Tumor-Specific Reactive Oxygen Species Accelerators Improve Chimeric Antigen Receptor T Cell Therapy in B Cell Malignancies



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The ROS Accelerator PipFcB Increases CART-Mediated Lysis in Lymphoma Cell Lines
2.2. The ROS Accelerator PipFcB Increases CART-Mediated Lysis in Primary CLL Cells
2.3. Pretreatment with the ROS Accelerator PipFcB Sensitizes Lymphoma Cells to CART-Mediated Lysis
2.4. Increased Tumor Lysis by Combination of Other ROS Accelerators with CARTs
2.5. Influence of PipFcB on Lysis by CARTs in CD19− Cells
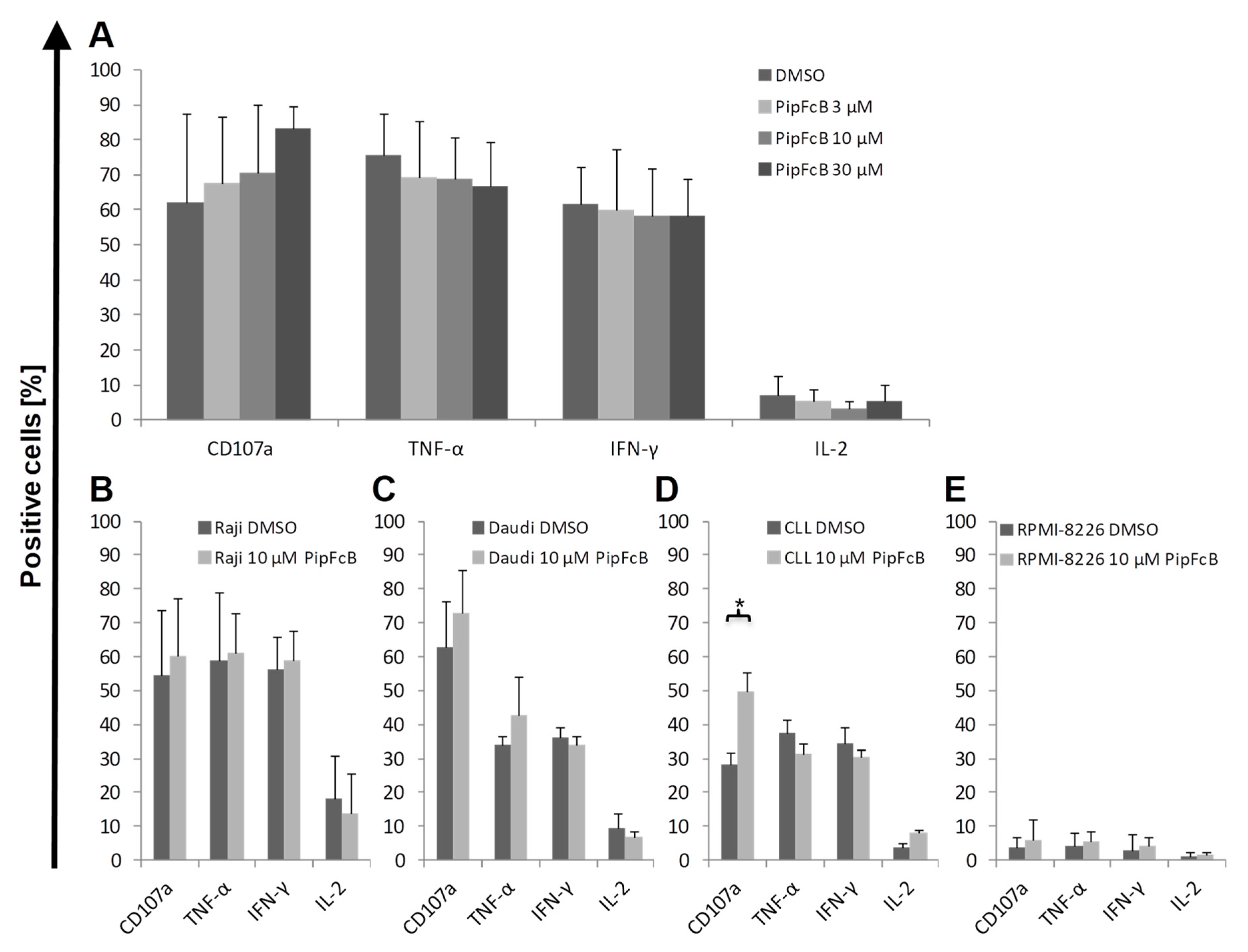
2.6. No Influence of PipFcB on Degranulation or Intracellular Cytokine Production of CARTs
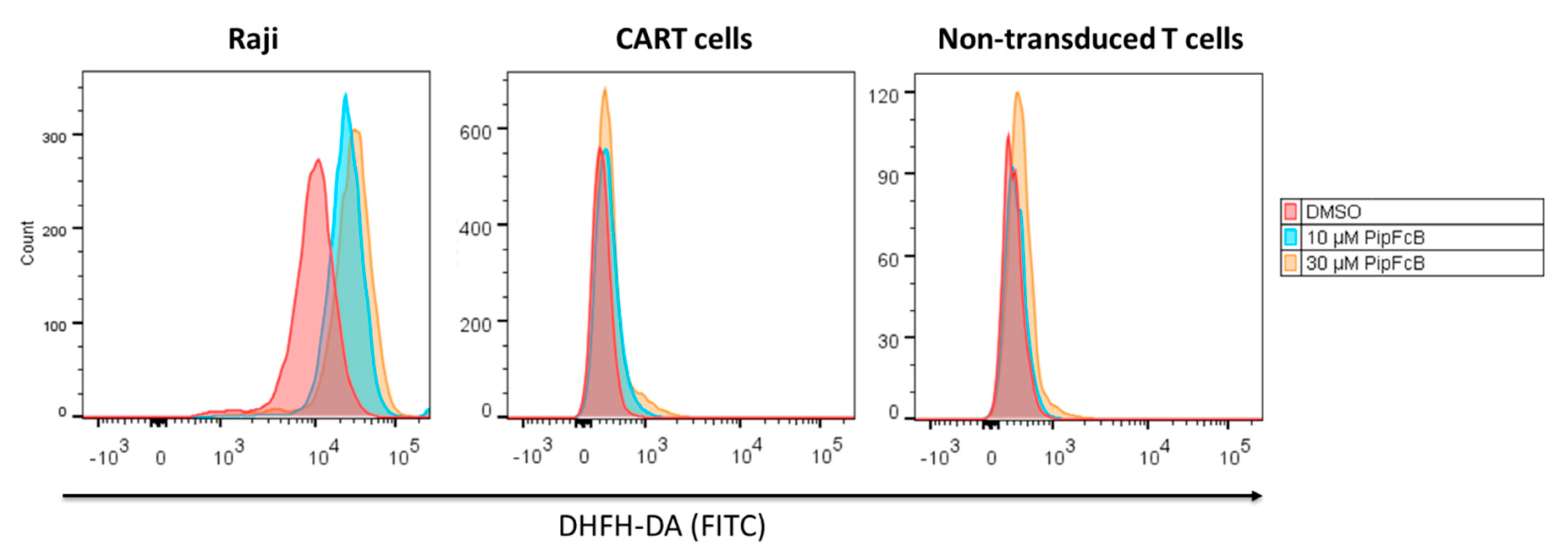
2.7. Malignant Lymphoma Cells are More Susceptible to PipFcB-Mediated Oxidative Stress
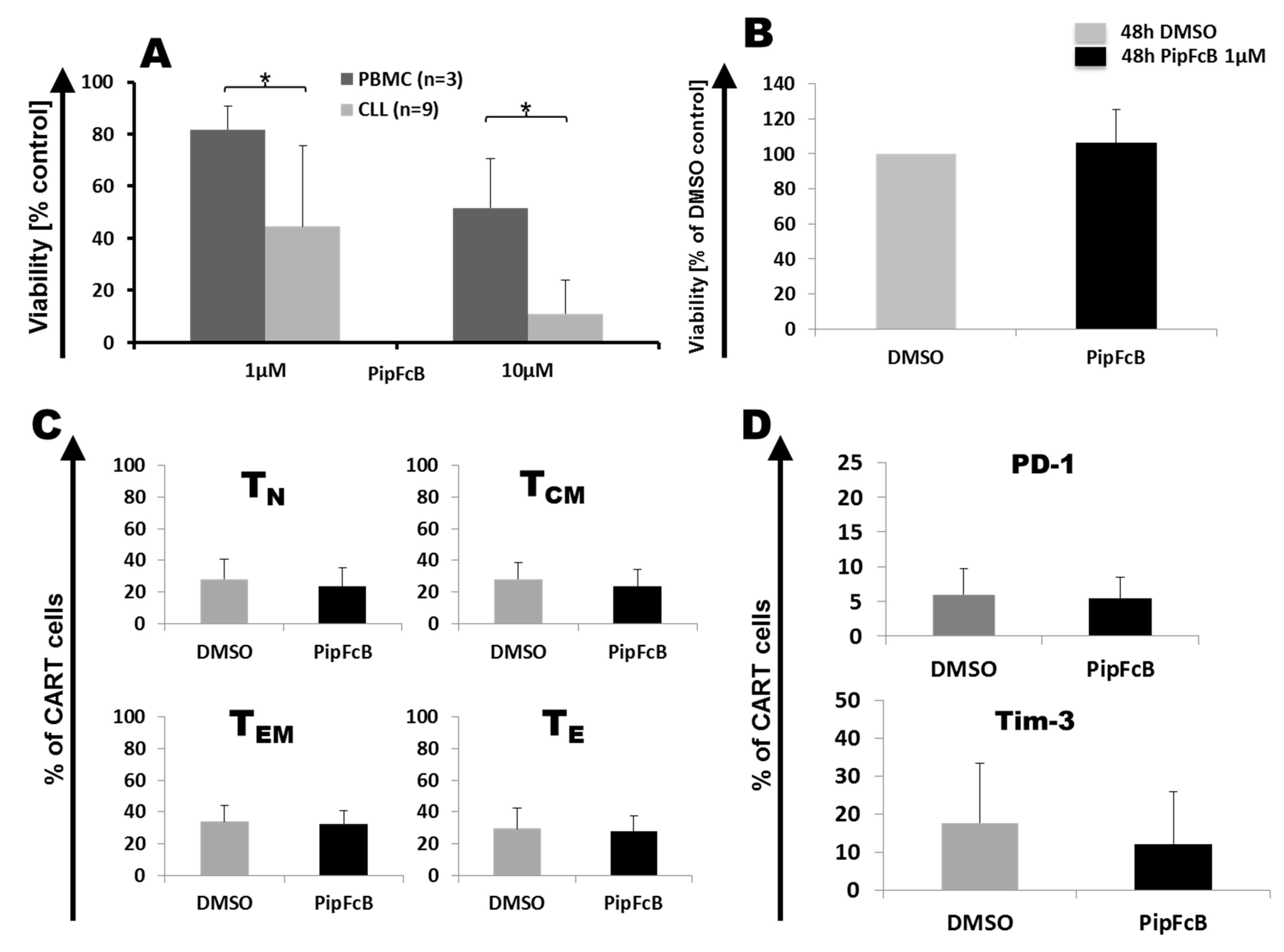
2.8. Influence of Long-Term PipFcB Exposure on CARTs
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Primary Cells
4.3. Retroviral Vector Production
4.4. CD19-CART Generation and Culture
4.5. Compounds
4.6. Chromium-51 Release Assay
4.7. Immunophenotyping and Intracellular Cytokine Staining
4.8. Evaluation of Intracellular Oxidative Stress
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 51Cr | Chromium-51 |
| BSO | Buthionine sulfoximine |
| BTK | Bruton’s tyrosine kinase |
| CAR | Chimeric antigen receptor |
| CART | Chimeric antigen receptor T cell |
| CLL | Chronic lymphocytic leukemia |
| CTLA4 | Cytotoxic T-lymphocyte-associated antigen 4 |
| DCFH-DA | Dichloro-dihydro-fluorescein diacetate |
| DMSO | dimethyl sulfoxide |
| FBS | Fetal bovine serum |
| IFN-γ | Interferon-gamma |
| IL-2 | Interleukin-2 |
| NT | Non-transduced T cells |
| PB | Peripheral blood |
| PBMC | Peripheral blood mononuclear cells |
| PD-1 | Programmed cell death-1 |
| PipFcB | N-(3-(piperidin-1-ylmethyl)benzyl)-4- (ferrocenylcarbamatmethyl)phenyl boronic acid pinacol ester |
| ROS | Reactive oxygen species |
| TCM | Central memory-like T cell |
| TE | Effector-like T cell |
| TEM | Effector memory-like T cell |
| TN | Naïve-like T cell |
| TIM-3 | T-cell immunoglobulin and mucin-domain containing-3 |
| TNF-α | Tumor necrosis factor-alpha |
References
- Schubert, M.L.; Huckelhoven, A.; Hoffmann, J.M.; Schmitt, A.; Wuchter, P.; Sellner, L.; Hofmann, S.; Ho, A.D.; Dreger, P.; Schmitt, M. Chimeric Antigen Receptor T Cell Therapy Targeting CD19-Positive Leukemia and Lymphoma in the Context of Stem Cell Transplantation. Hum. Gene Ther. 2016, 27, 758–771. [Google Scholar] [CrossRef]
- Jacoby, E.; Nguyen, S.M.; Fountaine, T.J.; Welp, K.; Gryder, B.; Qin, H.; Yang, Y.; Chien, C.D.; Seif, A.E.; Lei, H.; et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat. Commun. 2016, 7, 12320. [Google Scholar] [CrossRef] [Green Version]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Hu, Y.; Huang, H. Acute lymphoblastic leukemia relapse after CD19-targeted chimeric antigen receptor T cell therapy. J. Leukoc. Biol. 2017, 102, 1347–1356. [Google Scholar] [CrossRef]
- Wang, X.; Walter, M.; Urak, R.; Weng, L.; Huynh, C.; Lim, L.; Wong, C.W.; Chang, W.C.; Thomas, S.H.; Sanchez, J.F.; et al. Lenalidomide Enhances the Function of CS1 Chimeric Antigen Receptor-Redirected T Cells Against Multiple Myeloma. Clin. Cancer Res. 2018, 24, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Kenderian, S.S.; Shestova, O.; Fraietta, J.A.; Qayyum, S.; Zhang, Q.; Maus, M.V.; Liu, X.; Nunez-Cruz, S.; Klichinsky, M.; et al. The Addition of the BTK Inhibitor Ibrutinib to Anti-CD19 Chimeric Antigen Receptor T Cells (CART19) Improves Responses against Mantle Cell Lymphoma. Clin. Cancer Res. 2016, 22, 2684–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otahal, P.; Prukova, D.; Kral, V.; Fabry, M.; Vockova, P.; Lateckova, L.; Trneny, M.; Klener, P. Lenalidomide enhances antitumor functions of chimeric antigen receptor modified T cells. Oncoimmunology 2016, 5, e1115940. [Google Scholar] [CrossRef] [PubMed]
- Fowler, N.H.; Cheah, C.Y.; Gascoyne, R.D.; Gribben, J.; Neelapu, S.S.; Ghia, P.; Bollard, C.; Ansell, S.; Curran, M.; Wilson, W.H.; et al. Role of the tumor microenvironment in mature B-cell lymphoid malignancies. Haematologica 2016, 101, 531–540. [Google Scholar] [CrossRef] [Green Version]
- Andersen, M.H. The targeting of immunosuppressive mechanisms in hematological malignancies. Leukemia 2014, 28, 1784–1792. [Google Scholar] [CrossRef]
- Melero, I.; Berman, D.M.; Aznar, M.A.; Korman, A.J.; Perez Gracia, J.L.; Haanen, J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat. Rev. Cancer 2015, 15, 457–472. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative stress and cancer: Have we moved forward? Biochem. J. 2007, 401, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J. Immunol. 2009, 182, 5693–5701. [Google Scholar] [CrossRef]
- Schmielau, J.; Finn, O.J. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001, 61, 4756–4760. [Google Scholar]
- Daum, S.; Chekhun, V.F.; Todor, I.N.; Lukianova, N.Y.; Shvets, Y.V.; Sellner, L.; Putzker, K.; Lewis, J.; Zenz, T.; de Graaf, I.A.; et al. Improved synthesis of N-benzylaminoferrocene-based prodrugs and evaluation of their toxicity and antileukemic activity. J. Med. Chem. 2015, 58, 2015–2024. [Google Scholar] [CrossRef] [PubMed]
- Daum, S.; Reshetnikov, M.S.V.; Sisa, M.; Dumych, T.; Lootsik, M.D.; Bilyy, R.; Bila, E.; Janko, C.; Alexiou, C.; Herrmann, M.; et al. Lysosome-Targeting Amplifiers of Reactive Oxygen Species as Anticancer Prodrugs. Angew. Chem. Int. Ed. Engl. 2017, 56, 15545–15549. [Google Scholar] [CrossRef]
- Marzenell, P.; Hagen, H.; Sellner, L.; Zenz, T.; Grinyte, R.; Pavlov, V.; Daum, S.; Mokhir, A. Aminoferrocene-based prodrugs and their effects on human normal and cancer cells as well as bacterial cells. J. Med. Chem. 2013, 56, 6935–6944. [Google Scholar] [CrossRef]
- Doering, M.; Ba, L.A.; Lilienthal, N.; Nicco, C.; Scherer, C.; Abbas, M.; Zada, A.A.; Coriat, R.; Burkholz, T.; Wessjohann, L.; et al. Synthesis and selective anticancer activity of organochalcogen based redox catalysts. J. Med. Chem. 2010, 53, 6954–6963. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Cherian, S.; Chen, X.; Wood, B.; Lozanski, A.; Byrd, J.C.; Heimfeld, S.; et al. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated with CD19-Specific Chimeric Antigen Receptor-Modified T Cells After Failure of Ibrutinib. J. Clin. Oncol. 2017, 35, 3010–3020. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jager, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [Green Version]
- Fraietta, J.A.; Beckwith, K.A.; Patel, P.R.; Ruella, M.; Zheng, Z.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; McGettigan, S.E.; Cook, D.R.; et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016, 127, 1117–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Hileman, E.O.; Plunkett, W.; Keating, M.J.; Huang, P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood 2003, 101, 4098–4104. [Google Scholar] [CrossRef] [Green Version]
- Jitschin, R.; Hofmann, A.D.; Bruns, H.; Giessl, A.; Bricks, J.; Berger, J.; Saul, D.; Eckart, M.J.; Mackensen, A.; Mougiakakos, D. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood 2014, 123, 2663–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]
- Wuchter, P.; Saffrich, R.; Giselbrecht, S.; Nies, C.; Lorig, H.; Kolb, S.; Ho, A.D.; Gottwald, E. Microcavity arrays as an in vitro model system of the bone marrow niche for hematopoietic stem cells. Cell Tissue Res. 2016, 364, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef]
- Schubert, M.L.; Hoffmann, J.M.; Dreger, P.; Muller-Tidow, C.; Schmitt, M. Chimeric antigen receptor transduced T cells: Tuning up for the next generation. Int. J. Cancer 2018, 142, 1738–1747. [Google Scholar] [CrossRef]
- Stock, S.; Ubelhart, R.; Schubert, M.L.; Fan, F.; He, B.; Hoffmann, J.M.; Wang, L.; Wang, S.; Gong, W.; Neuber, B.; et al. Idelalisib for optimized CD19-specific chimeric antigen receptor T cells in chronic lymphocytic leukemia patients. Int. J. Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Stock, S.; Hoffmann, J.M.; Schubert, M.L.; Wang, L.; Wang, S.; Gong, W.; Neuber, B.; Gern, U.; Schmitt, A.; Muller-Tidow, C.; et al. Influence of Retronectin-Mediated T-Cell Activation on Expansion and Phenotype of CD19-Specific Chimeric Antigen Receptor T Cells. Hum. Gene Ther. 2018, 29, 1167–1182. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.M.; Schubert, M.L.; Wang, L.; Huckelhoven, A.; Sellner, L.; Stock, S.; Schmitt, A.; Kleist, C.; Gern, U.; Loskog, A.; et al. Differences in Expansion Potential of Naive Chimeric Antigen Receptor T Cells from Healthy Donors and Untreated Chronic Lymphocytic Leukemia Patients. Front. Immunol. 2017, 8, 1956. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoo, H.J.; Liu, Y.; Wang, L.; Schubert, M.-L.; Hoffmann, J.-M.; Wang, S.; Neuber, B.; Hückelhoven-Krauss, A.; Gern, U.; Schmitt, A.; et al. Tumor-Specific Reactive Oxygen Species Accelerators Improve Chimeric Antigen Receptor T Cell Therapy in B Cell Malignancies. Int. J. Mol. Sci. 2019, 20, 2469. https://doi.org/10.3390/ijms20102469
Yoo HJ, Liu Y, Wang L, Schubert M-L, Hoffmann J-M, Wang S, Neuber B, Hückelhoven-Krauss A, Gern U, Schmitt A, et al. Tumor-Specific Reactive Oxygen Species Accelerators Improve Chimeric Antigen Receptor T Cell Therapy in B Cell Malignancies. International Journal of Molecular Sciences. 2019; 20(10):2469. https://doi.org/10.3390/ijms20102469
Chicago/Turabian StyleYoo, Hyeon Joo, Yibin Liu, Lei Wang, Maria-Luisa Schubert, Jean-Marc Hoffmann, Sanmei Wang, Brigitte Neuber, Angela Hückelhoven-Krauss, Ulrike Gern, Anita Schmitt, and et al. 2019. "Tumor-Specific Reactive Oxygen Species Accelerators Improve Chimeric Antigen Receptor T Cell Therapy in B Cell Malignancies" International Journal of Molecular Sciences 20, no. 10: 2469. https://doi.org/10.3390/ijms20102469
APA StyleYoo, H. J., Liu, Y., Wang, L., Schubert, M. -L., Hoffmann, J. -M., Wang, S., Neuber, B., Hückelhoven-Krauss, A., Gern, U., Schmitt, A., Müller-Tidow, C., Dreger, P., Mokhir, A., Schmitt, M., & Sellner, L. (2019). Tumor-Specific Reactive Oxygen Species Accelerators Improve Chimeric Antigen Receptor T Cell Therapy in B Cell Malignancies. International Journal of Molecular Sciences, 20(10), 2469. https://doi.org/10.3390/ijms20102469