Transcriptome and Proteome Profiling of Different Colored Rice Reveals Physiological Dynamics Involved in the Flavonoid Pathway


 ,
,  ,
, 
Abstract
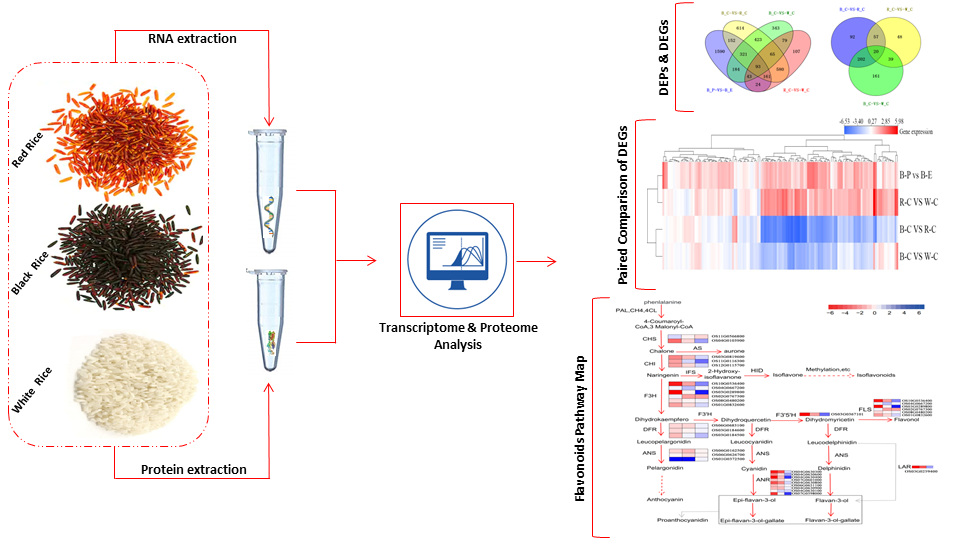
:
1. Introduction
2. Results
2.1. Overview of the Transcriptome and Proteome
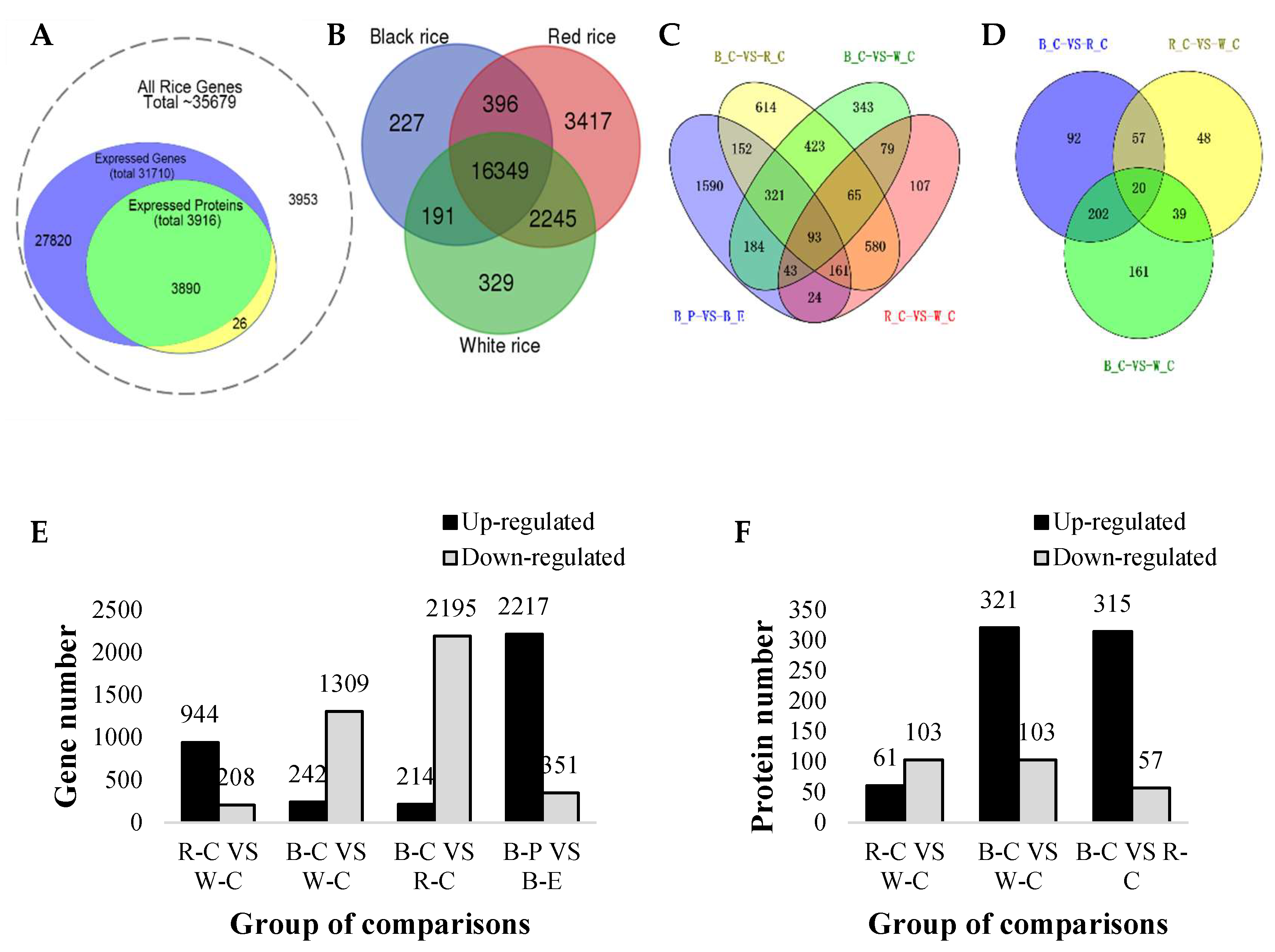
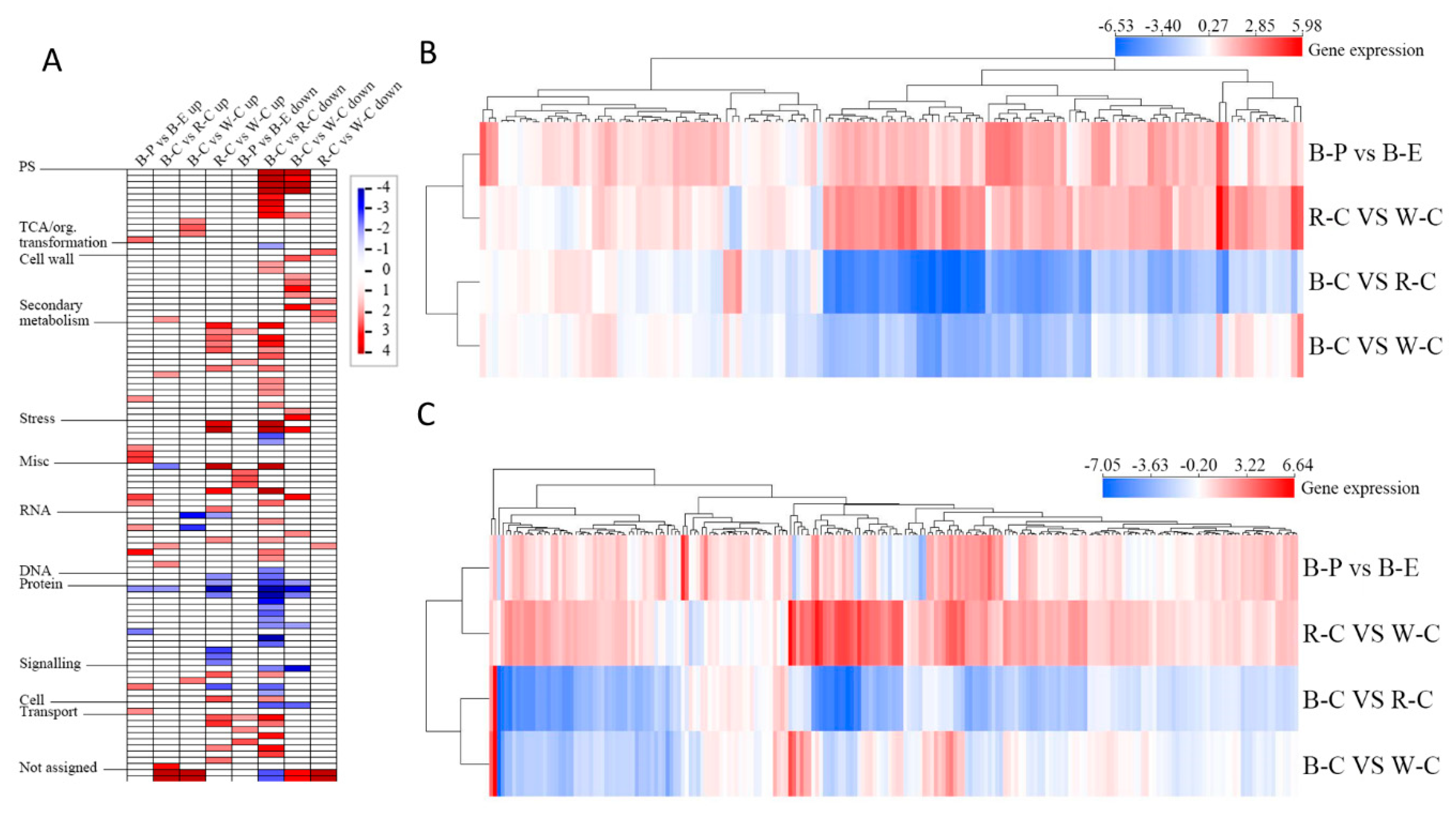
2.2. Differentially Expressed Genes (DEGs) and Differentially Expressed Proteins (DEPs)
2.3. Transcript Profiling of the Pericarp and Endosperm of Black Rice
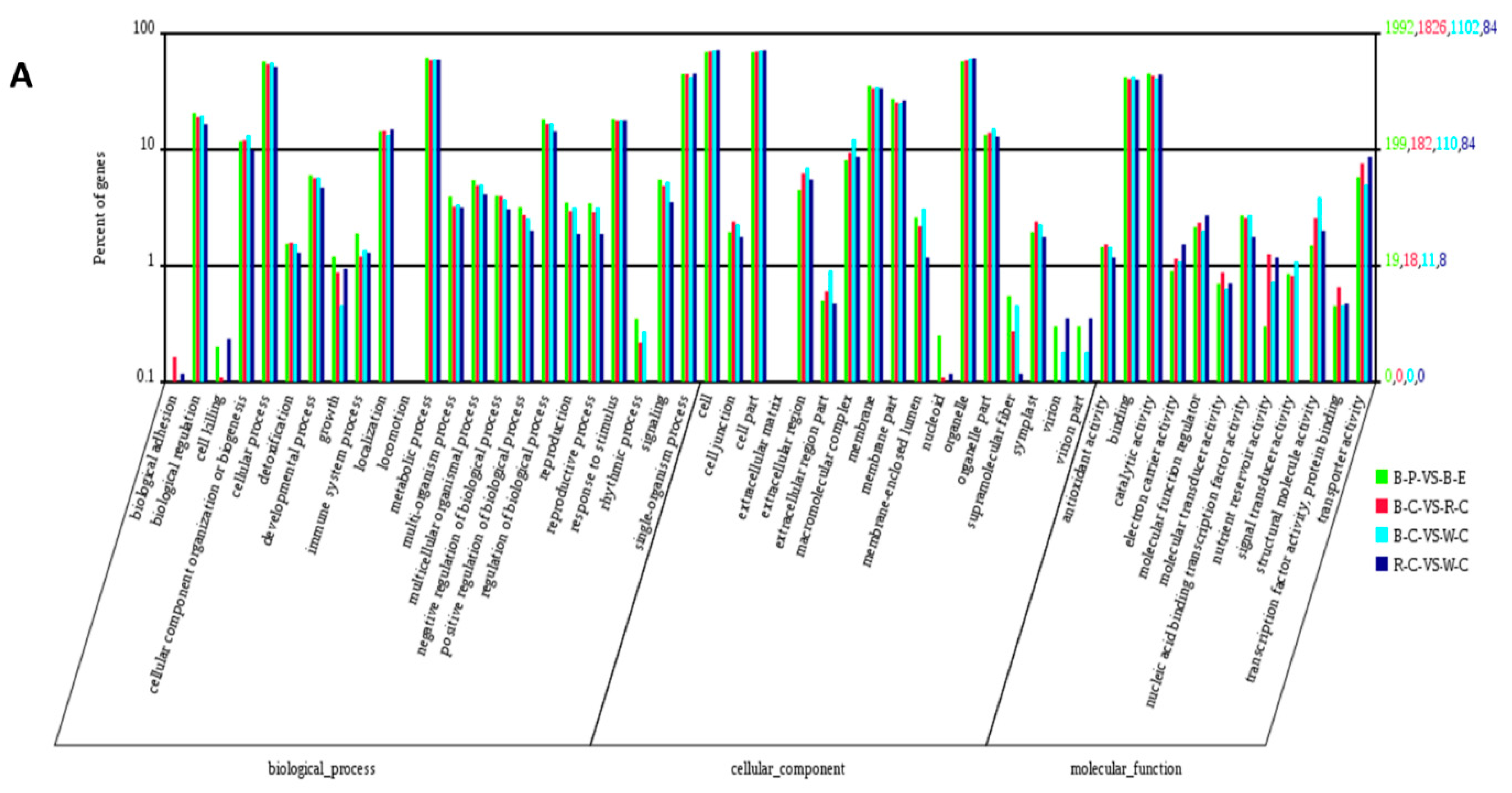
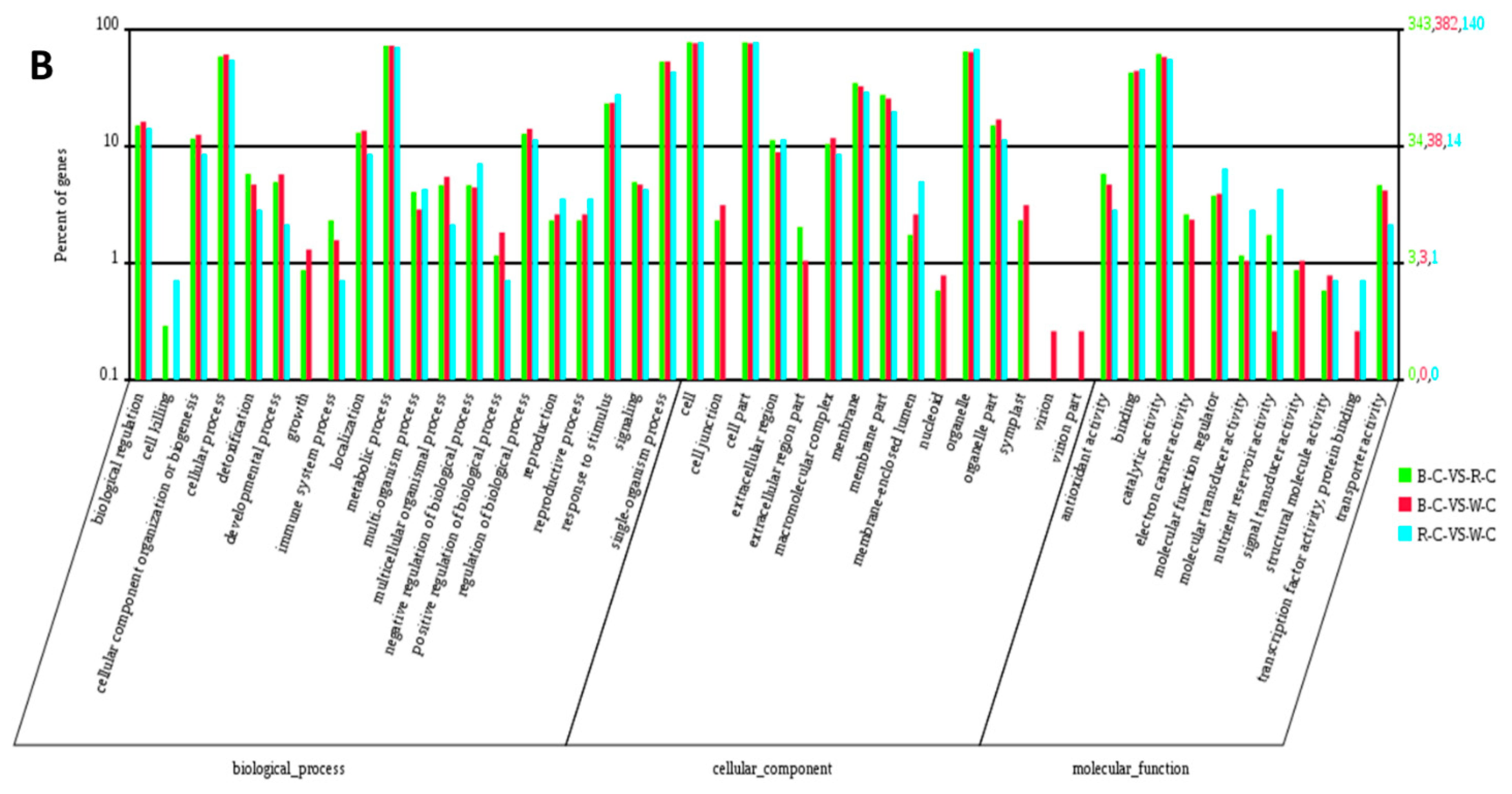
2.4. Gene Ontology (GO) Analysis of DEGs and DEPs in the Different Rice Cultivars
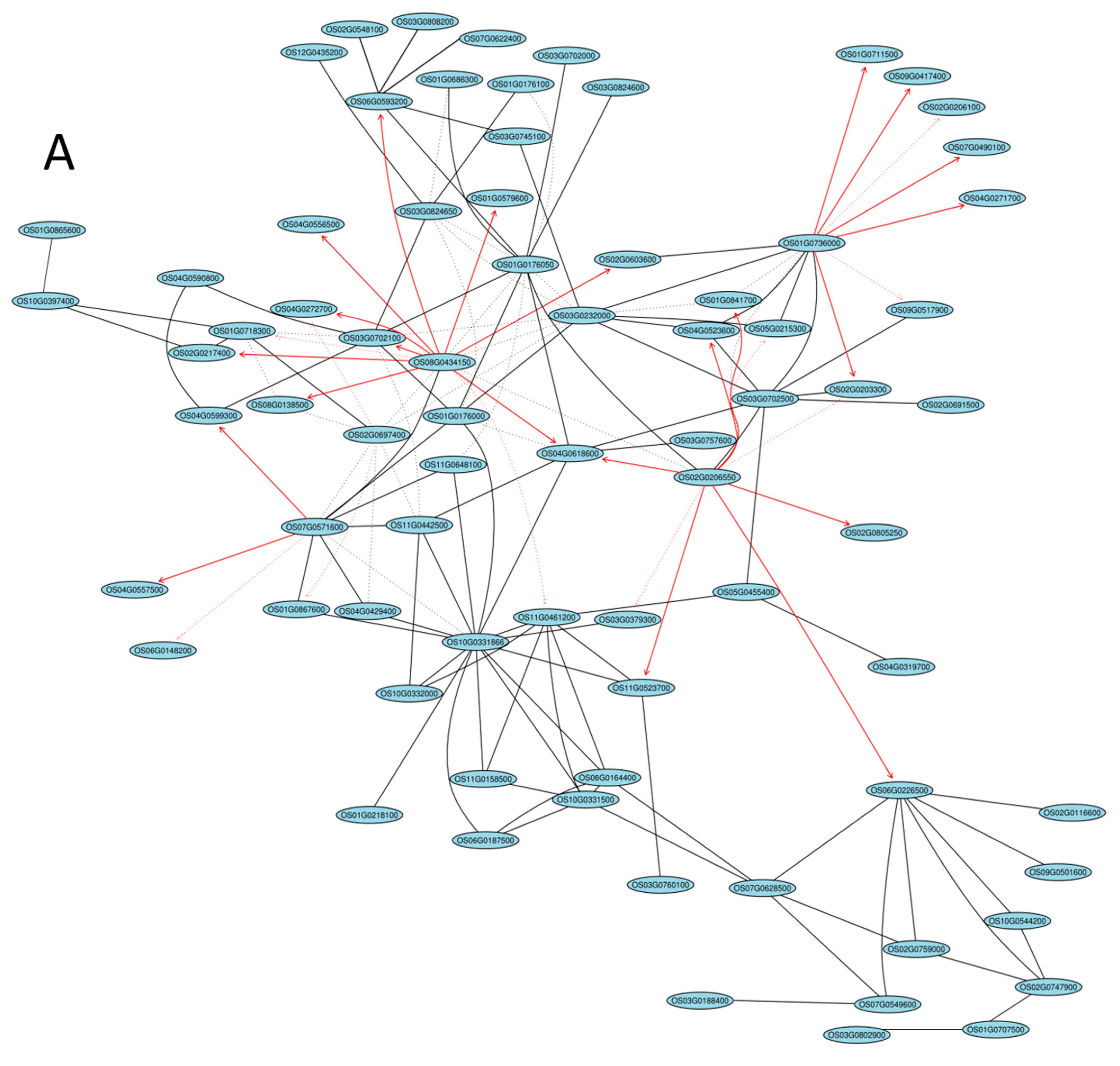
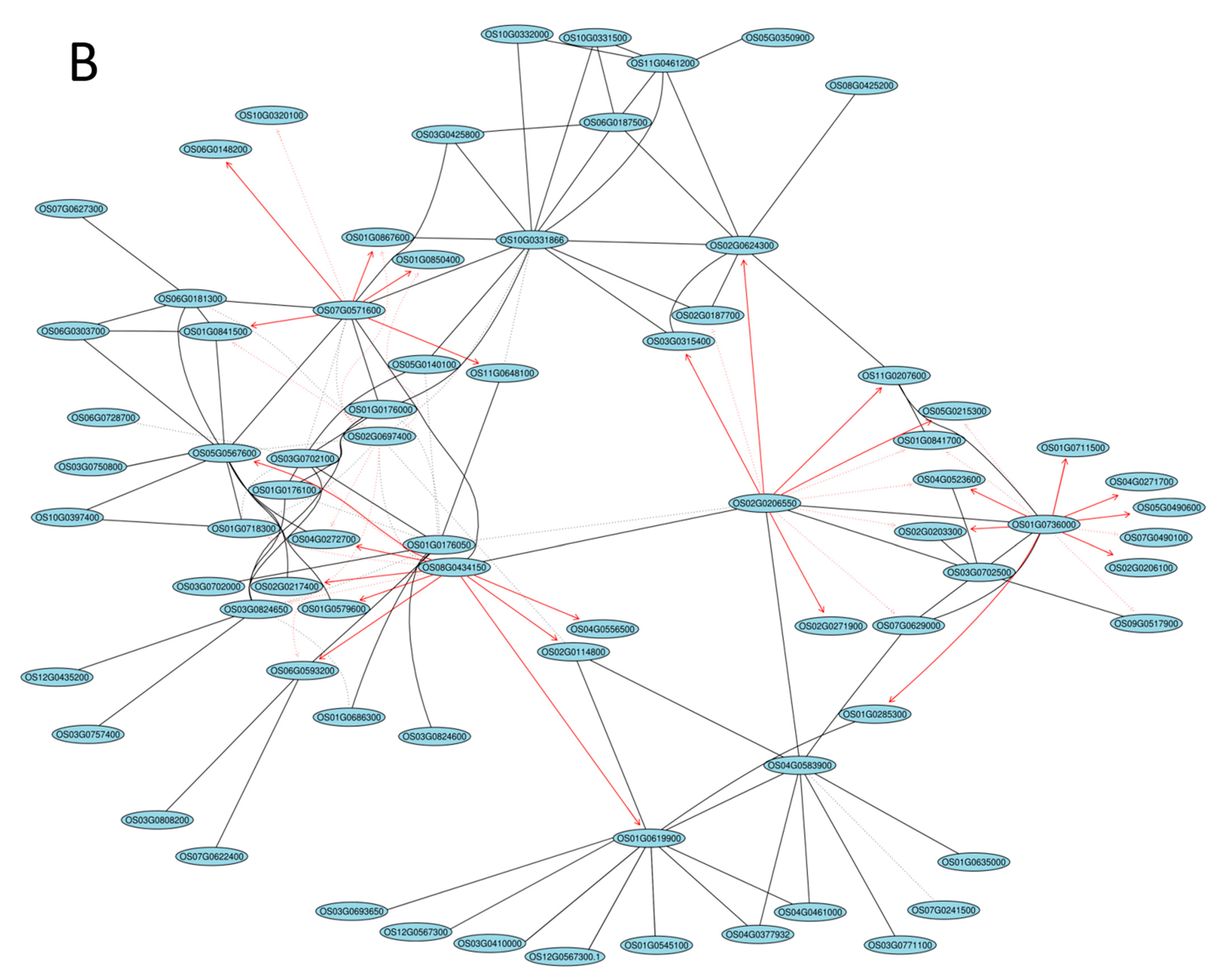
2.5. Gene Coexpression Network Analysis
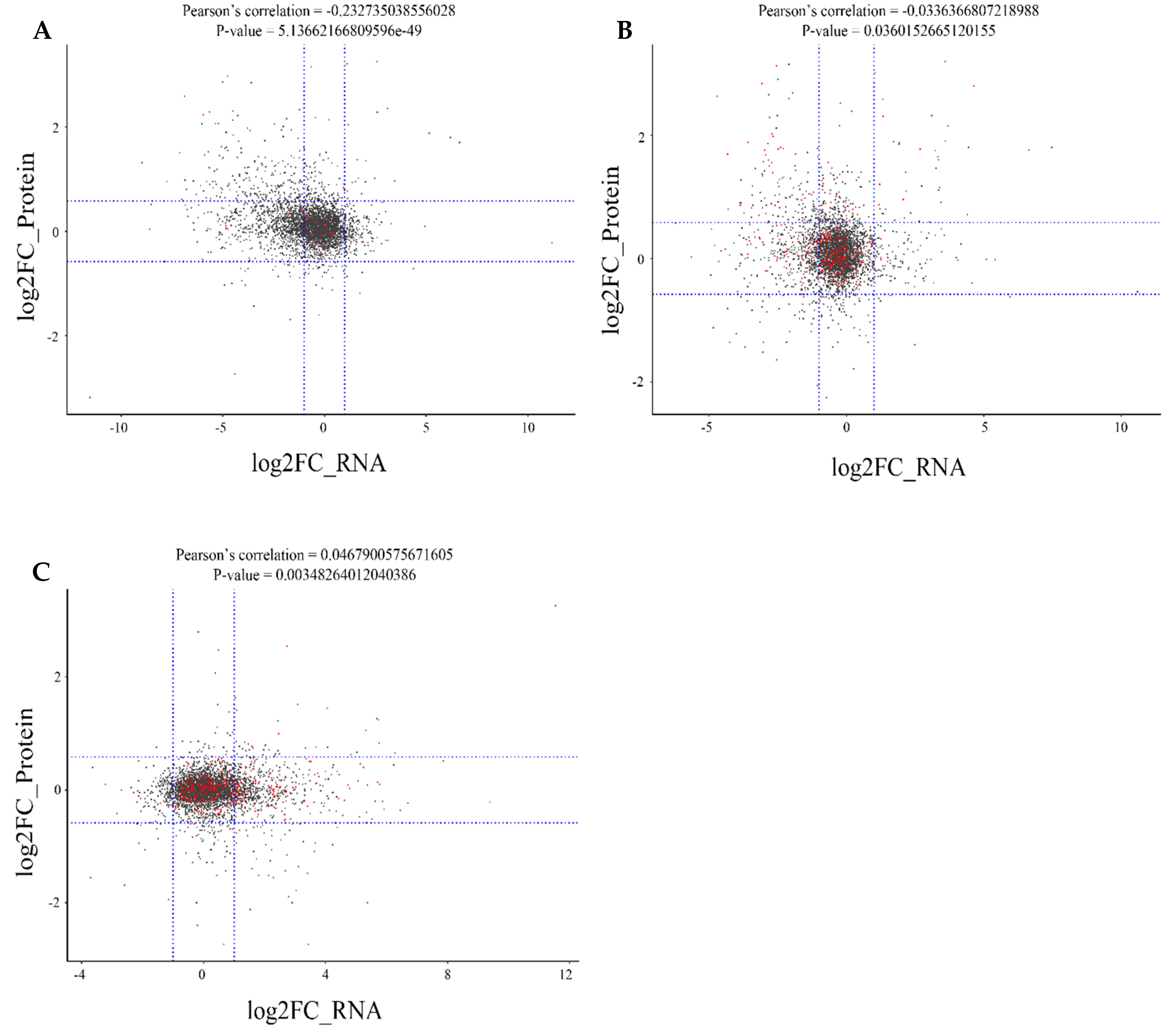
2.6. Correlation Analysis between the Proteome and Transcriptome of Different Rice Cultivars
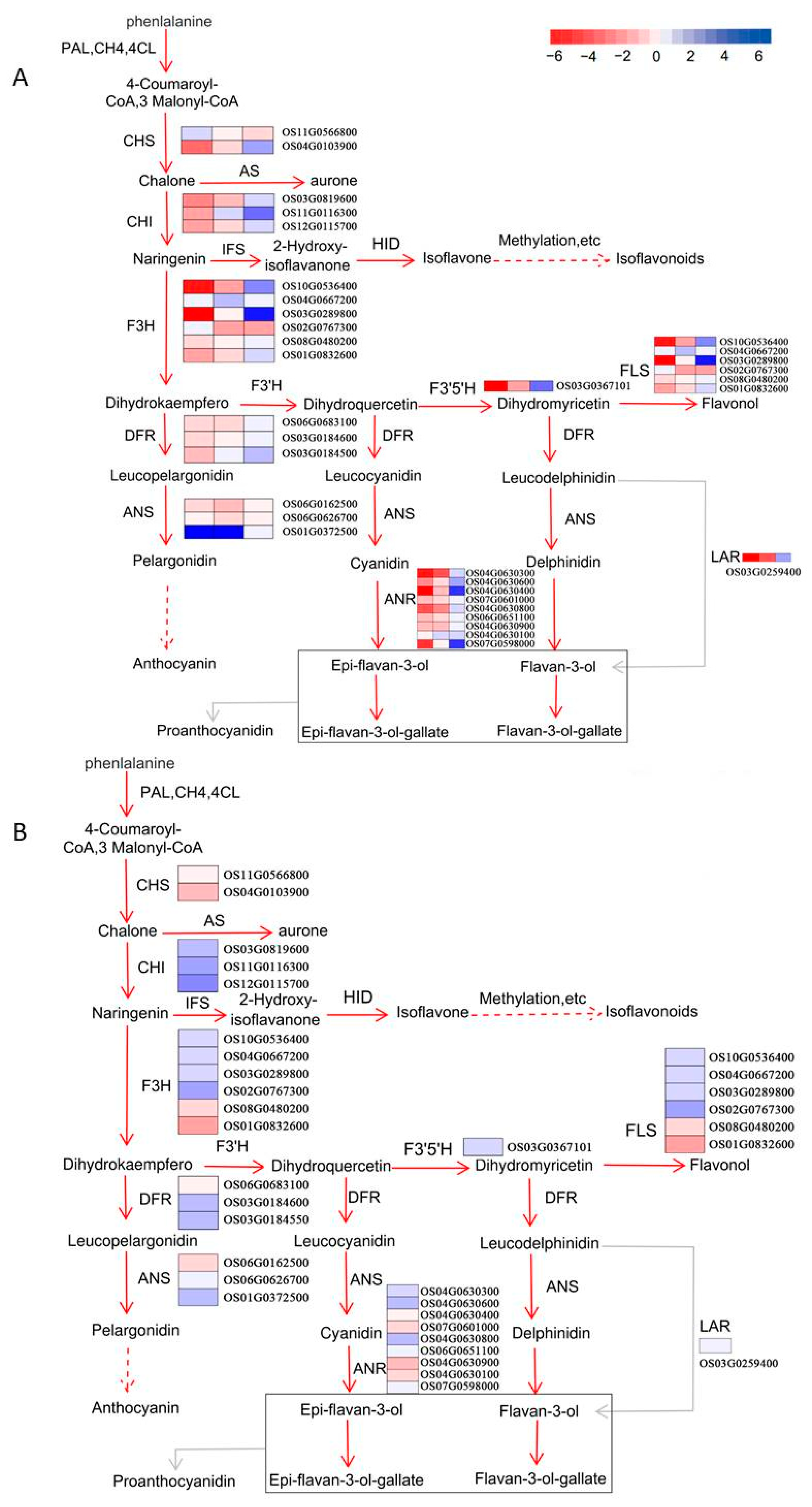
2.7. Evaluation of Data Related to the Flavonoid Biosynthesis Pathway
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. RNA Isolation and Library Preparation for Transcriptome Analysis
4.3. Transcriptome Analysis
4.4. Protein Preparation
4.5. iTRAQ Labeling, Strong Sation Exchnge Chromatography (SCX) Fractionation, and Liquid Chromatography-Electrospray Ionization-Tandem Mass Spectrometry (LC-ESI-MS/MS) Analysis
4.6. Proteome Data Analysis
4.7. Gene Coexpression Network Analysis
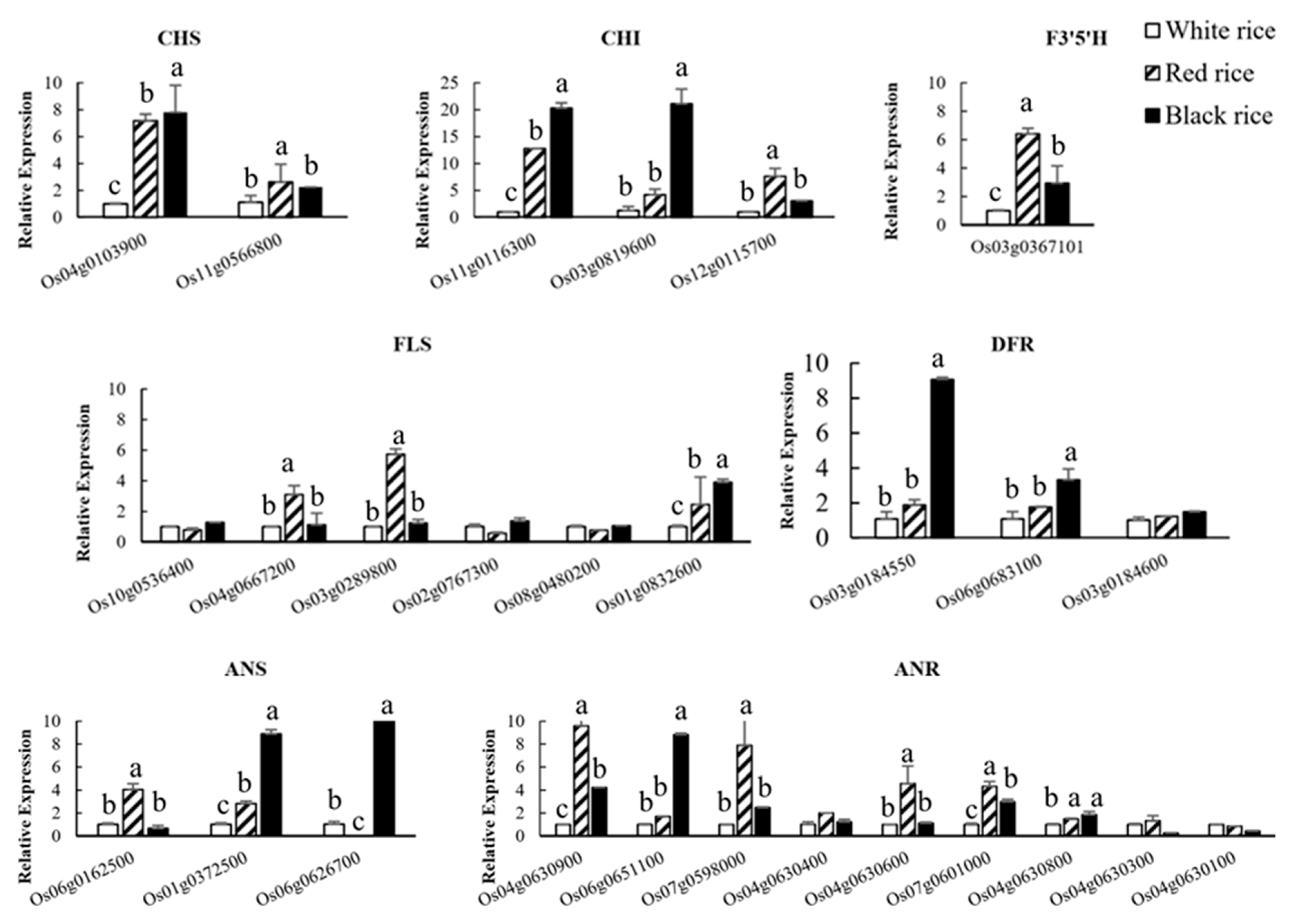
4.8. qRT-PCR Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sasaki, T.; Burr, B. International Rice Genome Sequencing Project: The effort to completely sequence the rice genome. Curr. Opin. Plant Biol. 2000, 3, 138–142. [Google Scholar] [CrossRef]
- Acquaviva, R.; Russo, A.; Gahlvano, F.; Galvano, G.; Barcellonam, L.; Li Volti, G.; Vanella, A. Cyanidin and cyanidin 3-O-beta-d-glucoside as DNA cleavage protectors and antioxidants. Cell Biol Toxicol. 2003, 19, 243–252. [Google Scholar] [CrossRef]
- Russo, A.; La Fauci, L.; Acquaviva, R.; Campisi, A.; Raciti, G.; Scifo, C.; Renis, M.; Galvano, G.; Vanella, A.; Galvano, F. Ochratoxin A-induced DNA damage in human fibroblast: Protective effect of cyanidin 3-O-beta-d-glucoside. J. Nutr. Biochem. 2005, 16, 31–37. [Google Scholar] [CrossRef]
- Chen, X.Q.; Nagao, N.; Itani, T.; Irifune, K. Anti-oxidative analysis, and identification and quantification of anthocyanin pigments in different colored rice. Food Chem. 2012, 135, 2783–2788. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.L.; Xu, Y.; Zhang, Y.Y.; Lu, Y.H. Black rice and anthocyanins induce inhibition of cholesterol absorption in vitro. Food Funct. 2013, 4, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Tahara, S.; Ibrahim, R.K. Prenylated isoflavonoids: An update. Phytochemistry 1995, 38, 1073–1094. [Google Scholar] [CrossRef]
- Abdel-Aal, E.S.M.; Young, J.C.; Rabalski, I. Anthocyanin composition in black, blue, pink, purple, and red cereal grains. J. Agric. Food Chem. 2006, 54, 4696–4704. [Google Scholar] [CrossRef] [PubMed]
- Nakornriab, M.; Sriseadka, T.; Wongpornchai, S. Quantification of carotenoid and flavonoid components in brans of some Thai black rice cultivars using supercritical fluid extraction and high performance liquid chromatography-mass spectrometry. J. Food Lipids 2008, 15, 488–503. [Google Scholar] [CrossRef]
- Kim, J.K.; Lee, S.Y.; Chu, S.M.; Lim, S.H.; Suh, S.C.; Lee, Y.T.; Cho, H.S.; Ha, S.H. Variation and correlation analysis of flavonoids and carotenoids in Korean pigmented rice (Oryza sativa L.) cultivars. J. Agric. Food Chem. 2010, 58, 12804–12809. [Google Scholar] [CrossRef]
- Pereira-Caro, G.; Watanabe, S.; Crozier, A.; Fujimura, T.; Yokota, T.; Ashihara, H. Phytochemical profile of a Japanese black-purple rice. Food Chem. 2013, 141, 2821–2827. [Google Scholar] [CrossRef] [PubMed]
- Bordiga, M.; Gomez-Alonso, S.; Locatelli, M.; Travaglia, F.; Coisson, J.D.; Hermosin--Gutierrez, I.; Arlorio, M. Phenolics characterization and antioxidant activity of six different pigmented Oryza sativa L. cultivars grown in Piedmont (Italy). Food Res. Int. 2014, 65, 282–290. [Google Scholar] [CrossRef]
- Grotewold, E. Plant metabolic diversity: A regulatory perspective. Trends Plant Sci. 2005, 10, 57–62. [Google Scholar] [CrossRef]
- Petroni, K.; Tonelli, C. Recent advances on the regulation of anthocyanin synthesis in reproductive organs. Plant Sci. 2011, 181, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.A.; Jakoby, M.; Werber, M.; Martin, C.; Weisshaar, B.; Bailey, P.C. The basic helix-loop-helix transcription factor family in plants: A genome-wide study of protein structure and functional diversity. Mole. Biol. Evol. 2003, 20, 735–747. [Google Scholar] [CrossRef]
- Toledo-Ortiz, G.; Huq, E.; Quail, P.H. The Arabidopsis basic/helix-loop-helix transcription factor family. Plant Cell 2003, 15, 1749–1770. [Google Scholar] [CrossRef]
- Li, X.; Duan, X.; Jiang, H.; Sun, Y.; Tang, Y.; Yuan, Z.; Guo, J.; Liang, W.; Chen, L.; Yin, J.; et al. Genome-wide analysis of basic/helix-loop-helix transcription factor family in rice and Arabidopsis. Plant Physiol. 2006, 141, 1167–1184. [Google Scholar] [CrossRef]
- Baier, M.; Hemmann, G.; Holman, R.; Corke, F.; Card, R.; Smith, C.; Rook, F.; Bevan, M. Characterization of mutants in Arabidopsis showing increased sugar-specific gene expression, growth, and developmental responses. Plant Physiol. 2004, 134, 81–91. [Google Scholar] [CrossRef]
- Tsukaya, H.; Ohshima, T.; Naito, S.; Chino, M.; Komeda, Y. Sugar-dependent expression of the CHS-A gene for chalcone synthase from petunia in transgenic Arabidopsis. Plant Physiol. 1991, 97, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Itani, T.; Wu, X.J.; Chikawa, Y.; Irifune, K. Physiological factors affecting transcription of genes involved in the flavonoid biosynthetic pathway in different rice cultivars. Plant Signal. Behav. 2013, 8, e27555. [Google Scholar] [CrossRef]
- Akashi, T.; Fukuchi-Mizutani, M.; Aoki, T.; Uyema, Y.; Yonekura-Sakakibara, K.; Tanaka, Y.; Kusumi, T.; Ayabe, S. Molecular cloning and biochemical characterization of a novel cytochrome P450, flavone synthase II, that catalyzes direct conversion of flavanones to flavones. Plant Cell Physiol. 1999, 40, 182–1186. [Google Scholar] [CrossRef]
- Lan, P.; Li, W.; Schmidt, W. Complementary proteome and transcriptome profiling in phosphate-deficient Arabidopsis roots reveals multiple levels of gene regulation. Mol. Cell Proteomics 2012, 11, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.L.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattant, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell Proteomics 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Risueno, M.A.; Van Norman, J.M.; Moreno, A.; Zhang, J.; Ahnert, S.E.; Benfey, P.N. Oscillating gene expression determines competence for periodic Arabidopsis root branching. Science 2010, 329, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.; Pakzad, M.; Taei, A.; Brink, T.C.; Pirhaji, L.; Ruiz, G. Comparative proteome and transcriptome analysis of embryonic stem cells during embryoid body-based differentiation. Proteomics 2009, 9, 4859–4870. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Gharib, T.G.; Huang, C.C.; Taylor, J.M.G.; Misek., D.E.; Kardia., S.L.R.; Giordano, T.J.; Iannettoni, M.D.; Orringer, M.B.; Hanash, S.M.; et al. Discordant protein and mRNA expression in lung adenocarcinomas. Mol. Cell Proteomics 2002, 1, 304–313. [Google Scholar] [CrossRef]
- Lan, P.; Li, W.F.; Wen, T.N.; Shiau, J.Y.; Wu, Y.C.; Lin, W.D.; Schmidt, W. iTRAQ protein profile analysis of Arabidopsis roots reveals new aspects critical for iron homeostasis. Plant Physiol. 2011, 155, 821–834. [Google Scholar] [CrossRef]
- Galland, M.; Huguet, R.; Arc, E.; Cueff, G.; Job, D.; Rajjou, L. Dynamic proteomics emphasizes the importance of selective mRNA translation and protein turnover during arabidopsis seed germination. Mol. Cell Proteomics 2014, 13, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Nakaminami, K.; Matsui, A.; Nakagami, H.; Minami, A.; Nomura, Y.; Tanaka, M.; Morosawa, T.; Ishida, J.; Takahashi, S.; Uemura, M.; et al. Analysis of different expression patterns of mRNA and protein during cold-acclimation and de-acclimation in Arabidopsis. Mol. Cell Proteomics 2014, 10, 3602–3611. [Google Scholar] [CrossRef]
- Lo, S.C.; Nicholson, R.L. Reduction of light-induced anthocyanin accumulation in inoculated sorghum mesocotyls. Implications for a compensatory role in the defense response. Plant Physiol. 1998, 116, 979–989. [Google Scholar] [CrossRef]
- Schulze-Lefert, P.; Dangl, J.L.; Becker-Andre, M.; Hahlbrock, K.; Schulz, W. Inducible in vivo DNA footprints define sequences necessary for UV light activation of the parsley chalcone synthase gene. EMBO J. 1989, 8, 651–656. [Google Scholar] [CrossRef]
- Christie, J.M.; Jenkins, G.I. Distinct UV-B and UV-A/blue light signal transduction pathways induce chalcone synthase gene expression in Arabidopsis cells. Plant Cell 1996, 8, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Rolland, F.; Baena-Gonzalez, E.; Sheen, J. Sugar sensing and signalling in plants: Conserved and novel mechanisms. Annu. Rev. Plant Biol. 2006, 57, 675–709. [Google Scholar] [CrossRef] [PubMed]
- Solfaneli, C.; Poggi, A.; Loreti, E.; Alpi, A.; Perata, P. Sucrose-specific induction of the anthocyanin biosynthetic pathway in Arabidopsis. J. Plant Physiol. 2006, 140, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Fini, A.; Brunetti, C.; di Ferdinando, M.; Ferrini, F.; Tattini, M. Stress-induced flavonoid biosynthesis and the antioxidant machinery of plants. Plant Signal. Behav. 2011, 6, 709–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treutter, D. Significance of flavonoids in plant resistance and enhancement of their biosynthesis. Plant Physiol. 2005, 7, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Tattini, M.; Galardi, C.; Pinelli, P.; Massai, R.; Remorini, D.; Agati, G. Differential accumulation of flavonoids and hydroxycinnamates in leaves of Ligustrum vulgare under excess light and drought stress. New Phytol. 2004, 163, 547–561. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Subramanian, S.; Zhang, Y.; Yu, O. Flavone synthase from Medicago truncatula are flavanone-2-hydroxylases and are important for nodulation. Plant Physiol. 2007, 144, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Lam, P.Y.; Zhu, F.Y.; Chan, W.L.; Liu, H.J.; Lo, C. Cytochrome P450 93G1 Is a Flavone Synthase II That Channels Flavanones to the Biosynthesis of Tricin O-Linked Conjugates in Rice. Plant Physiol. 2014, 165, 1315–1327. [Google Scholar] [CrossRef]
- Koskimäki, J.J.; Hokkanen, J.; Jaakola, L.; Suorsa, M.; Tolonen, A.; Mattila, S.; Pirttilä, A.M.; Hohtola, A. Flavonoid biosynthesis and degradation play a role in early defence responses of bilberry (Vaccinium myrtillus) against biotic stress. Eur. J. Plant Pathol. 2009, 125, 629–640. [Google Scholar] [CrossRef]
- Falcone-Ferreyra, M.L.; Rius, S.P.; Casati, P. Flavonoids: Biosynthesis, biological functions, and biotechnological applications. Front Plant Sci. 2012, 3, 222. [Google Scholar] [CrossRef]
- Sweeney, M.T.; Thomson, M.J.; Pfeil, B.E.; McCouch, S. Caught red-handed: Rc encodes a basic helix-loop-helix protein conditioning red pericarp in rice. Plant Cell 2006, 18, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Selinger, D.A.; Chandler, V.L. A mutation in the pale aleurone color1 gene identifies a novel regulator of the maize anthocyanin pathway. Plant Cell 1999, 11, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wessler, S.R. Inefficient re-initiation is responsible for upstream open reading frame-mediated translation repression of the maize R gene. Plant Cell 1998, 10, 1733–1745. [Google Scholar] [PubMed]
- Park, K.I.; Ishikawa, N.; Morita, Y.; Choi, J.D.; Hoshino, A.; Lida, S. A bHLH regulatory gene in the common morning glory, Ipomoea purpurea, controls anthocyanin biosynthesis in flowers, proanthocyanidin and phytomelanin pigmentation in seeds, and seed trichome formation. Plant J. 2007, 49, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Druka, A.; Kudrna, D.; Rostoks, N.; Brueggeman, R.; von Wettstein, D.; Kleinhofs, A. Chalcone isomerase gene from rice (Oryza sativa) and barley (Hordeum vulgare): Physical, genetic and mutation mapping. Gene 2003, 302, 171–178. [Google Scholar] [CrossRef]
- Reddy, A.R.; Scheffler, B.; Madhuri, G.; Srivastava, M.N.; Kumar, A.; Sathyanarayanan, P.V.; Nair, S.; Mohan, M. Chalcone synthase in rice (Oryza sativa L.): Detection of the CHS protein in seedlings and molecular mapping of the chs locus. Plant Mol. Biol. 1996, 32, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Maekawa, M.; Oki, T.; Suda, I.; Iida, S.; Shimada, H.; Takamure, I.; Kadowaki, K. The Rc and Rd genes are involved in proanthocyanidin synthesis in rice pericarp. Plant J. 2007, 49, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.M.; Reddy, V.S.; Scheffler, B.E.; Wienand, U.; Reddy, A.R. Novel transgenic rice overexpressing anthocyanidin synthase accumulates a mixture of flavonoids leading to an increased antioxidant potential. Metab. Eng. 2007, 9, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Chiu, L.W.; Zhou, X.J.; Burke, S.; Wu, X.L.; Prior, R.L.; Li, L. The purple cauliflower arises from activation of a MYB transcription factor. Plant Physiol. 2010, 154, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Spelt, C.; Quattrocchio, F.; Mol, J.N.M.; Koes, R. Anthocyanin1 of petunia encodes a basic helix-loop-helix protein that directly activates transcription of structural anthocyanin genes. Plant Cell 2000, 12, 1619–1632. [Google Scholar] [CrossRef] [PubMed]
- Nagao, S.; Takahashi, M.; Kinoshita, T. Genetical studies on rice plant, XXVI: Mode of inheritance and causal genes for one type of anthocyanin color character in foreign rice cultivars. J. Fac. Agric. Hokkaido Univ. 1962, 52, 20–50. [Google Scholar]
- Takahashi, M.E. Gene analysis and its related problems: Genetical studies on rice plant, LXXX. J. Fac. Agric. Hokkaido Univ. 1982, 61, 91–142. [Google Scholar]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, S.; Furio-Tari, P.; Turra, D.; Pietro, A.D.; Nueda, M.J.; Ferrer, A.; Conesa, A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015, 43, e140. [Google Scholar] [CrossRef]
- Usadel, B.; Nagel, A.; Steinhauser, D.; Gibon, Y.; Bläsing, O.E.; Redestig, H.; Sreenivasulu, N.; Krall, L.; Hannah, M.A.; Poree, F.; et al. PageMan: An interactive ontology tool to generate, display, and annotate overview graphs for profiling experiments. BMC Bioinformatics. 2006, 7, 535. [Google Scholar] [CrossRef]
- Glen, A.; Evans, C.A.; Gan, C.S.; Cross, S.S.; Hamdy, F.C.; Gibbins, J.; Lippitt, J.; Eaton, C.L.; Noirel, J.; Wright, P.; et al. Eight-Plex iTRAQ Analysis of Variant Metastatic Human Prostate Cancer Cells Identifies Candidate Biomarkers of Progression: An Exploratory Study. Prostate 2010, 70, 1313–1332. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D.; Bass, A.J.; Dabney, A.; Robinson, D. qvalue: Q-value Estimation for False Discovery Rate Control. R Package Version 2.0.0. Available online: github.com/jdstorey/qvalue (accessed on 14 April 2017).
- Schaefer, J.; Opgen-Rhein, R.; Strimmer, K. Reverse engineering genetic networks using the genenet package. R News 2006, 6, 50–53. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | log2FC(B-C/R-C) | log2FC(B-C/W-C) | log2FC(R-C/W-C) | Description |
|---|---|---|---|---|
| Os11G0530650 | −5.840043541 | - | 5.103077947 | - |
| Os11G0599200 | −5.734374274 | - | 3.273644684 | UDP-glycosyltransferase 72B3 |
| Os02G0188000 | −4.831826158 | - | - | cinnamate beta-D-glucosyltransferase |
| Os01G0179600 | −5.138751765 | - | 2.797789001 | crocetin glucosyltransferase, chloroplastic |
| Os03G0367150 | −5.252202169 | −2.676197753 | 2.576004415 | - |
| Os01G0850900 | - | - | 2.930368899 | heme-binding-like protein At3g10130, chloroplastic |
| Os08G0434100 | −5.040311748 | −3.361280923 | - | extracellular ribonuclease LE |
| Os10G0320100 | - | 2.681785408 | 3.706832427 | flavonoid 3′-monooxygenase |
| Os06G0608700 | −4.855820887 | −3.156281607 | - | fructose-bisphosphate aldolase, cytoplasmic isozyme 1 |
| Os01G0638000 | −5.070762648 | - | - | anthocyanin 3′-O-beta-glucosyltransferase |
| Os06G0593800 | −5.097684878 | - | 2.81431663 | crocetin glucosyltransferase, chloroplastic |
| Os11G0116300 | - | - | 3.4782357 | probable chalcone--flavonone isomerase 3 |
| Os02G0503100 | −6.152067498 | - | 3.052531824 | cytochrome P450 71A1 |
| Os09G0275400 | −5.060047384 | - | 5.773166236 | premnaspirodiene oxygenase |
| Os08G0547900 | - | - | 4.285402219 | cytochrome P450 76M5-like |
| Os03G0757000 | - | - | 5.039052777 | UDP-glycosyltransferase 83A1 |
| Os02G0697400 | - | 3.901446457 | - | probable 4-coumarate--CoA ligase 2 |
| Os11G0530600 | - | 4.641474522 | 6.273915733 | - |
| Os01G0372500 | 6.196397213 | 6.63840176 | - | leucoanthocyanidin dioxygenase |
| Os03G0757200 | −6.432291338 | - | 4.133633023 | - |
| Os03G0819600 | - | −1.607364477 | - | chalcone--flavonone isomerase |
| Os06G0165800 | - | −1.984442653 | - | caffeoyl-CoA O-methyltransferase 1 |
| Os02G0704000 | −6.716093942 | - | 4.661646158 | zeaxanthin 7,8(7′,8′)-cleavage dioxygenase, chromoplastic |
| Os03G0367101 | −5.912116705 | - | 3.786828232 | flavonoid 3′,5′-hydroxylase 1 |
| Os09G0441400 | −4.957665163 | - | - | cytochrome P450 71A1 |
| Os02G0207400 | - | - | 2.489120349 | UDP-glycosyltransferase 73C6-like |
| Os04G0320700 | −6.380423744 | - | 4.443988873 | 7-deoxyloganetin glucosyltransferase |
| Os07G0503300 | −6.050107771 | −4.496425826 | - | anthocyanidin 3-O-glucosyltransferase 2 |
| Os01G0906450 | −7.007961655 | −7.047123912 | - | - |
| Os06G0256500 | - | −1.379661054 | - | glucose-6-phosphate isomerase, cytosolic B |
| Os06G0288300 | −6.14198546 | - | 4.766118558 | UDP-glycosyltransferase 708A6 |
| Os10G0320201 | - | - | 4.19440866 | - |
| Protein ID | Mean Ratio B-C vs. R-C | Mean Ratio B-C vs. W-C | Mean Ratio R-C vs. W-C | Function |
|---|---|---|---|---|
| Os04G0206500 | - | - | 0.83 | crocetin glucosyltransferase 2 |
| Os01G0372500 | 3.48 | 3.38 | - | leucoanthocyanidin dioxygenase |
| Os04G0662600 | 2.32 | 2.4 | - | naringenin,2-oxoglutarate 3-dioxygenase |
| Os05G0527100 | 2.58 | 2.79 | - | UDP-glycosyltransferase 88F3-like |
| Os03G0819600 | 1.54 | 1.31 | - | chalcone--flavonone isomerase |
| Os09G0518000 | 1.39 | 1.49 | - | crocetin glucosyltransferase 2 |
| Os01G0686300 | - | 1.64 | 1.37 | cinnamate beta-D-glucosyltransferase |
| Os01G0850900 | 1.33 | 1.35 | - | heme-binding-like protein At3g10130, chloroplastic |
| OS11G0384789 | - | - | 0.83 | - |
| Os02G0816600 | - | 0.66 | 0.56 | uncharacterized protein sll0005 |
| Os07G0510500 | 4.54 | 3.62 | 0.8 | anthocyanidin 3-O-glucosyltransferase 2 |
| Os08G0434100 | - | 1.35 | 1.44 | extracellular ribonuclease LE |
| Os10G0320100 | 3.04 | 3.42 | - | flavonoid 3′-monooxygenase |
| Os01G0638000 | 1.68 | 1.46 | 0.87 | anthocyanin 3′-O-beta-glucosyltransferase |
| Os01G0805400 | 0.56 | 1.53 | 2.67 | UDP-glycosyltransferase 87A1 |
| Os04G0523600 | 0.87 | - | 1.5 | UDP-glycosyltransferase 73C7 |
| Os12G0115700 | 2.55 | 2.32 | - | probable chalcone--flavonone isomerase 3 |
| Os06G0192100 | 2.36 | 2.05 | - | anthocyanidin 3-O-glucosyltransferase |
| Os04G0206700 | 0.76 | - | 1.42 | UDP-glycosyltransferase 74F2 |
| Os07G0503500 | 2.47 | 2.3 | - | anthocyanidin 3-O-glucosyltransferase 2 |
| Os01G0176000 | 5.05 | 2.06 | 0.46 | UDP-glycosyltransferase 73C6 |
| Os06G0289900 | 1.32 | - | 0.77 | UDP-glycosyltransferase 708A6 |
| Os03G0693600 | - | 1.4 | 1.38 | indole-3-acetate beta-glucosyltransferase |
| Os06G0256500 | - | - | 0.78 | glucose-6-phosphate isomerase, cytosolic B |
| Os06G0282000 | - | - | 1.26 | UDP-glycosyltransferase 89B2 |
| Os11G0530600 | 3.86 | 6.95 | 1.57 | - |
| Os05G0133100 | 0.82 | - | - | nitrogen regulatory protein P-II homolog |
| Os03G0808200 | - | - | 1.14 | UDP-glucose flavonoid 3-O-glucosyltransferase 7 |
| Os03G0776000 | - | 1.27 | 1.24 | glucose-6-phosphate isomerase, cytosolic A |
| Os08G0547951 | 0.37 | - | 2.73 | - |
| Os08G0174300 | - | - | 0.78 | anthocyanin 5-aromatic acyltransferase |
| Gene ID | BR_RNA | BR_PEP | BW_RNA | BW_PEP | RW_RNA | RW_PEP | Description |
|---|---|---|---|---|---|---|---|
| Os01G0106400 | −5.30735 | 0.678072 | - | - | - | - | isoflavone reductase homolog IRL |
| Os01G0124650 | −6.88264 | 2.594549 | −4.70223 | 2.634593 | - | - | Bowman-Birk type bran trypsin inhibitor |
| Os01G0127600 | - | - | - | - | 5.728406 | 1.244887059 | Bowman-Birk type bran trypsin inhibitor |
| Os01G0180000 | - | - | −1.34699 | −0.73697 | - | - | leucine-rich repeat extensin-like protein 3 |
| Os01G0228600 | −2.30296 | 0.871844 | - | - | - | - | hydroxyphenylpyruvate reductase |
| Os01G0317800 | - | - | −1.58025 | 0.887525 | - | - | caffeoylshikimate esterase |
| Os01G0372500 | 6.196397 | 1.799087 | 6.638402 | 1.757023 | - | - | leucoanthocyanidin dioxygenase |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Tao, Y.; Ali, A.; Zhuang, Z.; Guo, D.; Guo, Q.; Riaz, A.; Zhang, H.; Xu, P.; Liao, Y.; et al. Transcriptome and Proteome Profiling of Different Colored Rice Reveals Physiological Dynamics Involved in the Flavonoid Pathway. Int. J. Mol. Sci. 2019, 20, 2463. https://doi.org/10.3390/ijms20102463
Chen X, Tao Y, Ali A, Zhuang Z, Guo D, Guo Q, Riaz A, Zhang H, Xu P, Liao Y, et al. Transcriptome and Proteome Profiling of Different Colored Rice Reveals Physiological Dynamics Involved in the Flavonoid Pathway. International Journal of Molecular Sciences. 2019; 20(10):2463. https://doi.org/10.3390/ijms20102463
Chicago/Turabian StyleChen, Xiaoqiong, Yu Tao, Asif Ali, Zhenhua Zhuang, Daiming Guo, Qiaoling Guo, Asad Riaz, Hongyu Zhang, Peizhou Xu, Yongxiang Liao, and et al. 2019. "Transcriptome and Proteome Profiling of Different Colored Rice Reveals Physiological Dynamics Involved in the Flavonoid Pathway" International Journal of Molecular Sciences 20, no. 10: 2463. https://doi.org/10.3390/ijms20102463
APA StyleChen, X., Tao, Y., Ali, A., Zhuang, Z., Guo, D., Guo, Q., Riaz, A., Zhang, H., Xu, P., Liao, Y., Wang, J., Sun, C., Xiang, Q., & Wu, X. (2019). Transcriptome and Proteome Profiling of Different Colored Rice Reveals Physiological Dynamics Involved in the Flavonoid Pathway. International Journal of Molecular Sciences, 20(10), 2463. https://doi.org/10.3390/ijms20102463