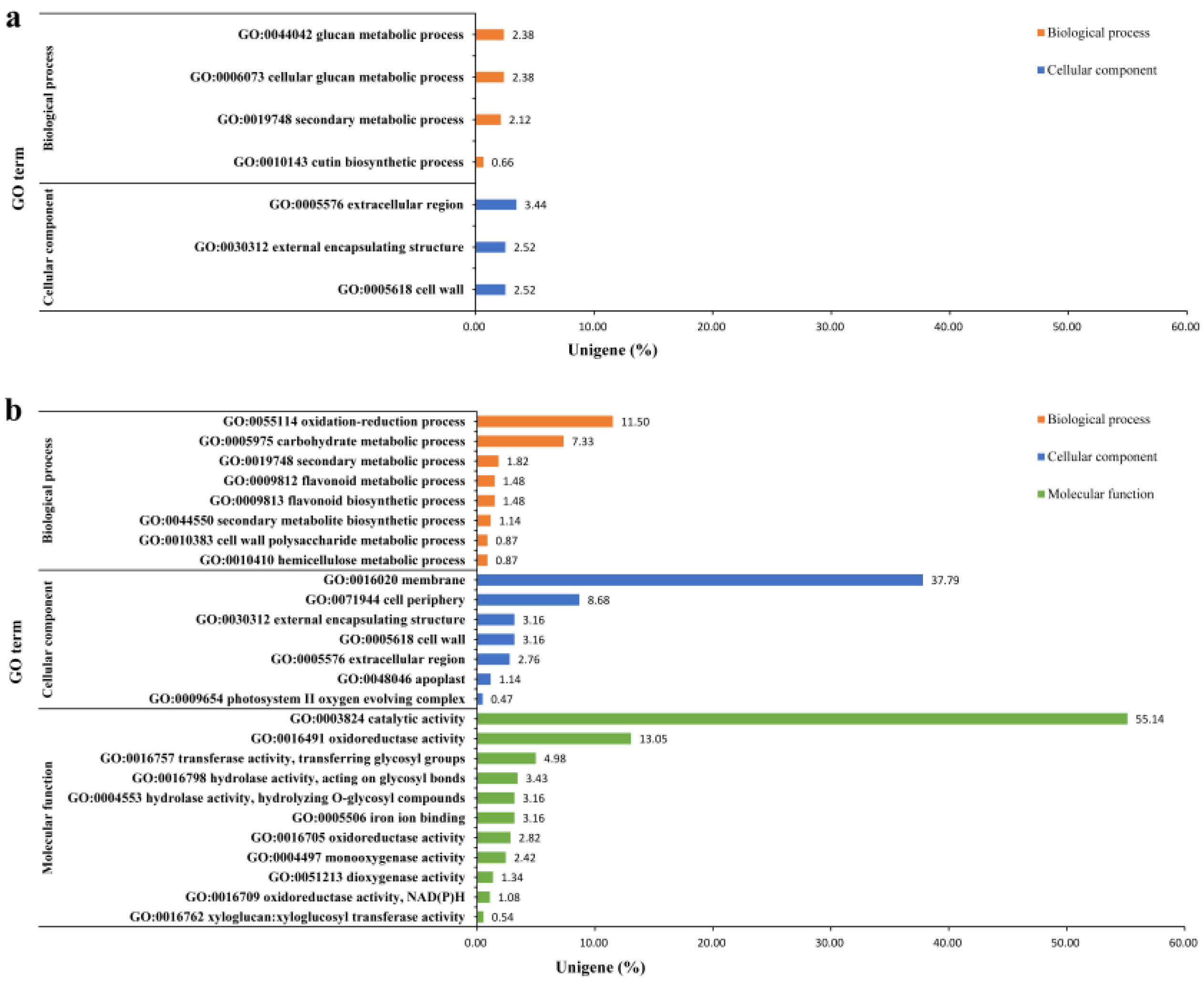
2.2.1. Sequencing Quality, Assembly Characterization and Functional Annotation
2.2.2. Differential Expression and Enrichment Analysis
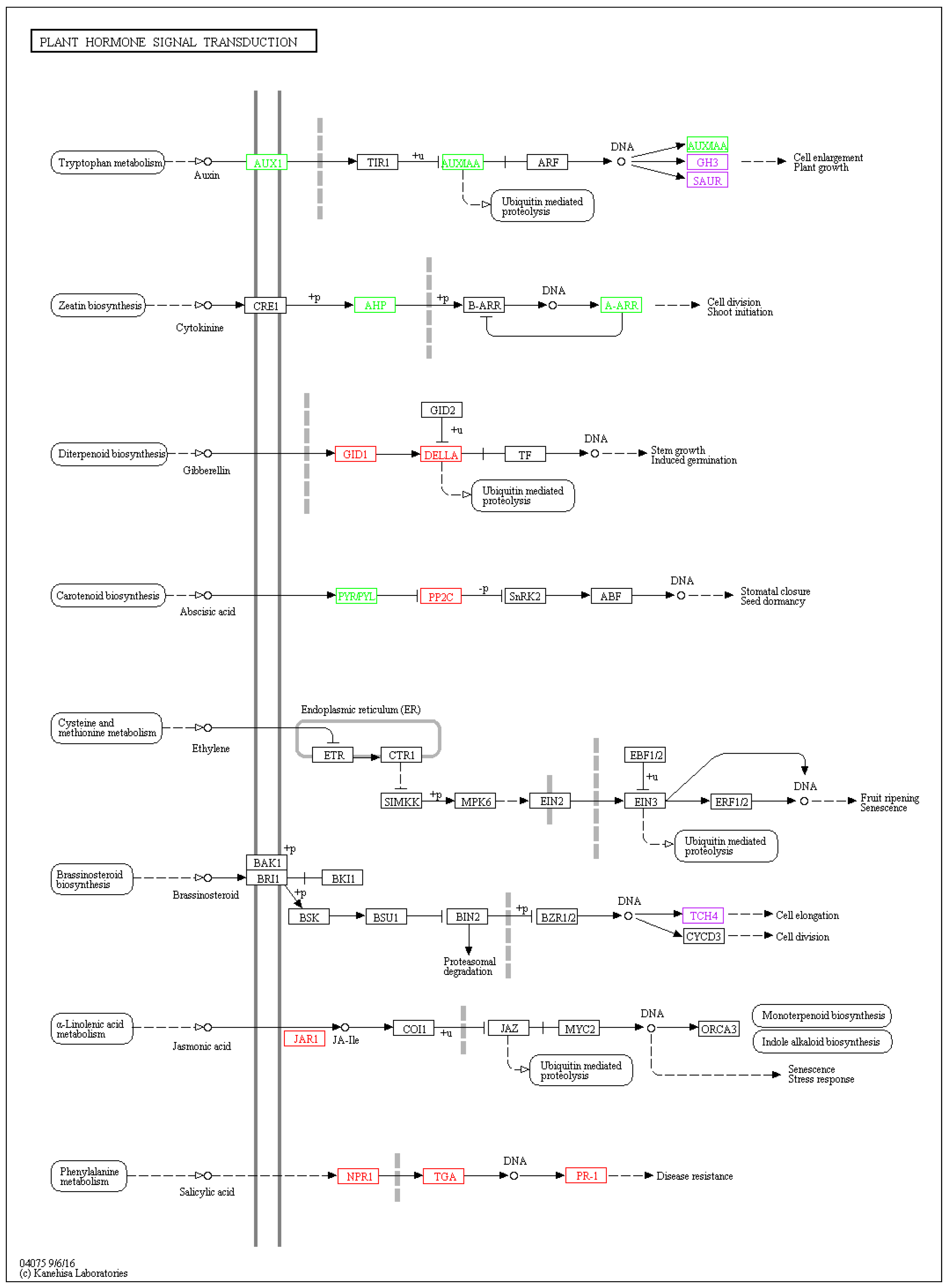
2.2.3. Visualization of Two Vital Plant Metabolic Pathways
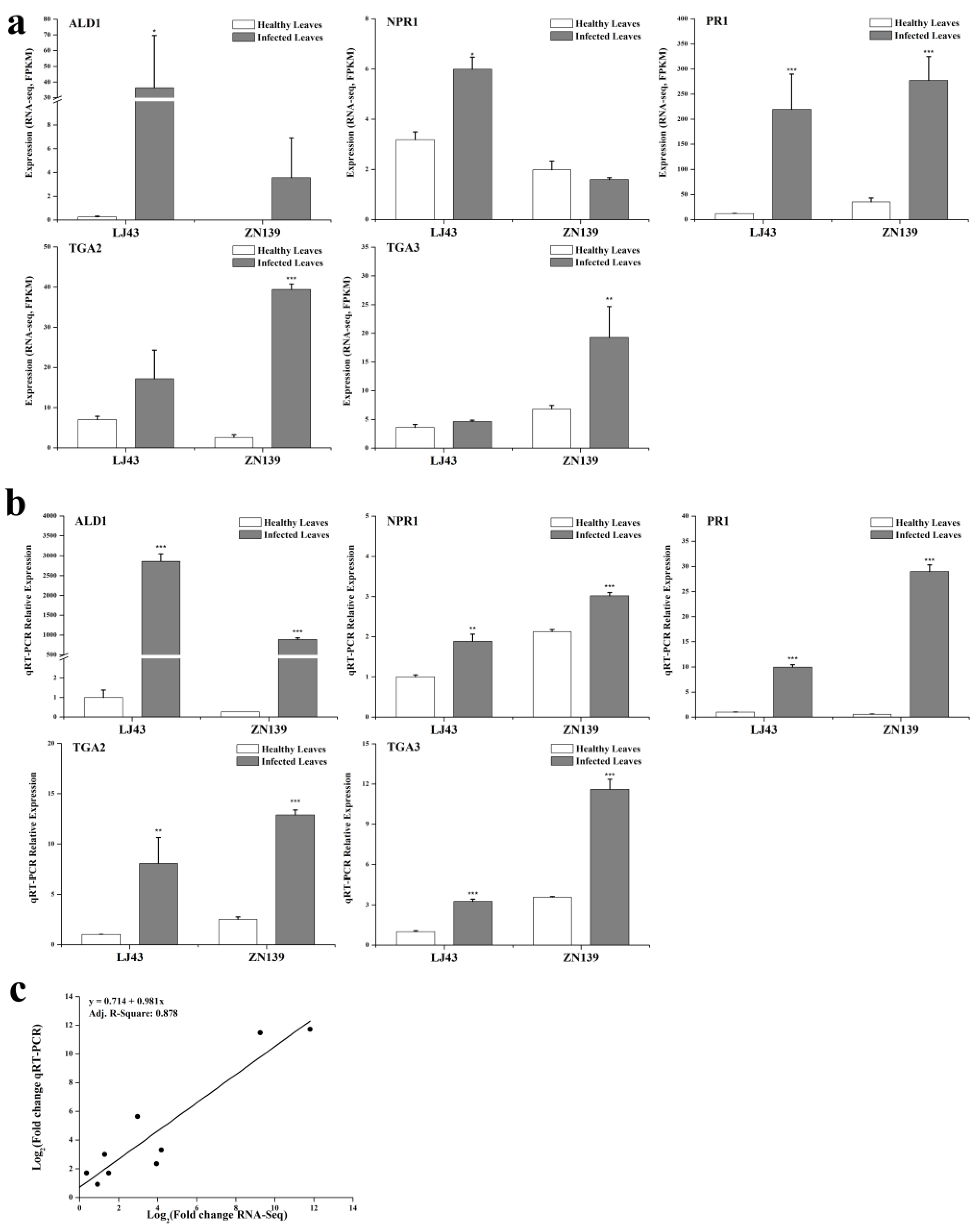
2.3. Quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT-PCR) Validation of Salicylic Acid Signaling Related Genes
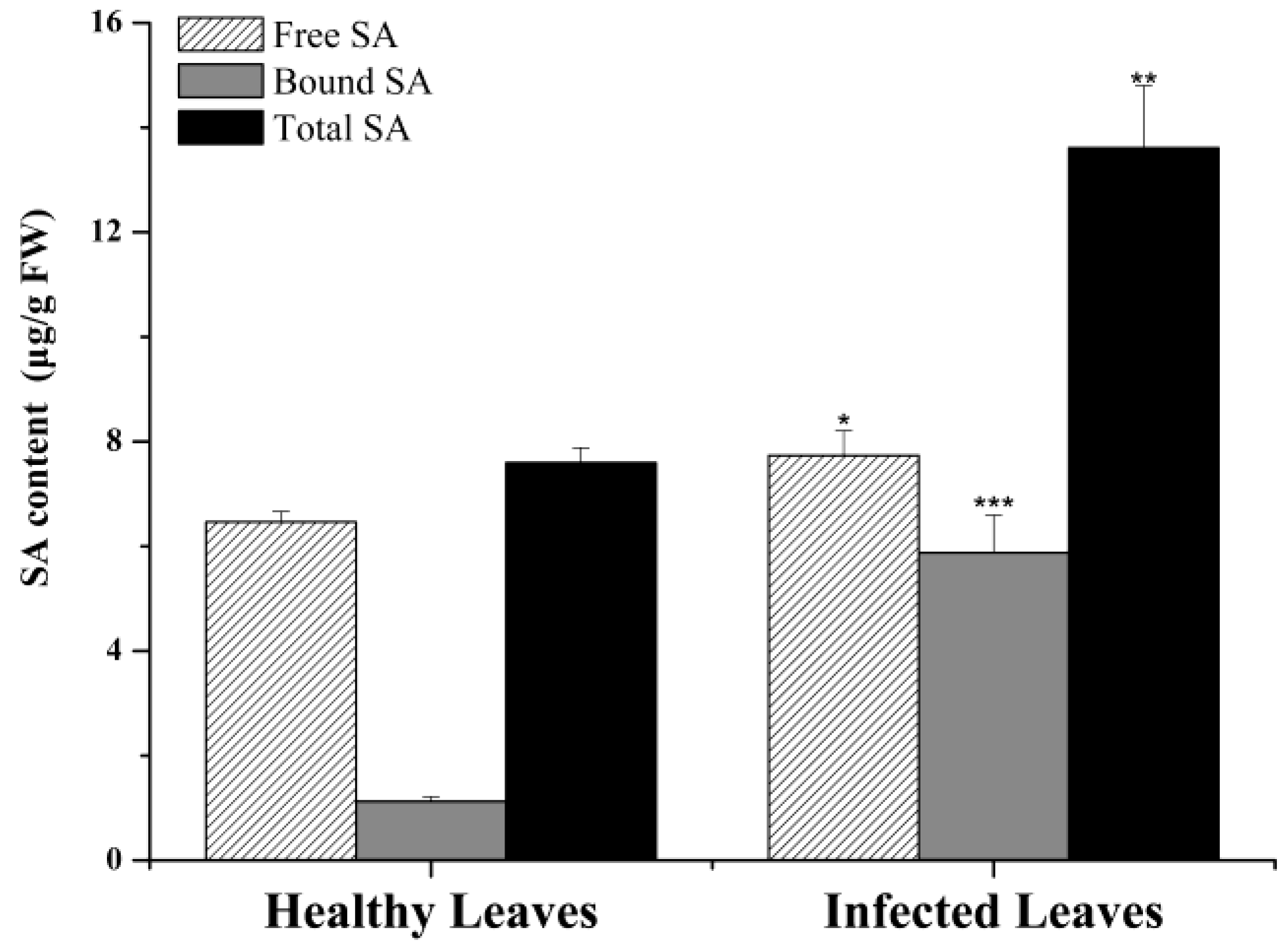
2.4. Salicylic Acid Content
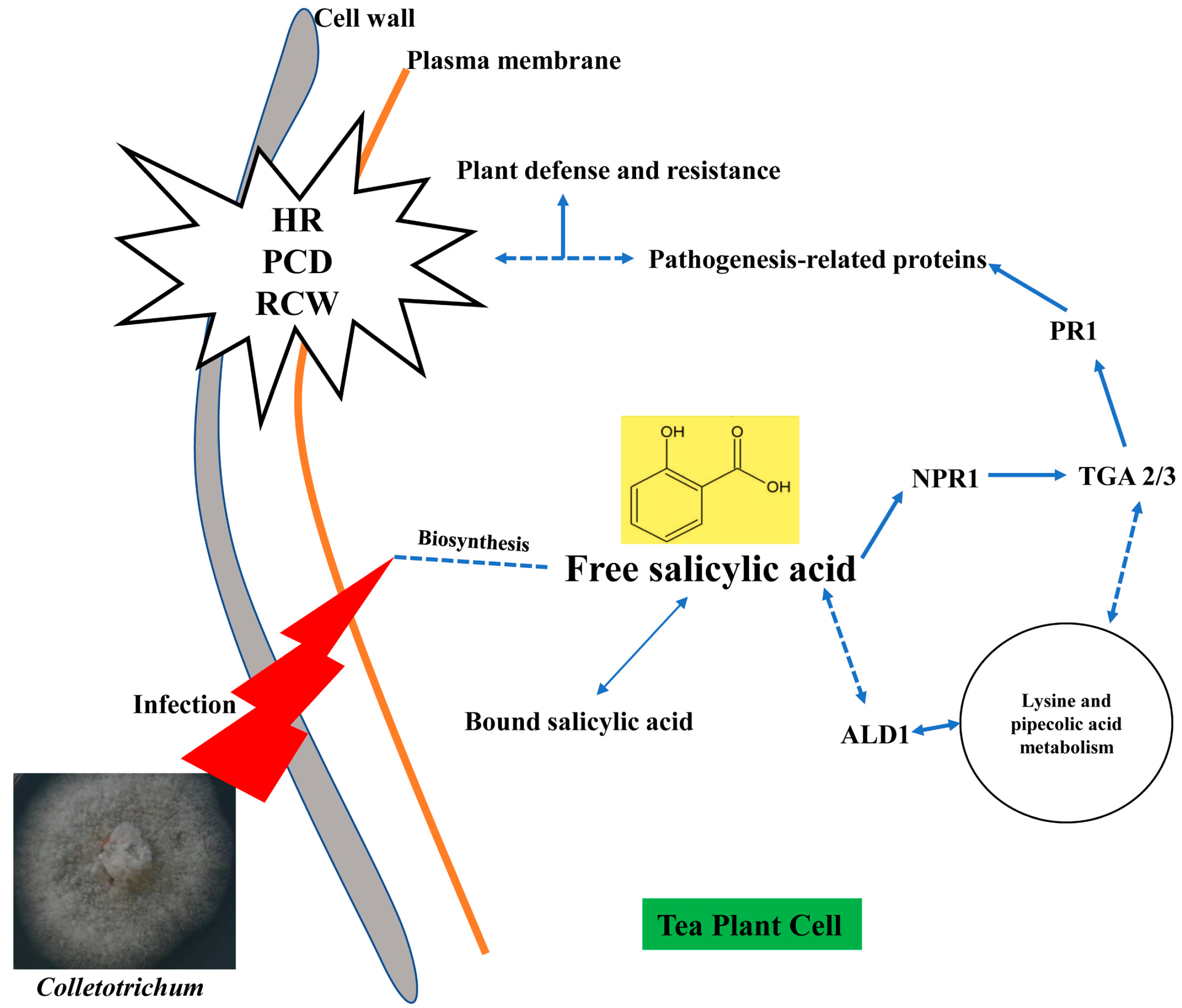
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Total RNA Extraction, cDNA Library Construction and Deep Sequencing
4.3. De Novo Assembly and Functional Annotation
4.4. Differential Expression Genes (DEGs) Analysis
4.5. qRT-PCR Validation
4.6. Salicylic Acid Extraction and High-Performance Liquid Chromatography (HPLC) Quantification
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALD1 | AGD2-like defense response gene 1 |
| DBG | De Bruijn Graph |
| DEGs | Differential expression genes |
| eggNOG | Evolutionary genealogy of genes: Non-supervised Orthologous Groups |
| ETI | Effector-triggered immunity |
| FDR | False discovery rate |
| FPKM | Expected number of Fragments Per Kilobase of transcript sequence per Millions base pairs sequenced |
| GO | Gene Ontology |
| HPLC | High-performance liquid chromatography |
| HR | Hypersensitive response |
| ISR | Immune system resistance |
| ITS | Internal transcribed spacer |
| KAAS | KEGG Automatic Annotation Server |
| KEGG | Kyoto Encyclopedia of Genes and Genome |
| MAS | Molecular assisted selection |
| NPR1 | Nonexpressor of pathogenesis-related gene 1 |
| Nr | NCBI non-redundant protein sequences |
| p | p-value |
| PCD | Programmed cell death |
| PR1 | Pathogenesis-related gene 1 |
| PTI | Pathogen-associated molecular patterns triggered immunity |
| qRT-PCR | Quantitative reverse transcriptase polymerase chain reaction |
| RCW | Reinforcement of the cell wall |
| ROS | Reactive oxygen species |
| SA | Salicylic acid |
| SAR | System acquired resistance |
| TGA | Trans-activating TGA Factors |
References
- Crous, P.W.; Gams, W.; Stalpers, J.A.; Robert, V.; Stegehuis, G. MycoBank: An online initiative to launch mycology into the 21st century. Stud. Mycol. 2004, 19–22. [Google Scholar]
- O’Connell, R.J.; Thon, M.R.; Hacquard, S.; Amyotte, S.G.; Kleemann, J.; Torres, M.F.; Damm, U.; Buiate, E.A.; Epstein, L.; Alkan, N.; et al. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 2012, 44, 1060. [Google Scholar] [CrossRef]
- Cannon, P.F.; Damm, U.; Johnston, P.R.; Weir, B.S. Colletotrichum—Current status and future directions. Stud. Mycol. 2012, 73, 181–213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Hao, X.Y.; Wang, L.; Xiao, B.; Wang, X.C.; Yang, Y.J. Diverse Colletotrichum species cause anthracnose of tea plants (Camellia sinensis (L.) O. Kuntze) in China. Sci. Rep. 2016, 6, 35287. [Google Scholar] [CrossRef]
- Liu, F.; Weir, B.S.; Damm, U.; Crous, P.W.; Wang, Y.; Liu, B.; Wang, M.; Zhang, M.; Cai, L. Unravelling Colletotrichum species associated with Camellia: Employing ApMat and GS loci to resolve species in the C. gloeosporioides complex. Persoonia 2015, 35, 63–86. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.C.; Cao, H.L.; Hao, X.Y.; Zeng, J.M.; Yang, Y.J.; Wang, X.C. Transcriptome analysis of an anthracnose-resistant tea plant cultivar reveals genes associated with resistance to Colletotrichum camelliae. PLoS ONE 2016, 11, e0148535. [Google Scholar] [CrossRef]
- Guo, M.; Pan, Y.M.; Dai, Y.L.; Gao, Z.M. First report of brown blight disease caused by Colletotrichum gloeosporioides on Camellia sinensis in Anhui Province, China. Plant Dis. 2014, 98, 284. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Takeda, Y. Evaluation of anthracnose resistance among tea genetic resources by wound-inoculation assay. JARQ-Jpn. Agr. Res. Quart. 2006, 40, 379–386. [Google Scholar] [CrossRef]
- Yamada, K.; Sonoda, R. A fluorescence microscopic study of the infection process of Discula theae-sinensis in Tea. JARQ-Jpn. Agr. Res. Quart. 2014, 48, 399–402. [Google Scholar] [CrossRef]
- Yoshida, K.; Ogino, A.; Yamada, K.; Sonoda, R. Induction of disease resistance in tea (Camellia sinensis L.) by plant activators. JARQ-Jpn. Agr. Res. Quart. 2010, 44, 391–398. [Google Scholar] [CrossRef]
- Kim, G.H.; Lim, M.T.; Hur, J.S.; Yum, K.J.; Koh, Y.J. Biological control of tea anthracnose using an antagonistic bacterium of Bacillus subtilis isolated from tea leaves. Plant Pathol. J. 2009, 25, 99–102. [Google Scholar] [CrossRef]
- Wang, Y.C.; Hao, X.Y.; Lu, Q.H.; Wang, L.; Qian, W.J.; Li, N.N.; Ding, C.Q.; Wang, X.C.; Yang, Y.J. Transcriptional analysis and histochemistry reveal that hypersensitive cell death and H2O2 have crucial roles in the resistance of tea plant (Camellia sinensis (L.) O. Kuntze) to anthracnose. Horticul. Res. 2018, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Qian, W.J.; Li, N.N.; Hao, X.Y.; Wang, L.; Xiao, B.; Wang, X.C.; Yang, Y.J. Metabolic changes of caffeine in tea plant (Camellia sinensis (L.) O. Kuntze) as defense response to Colletotrichum fructicola. J. Agric. Food Chem. 2016, 64, 6685–6693. [Google Scholar] [CrossRef]
- Lu, Q.; Wang, Y.; Li, N.; Ni, D.; Yang, Y.; Wang, X. Differences in the characteristics and pathogenicity of Colletotrichum camelliae and C. fructicola isolated from the tea plant [Camellia sinensis (L.) O. Kuntze]. Front. Microbiol. 2018, 9, 3060. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Qiao, W.J.; Zeng, L.; Shen, D.H.; Liu, Z.; Wang, X.S.; Tong, H.R. Characterization, pathogenicity, and phylogenetic analyses of Colletotrichum species associated with brown blight disease on Camellia sinensis in China. Plant Dis. 2017, 101, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Vlot, A.C.; Dempsey, D.A.; Klessig, D.F. Salicylic acid, a multifaceted hormone to combat disease. Annu. Rev. Phytopathol. 2009, 47, 177–206. [Google Scholar] [CrossRef]
- An, C.F.; Mou, Z.L. Salicylic acid and its function in plant immunity. J. Integr. Plant Biol. 2011, 53, 412–428. [Google Scholar] [CrossRef]
- Pal, M.; Kovacs, V.; Szalai, G.; Soos, V.; Ma, X.; Liu, H.; Mei, H.; Janda, T. Salicylic acid and abiotic stress responses in rice. J. Agron. Crop. Sci. 2014, 200, 1–11. [Google Scholar] [CrossRef]
- Kovacs, V.; Gondor, O.K.; Szalai, G.; Darko, E.; Majlath, I.; Janda, T.; Pal, M. Synthesis and role of salicylic acid in wheat varieties with different levels of cadmium tolerance. J. Hazard. Mater. 2014, 280, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Yalpani, N.; Enyedi, A.J.; Leon, J.; Raskin, I. Ultraviolet-Light And Ozone Stimulate accumulation of salicylic-acid, pathogenesis-related proteins and virus-resistance in tobacco. Planta 1994, 193, 372–376. [Google Scholar] [CrossRef]
- Zhou, N.; Tootle, T.L.; Tsui, F.; Klessig, D.F.; Glazebrook, J. PAD4 functions upstream from salicylic acid to control defense responses in Arabidopsis. Plant Cell 1998, 10, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Nawrath, C.; Metraux, J.P. Salicylic acid induction-deficient mutants of Arabidopsis express PR-2 and PR-5 and accumulate high levels of camalexin after pathogen inoculation. Plant Cell 1999, 11, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Shirasu, K.; Nakajima, H.; Rajasekhar, V.K.; Dixon, R.A.; Lamb, C. Salicylic acid potentiates an agonist-dependent gain control that amplifies pathogen signals in the activation of defense mechanisms. Plant Cell 1997, 9, 261–270. [Google Scholar] [CrossRef]
- Loake, G.; Grant, M. Salicylic acid in plant defence-the players and protagonists. Curr. Opin. Plant Biol. 2007, 10, 466–472. [Google Scholar] [CrossRef]
- Gaffney, T.; Friedrich, L.; Vernooij, B.; Negrotto, D.; Nye, G.; Uknes, S.; Ward, E.; Kessmann, H.; Ryals, J. Requirement of salicylic-acid for the induction of systemic acquired-resistance. Science 1993, 261, 754–756. [Google Scholar] [CrossRef]
- Hammerschmidt, R. Systemic acquired resistance. Adv. Bot. Res. 2009, 51, 173–222. [Google Scholar] [CrossRef]
- Westermann, A.J.; Gorski, S.A.; Vogel, J. Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 2012, 10, 618–630. [Google Scholar] [CrossRef] [Green Version]
- Marguerat, S.; Bahler, J. RNA-seq: From technology to biology. Cell. Mol. Life Sci. 2010, 67, 569–579. [Google Scholar] [CrossRef]
- Torres, T.T.; Metta, M.; Ottenwalder, B.; Schlotterer, C. Gene expression profiling by massively parallel sequencing. Genome Res. 2008, 18, 172–177. [Google Scholar] [CrossRef]
- Zhang, J.A.; Liang, S.; Duan, J.L.; Wang, J.; Chen, S.L.; Cheng, Z.S.; Zhang, Q.; Liang, X.Q.; Li, Y.R. De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in peanut (Arachis hypogaea L.). BMC Genom. 2012, 13, 90. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.L.; Yang, H.; Wang, S.B.; Zhao, J.; Liu, C.; Gao, L.P.; Xia, E.H.; Lu, Y.; Tai, Y.L.; She, G.B.; et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. USA 2018, 115, E4151–E4158. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J.; Liang, Y.R. Clonal Tea Cultivars Records in China; Yang, Y.J., Liang, Y.R., Eds.; Shanghai Scientific and Technical Publishers: Shanghai, China, 2014; pp. 27–98. (In Chinese) [Google Scholar]
- Mur, L.A.J.; Kenton, P.; Atzorn, R.; Miersch, O.; Wasternack, C. The outcomes of concentration-specific interactions between salicylate and jasmonate signaling include synergy, antagonism, and oxidative stress leading to cell death. Plant Physiol. 2006, 140, 249–262. [Google Scholar] [CrossRef]
- Verberne, M.C.; Brouwer, N.; Delbianco, F.; Linthorst, H.J.M.; Bol, J.F.; Verpoorte, R. Method for the extraction of the volatile compound salicylic acid from tobacco leaf material. Phytochem. Anal. 2002, 13, 45–50. [Google Scholar] [CrossRef]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Rastogi, G.; Coaker, G.L.; Leveau, J.H.J. New insights into the structure and function of phyllosphere microbiota through high-throughput molecular approaches. FEMS Microbiol. Lett. 2013, 348, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Yuan, D.; Yin, P.; Wang, Z.H.; Guo, G.Y. Research progress on anthracnose of tea plant. Chin. J. Trop. Agric. 2016, 36, 20–26. (In Chinese) [Google Scholar] [CrossRef]
- Bolwell, G.P. Role of active oxygen species and NO in plant defence responses. Curr. Opin. Plant Biol. 1999, 2, 287–294. [Google Scholar] [CrossRef]
- Banerjee, A.K.; Mandal, A.; Chanda, D.; Chakraborti, S. Oxidant, antioxidant and physical exercise. Mol. Cell. Biochem. 2003, 253, 307–312. [Google Scholar] [CrossRef]
- Doke, N. Generation of superoxide anion by potato-tuber protoplasts during the hypersensitive response to hyphal wall components of phytophthora-infestans and specific-inhibition of the reaction by suppressors of hypersensitivity. Physiol. Plant Pathol. 1983, 23, 359–367. [Google Scholar] [CrossRef]
- Bolwell, P.P.; Page, A.; Pislewska, M.; Wojtaszek, P. Pathogenic infection and the oxidative defences in plant apoplast. Protoplasma 2001, 217, 20–32. [Google Scholar] [CrossRef]
- Lamb, C.; Dixon, R.A. The oxidative burst in plant disease resistance. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1997, 48, 251–275. [Google Scholar] [CrossRef]
- Grant, J.J.; Loake, G.J. Role of reactive oxygen intermediates and cognate redox signaling in disease resistance. Plant Physiol. 2000, 124, 21–29. [Google Scholar] [CrossRef]
- Ellinger, D.; Naumann, M.; Falter, C.; Zwikowics, C.; Jamrow, T.; Manisseri, C.; Somerville, S.C.; Voigt, C.A. Elevated early callose deposition results in complete penetration resistance to powdery mildew in Arabidopsis. Plant Physiol. 2013, 161, 1433–1444. [Google Scholar] [CrossRef]
- ThordalChristensen, H.; Zhang, Z.G.; Wei, Y.D.; Collinge, D.B. Subcellular localization of H2O2 in plants. H2O2 accumulation in papillae and hypersensitive response during the barley-powdery mildew interaction. Plant J. 1997, 11, 1187–1194. [Google Scholar] [CrossRef]
- Denness, L.; McKenna, J.F.; Segonzac, C.; Wormit, A.; Madhou, P.; Bennett, M.; Mansfield, J.; Zipfel, C.; Hamann, T. Cell wall damage-induced lignin biosynthesis is regulated by a reactive oxygen species- and jasmonic acid-dependent process in Arabidopsis. Plant Physiol. 2011, 156, 1364–1374. [Google Scholar] [CrossRef]
- Bellincampi, D.; Cervone, F.; Lionetti, V. Plant cell wall dynamics and wall-related susceptibility in plant-pathogen interactions. Front. Plant Sci. 2014, 5, 228. [Google Scholar] [CrossRef]
- Torres, M.A.; Dangl, J.L.; Jones, J.D.G. Arabidopsis gp91(phox) homologues AtrbohD and AtrbohF are required for accumulation of reactive oxygen intermediates in the plant defense response. Proc. Natl. Acad. Sci. USA 2002, 99, 517–522. [Google Scholar] [CrossRef]
- Marino, D.; Dunand, C.; Puppo, A.; Pauly, N. A burst of plant NADPH oxidases. Trends Plant Sci. 2012, 17, 9–15. [Google Scholar] [CrossRef]
- Kadota, Y.; Sklenar, J.; Derbyshire, P.; Stransfeld, L.; Asai, S.; Ntoukakis, V.; Jones, J.D.G.; Shirasu, K.; Menke, F.; Jones, A.; et al. Direct regulation of the NADPH oxidase RBOHD by the PRR-associated kinase BIK1 during plant immunity. Mol. Cell 2014, 54, 43–55. [Google Scholar] [CrossRef]
- Kobayashi, M.; Ohura, I.; Kawakita, K.; Yokota, N.; Fujiwara, M.; Shimamoto, K.; Doke, N.; Yoshioka, H. Calcium-dependent protein kinases regulate the production of reactive oxygen species by potato NADPH oxidase. Plant Cell 2007, 19, 1065–1080. [Google Scholar] [CrossRef]
- Kim, M.C.; Panstruga, R.; Elliott, C.; Muller, J.; Devoto, A.; Yoon, H.W.; Park, H.C.; Cho, M.J.; Schulze-Lefert, P. Calmodulin interacts with MLO protein to regulate defence against mildew in barley. Nature 2002, 416, 447–450. [Google Scholar] [CrossRef]
- Sun, D.Y.; Bian, Y.Q.; Zhao, B.H.; Zhao, L.Y.; Yu, X.M.; Shengjun, D. The effects of extracellular calmodulin on cell-wall regeneration of protoplasts and cell-division. Plant Cell Physiol. 1995, 36, 133–138. [Google Scholar]
- Lecourieux, D.; Raneva, R.; Pugin, A. Calcium in plant defence-signalling pathways. New Phytol. 2006, 171, 249–269. [Google Scholar] [CrossRef] [Green Version]
- Moffett, P. Mechanisms of recognition in dominant r gene mediated resistance. Adv. Virus Res. 2009, 75, 1–299. [Google Scholar] [CrossRef]
- Hubert, D.A.; Tornero, P.; Belkhadir, Y.; Krishna, P.; Takahashi, A.; Shirasu, K.; Dangl, J.L. Cytosolic HSP90 associates with and modulates the Arabidopsis RPM1 disease resistance protein. EMBO J. 2003, 22, 5679–5689. [Google Scholar] [CrossRef]
- Arofatullah, N.A.; Hasegawa, M.; Tanabata, S.; Ogiwara, I.; Sato, T. Heat shock-induced resistance against pseudomonas syringae pv. tomato (Okabe) Young et al. via Heat shock transcription factors in tomato. Agronomy 2019, 9, 2. [Google Scholar] [CrossRef]
- Glazebrook, J. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 2005, 43, 205–227. [Google Scholar] [CrossRef]
- Tsuda, K.; Mine, A.; Bethke, G.; Igarashi, D.; Botanga, C.J.; Tsuda, Y.; Glazebrook, J.; Sato, M.; Katagiri, F. Dual regulation of gene expression mediated by extended MAPK activation and salicylic acid contributes to robust innate immunity in Arabidopsis thaliana. PLoS Genet. 2013, 9, e1004015. [Google Scholar] [CrossRef]
- Abad, L.R.; DUrzo, M.P.; Liu, D.; Narasimhan, M.L.; Reuveni, M.; Zhu, J.K.; Niu, X.M.; Singh, N.K.; Hasegawa, P.M.; Bressan, R.A. Antifungal activity of tobacco osmotin has specificity and involves plasma membrane permeabilization. Plant Sci. 1996, 118, 11–23. [Google Scholar] [CrossRef]
- Song, J.T.; Lu, H.; McDowell, J.M.; Greenberg, J.T. A key role for ALD1 in activation of local and systemic defenses in Arabidopsis. Plant J. 2004, 40, 200–212. [Google Scholar] [CrossRef]
- Mou, Z.; Fan, W.H.; Dong, X.N. Inducers of plant systemic acquired resistance regulate NPR1 function through redox changes. Cell 2003, 113, 935–944. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, D.; Chu, J.Y.; Boyle, P.; Wang, Y.; Brindle, I.D.; De Luca, V.; Despres, C. The Arabidopsis NPR1 protein is a receptor for the plant defense hormone salicylic acid. Cell Rep. 2012, 1, 639–647. [Google Scholar] [CrossRef]
- Zhou, J.M.; Trifa, Y.; Silva, H.; Pontier, D.; Lam, E.; Shah, J.; Klessig, D.F. NPR1 differentially interacts with members of the TGA/OBF family of transcription factors that bind an element of the PR-1 gene required for induction by salicylic acid. Mol. Plant-Microbe Interact. 2000, 13, 191–202. [Google Scholar] [CrossRef]
- Klessig, D.F.; Durner, J.; Noad, R.; Navarre, D.A.; Wendehenne, D.; Kumar, D.; Zhou, J.M.; Shah, J.; Zhang, S.Q.; Kachroo, P.; et al. Nitric oxide and salicylic acid signaling in plant defense. Proc. Natl. Acad. Sci. USA 2000, 97, 8849–8855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Amornsiripanitch, N.; Dong, X.N. A genomic approach to identify regulatory nodes in the transcriptional network of systemic acquired resistance in plants. PLoS Path. 2006, 2, 1042–1050. [Google Scholar] [CrossRef]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.X.J. WRKY transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Thomma, B.P.H.J.; Eggermont, K.; Penninckx, I.A.M.A.; Mauch-Mani, B.; Vogelsang, R.; Cammue, B.P.A.; Broekaert, W.F. Separate jasmonate-dependent and salicylate-dependent defense-response pathways in Arabidopsis are essential for resistance to distinct microbial pathogens. Proc. Natl. Acad. Sci. USA 1998, 95, 15107–15111. [Google Scholar] [CrossRef]
- Hartmann, M.; Zeier, T.; Bernsdorff, F.; Reichel-Deland, V.; Kim, D.; Hohmann, M.; Scholten, N.; Schuck, S.; Brautigam, A.; Holzel, T.; et al. Flavin monooxygenase-generated n-hydroxypipecolic acid is a critical element of plant systemic immunity. Cell 2018, 173, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Pirrung, M.; McCue, L.A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.S.; Li, X.M.; Qiao, R.Y.; Shen, E.H.; Lin, X.M.; Lu, J.L.; Ye, J.H.; Liang, Y.R.; Zheng, X.Q. De novo transcriptome assembly of fluorine accumulator tea plant Camellia sinensis with fluoride treatments. Sci. Data 2018, 5, 180194. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST plus: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007, 35, D61–D65. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, S.; Forslund, K.; Szklarczyk, D.; Trachana, K.; Roth, A.; Huerta-Cepas, J.; Gabaldon, T.; Rattei, T.; Creevey, C.; Kuhn, M.; et al. eggNOG v4.0: Nested orthology inference across 3686 organisms. Nucleic Acids Res. 2014, 42, D231–D239. [Google Scholar] [CrossRef] [PubMed]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.S.; Lin, X.M.; Qiao, R.Y.; Zheng, X.Q.; Lu, J.L.; Ye, J.H.; Liang, Y.R. Effect of fluoride treatment on gene expression in tea plant (Camellia sinensis). Sci. Rep. 2017, 7, 9847. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Zawoznik, M.S.; Groppa, M.D.; Tomaro, M.L.; Benavides, M.P. Endogenous salicylic acid potentiates cadmium-induced oxidative stress in Arabidopsis thaliana. Plant Sci. 2007, 173, 190–197. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | LJ43_H | LJ43_D | ZN139_H | ZN139_D | All Samples |
|---|---|---|---|---|---|
| Raw Reads Number (million) | 38.86 ± 1.19 | 42.94 ± 0.03 | 40.06 ± 3.52 | 37.40 ± 2.27 | 318.52 |
| Total Bases (billion bp) | 5.83 ± 0.18 | 6.44 ± 0.00 | 6.01 ± 0.53 | 5.61 ± 0.34 | 47.78 |
| N (%) | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | |
| Q20 (%) | 95.29 ± 0.38 | 95.43 ± 0.13 | 95.06 ± 0.24 | 95.32 ± 0.32 | |
| Q30 (%) | 89.38 ± 0.69 | 89.67 ± 0.27 | 88.89 ± 0.40 | 89.47 ± 0.57 | |
| GC (%) | 48.72 ± 0.01 | 49.07 ± 0.13 | 48.79 ± 0.30 | 48.41 ± 0.72 | |
| Clean Reads (%) | 98.04 ± 0.28 | 98.11 ± 0.05 | 97.99 ± 0.17 | 97.99 ± 0.25 |
| Cultivar | LJ43 | ZN139 | |
|---|---|---|---|
| Contig | Total Length (bp) | 102,521,451 | 107,758,129 |
| Sequence Number | 335,186 | 352,038 | |
| Mean Length (bp) | 306 | 306 | |
| N50 (bp) | 404 | 405 | |
| N50 Sequence No. | 54,939 | 58,050 | |
| N90 (bp) | 150 | 150 | |
| N90 Sequence No. | 251,498 | 264,172 | |
| Unigene | Total Length (bp) | 65,163,329 | 68,536,138 |
| Sequence Number | 109,316 | 115,953 | |
| Mean Length (bp) | 596 | 591 | |
| N50 (bp) | 821 | 803 | |
| N50 Sequence No. | 20,020 | 21,541 | |
| N90 (bp) | 261 | 260 | |
| N90 Sequence No. | 81,051 | 86,082 | |
| Annotated Unigene Number in Database | Nr | 45,230 (41.38%) | 46,383 (40%) |
| GO | 24,892 (22.77%) | 25,168 (21.71%) | |
| KEGG | 5799 (5.3%) | 5873 (5.06%) | |
| eggNOG | 43,025 (39.36%) | 44,067 (38%) | |
| Swiss-Prot | 33,895 (31.01%) | 34,860 (30.06%) | |
| In all database | 4605 (4.21%) | 4677 (4.03%) | |
| Control | Case | Up-Regulated DEGs | Down-Regulated DEGs | Total DEGs |
|---|---|---|---|---|
| LJ43_H | LJ43_D | 1082 | 539 | 1621 |
| ZN139_H | ZN139_D | 1527 | 1562 | 3089 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Y.-L.; Sheng, Y.-Y.; Cai, Z.-Y.; Yang, R.; Li, Q.-S.; Li, X.-M.; Li, D.; Guo, X.-Y.; Lu, J.-L.; Ye, J.-H.; et al. Involvement of Salicylic Acid in Anthracnose Infection in Tea Plants Revealed by Transcriptome Profiling. Int. J. Mol. Sci. 2019, 20, 2439. https://doi.org/10.3390/ijms20102439
Shi Y-L, Sheng Y-Y, Cai Z-Y, Yang R, Li Q-S, Li X-M, Li D, Guo X-Y, Lu J-L, Ye J-H, et al. Involvement of Salicylic Acid in Anthracnose Infection in Tea Plants Revealed by Transcriptome Profiling. International Journal of Molecular Sciences. 2019; 20(10):2439. https://doi.org/10.3390/ijms20102439
Chicago/Turabian StyleShi, Yun-Long, Yue-Yue Sheng, Zhuo-Yu Cai, Rui Yang, Qing-Sheng Li, Xu-Min Li, Da Li, Xiao-Yuan Guo, Jian-Liang Lu, Jian-Hui Ye, and et al. 2019. "Involvement of Salicylic Acid in Anthracnose Infection in Tea Plants Revealed by Transcriptome Profiling" International Journal of Molecular Sciences 20, no. 10: 2439. https://doi.org/10.3390/ijms20102439
APA StyleShi, Y. -L., Sheng, Y. -Y., Cai, Z. -Y., Yang, R., Li, Q. -S., Li, X. -M., Li, D., Guo, X. -Y., Lu, J. -L., Ye, J. -H., Wang, K. -R., Zhang, L. -J., Liang, Y. -R., & Zheng, X. -Q. (2019). Involvement of Salicylic Acid in Anthracnose Infection in Tea Plants Revealed by Transcriptome Profiling. International Journal of Molecular Sciences, 20(10), 2439. https://doi.org/10.3390/ijms20102439