Adult Stem Cell Functioning in the Tumor Micro-Environment


 , ,
, , 

Abstract
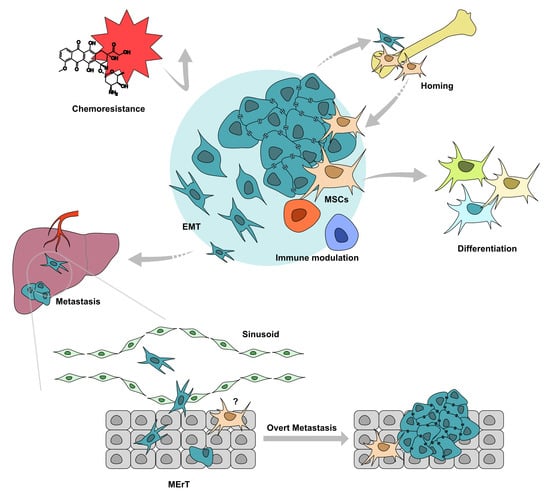
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Characterization of Human Mesenchymal Stem Cells/Multipotent Stromal Cells (MSCs)
3. Mutual Homing between Tumors and MSCs
4. MSCs in Tumor Metastasis
4.1. Soluble Factors from MSCs Contribute in Tumor EMT
4.2. MSCs Influence Tumor EMT via Multiple Signals
4.3. MSCs in Tumor Chemoresistance
4.4. Immune Modulation of MSCs in TME
5. MSCs Differentiation in TME
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gunasinghe, N.P.A.D.; Wells, A.; Thompson, E.W.; Hugo, H.J. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metast. Rev. 2012, 31, 469–478. [Google Scholar] [CrossRef]
- Thompson, E.W.; Haviv, I. The social aspects of EMT-MET plasticity. Nat. Med. 2011, 17, 1048–1049. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.; Yates, C.; Shepard, C.R. E-cadherin as an indicator of mesenchymal to epithelial reverting transitions during the metastatic seeding of disseminated carcinomas. Clin. Exp. Metastasis 2008, 25, 621–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, A.; Clark, A.; Bradshaw, A.; Ma, B.; Edington, H. The great escape: How metastases of melanoma, and other carcinomas, avoid elimination. Exp. Biol. Med. (Maywood) 2018, 243, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.M.; Wheeler, S.E.; Young, C.L.; Stockdale, L.; Shepard Neiman, J.; Zhao, W.; Stolz, D.B.; Venkataramanan, R.; Lauffenburger, D.; Griffith, L.; et al. A liver microphysiological system of tumor cell dormancy and inflammatory responsiveness is affected by scaffold properties. Lab. Chip. 2016, 17, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Wheeler, S.E.; Clark, A.M.; Whaley, D.L.; Yang, M.; Wells, A. Liver protects metastatic prostate cancer from induced death by activating E-cadherin signaling. Hepatology 2016, 64, 1725–1742. [Google Scholar] [CrossRef] [Green Version]
- Brodt, P. Role of the Microenvironment in Liver Metastasis: From Pre- to Prometastatic Niches. Clin. Cancer Res. 2016, 22, 5971–5982. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Rafii, S.; Lyden, D. Preparing the “soil”: The premetastatic niche. Cancer Res. 2006, 66, 11089–11093. [Google Scholar] [CrossRef]
- Caplan, A.I. Mesenchymal Stem-Cells. J. Orthop. Res. 1991, 9, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; Kasper, C.; Bohm, S.; Jacobs, R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun. Signal 2011, 9. [Google Scholar] [CrossRef]
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells - current trends and future prospective. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef] [PubMed]
- DiMarino, A.M.; Caplan, A.I.; Bonfield, T.L. Mesenchymal stem cells in tissue repair. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef]
- Atsma, D.E.; Fibbe, W.E.; Rabelink, T.J. Opportunities and challenges for mesenchymal stem cell-mediated heart repair. Curr. Opin. Lipidol. 2007, 18, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Fathi, S.S.; Zaminy, A. Stem cell therapy for nerve injury. World J. Stem Cells 2017, 9, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Prockop, D.J. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science 1997, 276, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I.; Dennis, J.E. Mesenchymal stem cells as trophic mediators. J. Cell Biochem. 2006, 98, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Phinney, D.G.; Prockop, D.J. Concise review: Mesenchymal stem/multipotent stromal cells: The state of transdifferentiation and modes of tissue repair--current views. Stem Cells 2007, 25, 2896–2902. [Google Scholar] [CrossRef]
- Xi, J.; Yan, X.; Zhou, J.; Yue, W.; Pei, X. Mesenchymal stem cells in tissue repairing and regeneration: Progress and future. Burn. Trauma 2013, 1, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Lv, F.J.; Tuan, R.S.; Cheung, K.M.; Leung, V.Y. Concise review: The surface markers and identity of human mesenchymal stem cells. Stem Cells 2014, 32, 1408–1419. [Google Scholar] [CrossRef]
- Yang, Z.X.; Han, Z.B.; Ji, Y.R.; Wang, Y.W.; Liang, L.; Chi, Y.; Yang, S.G.; Li, L.N.; Luo, W.F.; Li, J.P.; et al. CD106 identifies a subpopulation of mesenchymal stem cells with unique immunomodulatory properties. PLoS ONE 2013, 8, e59354. [Google Scholar] [CrossRef]
- Buhring, H.J.; Battula, V.L.; Treml, S.; Schewe, B.; Kanz, L.; Vogel, W. Novel markers for the prospective isolation of human MSC. Ann. Ny. Acad. Sci. 2007, 1106, 262–271. [Google Scholar] [CrossRef]
- Rasini, V.; Dominici, M.; Kluba, T.; Siegel, G.; Lusenti, G.; Northoff, H.; Horwitz, E.M.; Schafer, R. Mesenchymal stromal/stem cells markers in the human bone marrow. Cytotherapy 2013, 15, 292–306. [Google Scholar] [CrossRef]
- Nadri, S.; Soleimani, M. Isolation murine mesenchymal stem cells by positive selection. In Vitro Cell Dev-An 2007, 43, 276–282. [Google Scholar] [CrossRef]
- Soleimani, M.; Nadri, S. A protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. Nat. Protoc. 2009, 4, 102–106. [Google Scholar] [CrossRef]
- Bourin, P.; Bunnell, B.A.; Casteilla, L.; Dominici, M.; Katz, A.J.; March, K.L.; Redl, H.; Rubin, J.P.; Yoshimura, K.; Gimble, J.M. Stromal cells from the adipose tissue-derived stromal vascular fraction and culture expanded adipose tissue-derived stromal/stem cells: A joint statement of the International Federation for Adipose Therapeutics and Science (IFATS) and the International Society for Cellular Therapy (ISCT). Cytotherapy 2013, 15, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Gimble, J.M.; Katz, A.J.; Bunnell, B.A. Adipose-derived stem cells for regenerative medicine. Circ. Res. 2007, 100, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Zimmerlin, L.; Donnenberg, V.S.; Rubin, J.P.; Donnenberg, A.D. Mesenchymal markers on human adipose stem/progenitor cells. Cytom. A 2013, 83, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Ridge, S.M.; Sullivan, F.J.; Glynn, S.A. Mesenchymal stem cells: Key players in cancer progression. Mol. Cancer 2017, 16, 10. [Google Scholar] [CrossRef]
- Direkze, N.C.; Hodivala-Dilke, K.; Jeffery, R.; Hunt, T.; Poulsom, R.; Oukrif, D.; Alison, M.R.; Wright, N.A. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004, 64, 8492–8495. [Google Scholar] [CrossRef]
- Ishii, G.; Sangai, T.; Oda, T.; Aoyagi, Y.; Hasebe, T.; Kanomata, N.; Endoh, Y.; Okumura, C.; Okuhara, Y.; Magae, J.; et al. Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem. Biophys. Res. Commun. 2003, 309, 232–240. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, S.H.; Zhao, R.C. The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J. Hematol. Oncol. 2014, 7, 10. [Google Scholar] [CrossRef]
- Clark, A.M.; Kumar, M.P.; Wheeler, S.E.; Young, C.L.; Venkataramanan, R.; Stolz, D.B.; Griffith, L.G.; Lauffenburger, D.A.; Wells, A. A Model of Dormant-Emergent Metastatic Breast Cancer Progression Enabling Exploration of Biomarker Signatures. Mol. Cell Proteom. 2018, 17, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, C.B.; Porter, R.M.; Evans, C.H.; Ritter, T.; Shaw, G.; Barry, F.; Murphy, J.M. TNF alpha and IL-1 beta influence the differentiation and migration of murine MSCs independently of the NF-kappa B pathway. Stem Cell Res. Ther. 2014, 5. [Google Scholar] [CrossRef]
- Uchibori, R.; Tsukahara, T.; Mizuguchi, H.; Saga, Y.; Urabe, M.; Mizukami, H.; Kume, A.; Ozawa, K. NF-kappa B Activity Regulates Mesenchymal Stem Cell Accumulation at Tumor Sites. Cancer Res. 2013, 73, 364–372. [Google Scholar] [CrossRef]
- Rattigan, Y.; Hsu, J.M.; Mishra, P.J.; Glod, J.; Banerjee, D. Interleukin 6 mediated recruitment of mesenchymal stem cells to the hypoxic tumor milieu. Exp. Cell Res. 2010, 316, 3417–3424. [Google Scholar] [CrossRef]
- Dubon, M.J.; Yu, J.; Choi, S.; Park, K.S. Transforming growth factor beta induces bone marrow mesenchymal stem cell migration via noncanonical signals and N-cadherin. J. Cell. Physiol. 2018, 233, 201–213. [Google Scholar] [CrossRef]
- Xu, W.T.; Bian, Z.Y.; Fan, Q.M.; Li, G.; Tang, T.T. Human mesenchymal stem cells (hMSCs) target osteosarcoma and promote its growth and pulmonary metastasis. Cancer Lett. 2009, 281, 32–41. [Google Scholar] [CrossRef]
- Jung, Y.H.; Kim, J.K.; Shiozawa, Y.; Wang, J.C.; Mishra, A.; Joseph, J.; Berry, J.E.; McGee, S.; Lee, E.; Sun, H.L.; et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat. Commun. 2013, 4, 11. [Google Scholar] [CrossRef]
- Cooper, C.R.; Chay, C.H.; Gendernalik, J.D.; Lee, H.L.; Bhatia, J.; Taichman, R.S.; McCauley, L.K.; Keller, E.T.; Pienta, K.J. Stromal factors involved in prostate carcinoma metastasis to bone. Cancer 2003, 97, 739–747. [Google Scholar] [CrossRef] [Green Version]
- Taichman, R.S.; Cooper, C.; Keller, E.T.; Pienta, K.J.; Taichman, N.S.; McCauley, L.K. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002, 62, 1832–1837. [Google Scholar]
- Rossnagl, S.; Ghura, H.; Groth, C.; Altrock, E.; Jakob, F.; Schott, S.; Wimberger, P.; Link, T.; Kuhlmann, J.D.; Stenzl, A.; et al. A Subpopulation of Stromal Cells Controls Cancer Cell Homing to the Bone Marrow. Cancer Res. 2018, 78, 129–142. [Google Scholar] [CrossRef]
- Bastid, J. EMT in carcinoma progression and dissemination: Facts, unanswered questions, and clinical considerations. Cancer Metastasis Rev. 2012, 31, 277–283. [Google Scholar] [CrossRef]
- Chao, Y.; Wu, Q.; Acquafondata, M.; Dhir, R.; Wells, A. Partial mesenchymal to epithelial reverting transition in breast and prostate cancer metastases. Cancer Microenviron. 2012, 5, 19–28. [Google Scholar] [CrossRef]
- Bian, Z.Y.; Fan, Q.M.; Li, G.; Xu, W.T.; Tang, T.T. Human mesenchymal stem cells promote growth of osteosarcoma: Involvement of interleukin-6 in the interaction between human mesenchymal stem cells and Saos-2. Cancer Sci. 2010, 101, 2554–2560. [Google Scholar] [CrossRef]
- Li, H.; Feng, Z.; Tsang, T.C.; Tang, T.; Jia, X.; He, X.; Pennington, M.E.; Badowski, M.S.; Liu, A.K.; Chen, D.; et al. Fusion of HepG2 cells with mesenchymal stem cells increases cancerassociated and malignant properties: An in vivo metastasis model. Oncol. Rep. 2014, 32, 539–547. [Google Scholar] [CrossRef]
- Liu, S.L.; Ginestier, C.; Ou, S.J.; Clouthier, S.G.; Patel, S.H.; Monville, F.; Korkaya, H.; Heath, A.; Dutcher, J.; Kleer, C.G.; et al. Breast Cancer Stem Cells Are Regulated by Mesenchymal Stem Cells through Cytokine Networks. Cancer Res. 2011, 71, 614–624. [Google Scholar] [CrossRef] [Green Version]
- Takigawa, H.; Kitadai, Y.; Shinagawa, K.; Yuge, R.; Higashi, Y.; Tanaka, S.; Yasui, W.; Chayama, K. Mesenchymal Stem Cells Induce Epithelial to Mesenchymal Transition in Colon Cancer Cells through Direct Cell-to-Cell Contact. Neoplasia 2017, 19, 429–438. [Google Scholar] [CrossRef]
- Xie, C.; Yang, Z.; Suo, Y.; Chen, Q.; Wei, D.; Weng, X.; Gu, Z.; Wei, X. Systemically Infused Mesenchymal Stem Cells Show Different Homing Profiles in Healthy and Tumor Mouse Models. Stem Cells Transl. Med. 2017, 6, 1120–1131. [Google Scholar] [CrossRef]
- Hill, B.S.; Pelagalli, A.; Passaro, N.; Zannetti, A. Tumor-educated mesenchymal stem cells promote pro-metastatic phenotype. Oncotarget 2017, 8, 73296–73311. [Google Scholar] [CrossRef] [Green Version]
- Fontanella, R.; Pelagalli, A.; Nardelli, A.; D’Alterio, C.; Ierano, C.; Cerchia, L.; Lucarelli, E.; Scala, S.; Zannetti, A. A novel antagonist of CXCR4 prevents bone marrow-derived mesenchymal stem cell-mediated osteosarcoma and hepatocellular carcinoma cell migration and invasion. Cancer Lett. 2016, 370, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Halpern, J.L.; Kilbarger, A.; Lynch, C.C. Mesenchymal stem cells promote mammary cancer cell migration in vitro via the CXCR2 receptor. Cancer Lett. 2011, 308, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Dong, L.; Yan, K.; Long, H.; Yang, T.T.; Dong, M.Q.; Zhou, Y.; Fan, Q.Y.; Ma, B.A. CXCR4-mediated osteosarcoma growth and pulmonary metastasis is promoted by mesenchymal stem cells through VEGF. Oncol. Rep. 2013, 30, 1753–1761. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Khazali, A.; Wells, A. CXCR3 in carcinoma progression. Histol. Histopathol. 2015, 30, 781–792. [Google Scholar] [CrossRef]
- Shin, S.Y.; Nam, J.S.; Lim, Y.; Lee, Y.H. TNF alpha-exposed Bone Marrow-derived Mesenchymal Stem Cells Promote Locomotion of MDA-MB-231 Breast Cancer Cells through Transcriptional Activation of CXCR3 Ligand Chemokines. J. Biol. Chem. 2010, 285, 30731–30740. [Google Scholar] [CrossRef]
- Anton, K.; Banerjee, D.; Glod, J. Macrophage-Associated Mesenchymal Stem Cells Assume an Activated, Migratory, Pro-Inflammatory Phenotype with Increased IL-6 and CXCL10 Secretion. PLoS ONE 2012, 7, 10. [Google Scholar] [CrossRef]
- Chen, D.M.; Liu, S.D.; Ma, H.M.; Liang, X.Y.; Ma, H.B.; Yan, X.R.; Yang, B.; Wei, J.; Liu, X.M. Paracrine factors from adipose-mesenchymal stem cells enhance metastatic capacity through Wnt signaling pathway in a colon cancer cell co-culture model. Cancer Cell Int. 2015, 15, 13. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Marini, F.C.; Watson, K.; Zwezdaryk, K.J.; Dembinski, J.L.; LaMarca, H.L.; Tomchuck, S.L.; Bentrup, K.H.Z.; Danka, E.S.; Henkle, S.L.; et al. The pro-inflammatory peptide LL-37 promotes ovarian tumor progression through recruitment of multipotent mesenchymal stromal cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3806–3811. [Google Scholar] [CrossRef] [Green Version]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.C.; Hu, F.Y.; Li, G.; Li, G.D.; Yang, X.; Liu, L.; Zhang, R.S.; Zhang, B.X.; Feng, Y.D. Human colorectal cancer-derived mesenchymal stem cells promote colorectal cancer progression through IL-6/JAK2/STAT3 signaling. Cell Death Dis. 2018, 9, 13. [Google Scholar] [CrossRef]
- Wolfe, A.R.; Trenton, N.J.; Debeb, B.G.; Larson, R.; Ruffell, B.; Chu, K.; Hittelman, W.; Diehl, M.; Reuben, J.M.; Ueno, N.T.; et al. Mesenchymal stem cells and macrophages interact through IL-6 to promote inflammatory breast cancer in pre-clinical models. Oncotarget 2016, 7, 82482–82492. [Google Scholar] [CrossRef]
- Zavadil, J.; Bottinger, E.P. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef]
- Trivanovic, D.; Jaukovic, A.; Krstic, J.; Nikolic, S.; Djordjevic, I.O.; Kukolj, T.; Obradovic, H.; Mojsilovic, S.; Ilic, V.; Santibanez, J.F.; et al. Inflammatory Cytokines Prime Adipose Tissue Mesenchymal Stem Cells to Enhance Malignancy of MCF-7 Breast Cancer Cells via Transforming Growth Factor-beta 1. IUBMB Life 2016, 68, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Dai, H.Y.; Sun, M.Y.; Zhao, H.; Wu, K.; Zhu, J.; Wang, Y.C.; Cao, X.; Xia, Z.F.; Xue, C.Y. Mesenchymal stem cells induce epithelial mesenchymal transition in melanoma by paracrine secretion of transforming growth factor. Melanoma Res. 2017, 27, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Huang, L.; Li, Y.H.; Qian, H.; Shan, X.H.; Yan, Y.M.; Mao, F.; Wu, X.S.; Xu, W.R. Mesenchymal stem cell-secreted soluble signaling molecules potentiate tumor growth. Cell Cycle 2011, 10, 10. [Google Scholar] [CrossRef]
- Yan, X.L.; Fu, C.J.; Chen, L.; Qin, J.H.; Zeng, Q.; Yuan, H.F.; Nan, X.; Chen, H.X.; Zhou, J.N.; Lin, Y.L.; et al. Mesenchymal stem cells from primary breast cancer tissue promote cancer proliferation and enhance mammosphere formation partially via EGF/EGFR/Akt pathway. Breast Cancer Res. Treat. 2012, 132, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga-Kalabis, M.; Martinez, G.; Nguyen, T.K.; Kim, D.; Santiago-Walker, A.; Roesch, A.; Herlyn, M. Tenascin-C promotes melanoma progression by maintaining the ABCB5-positive side population. Oncogene 2010, 29, 6115–6124. [Google Scholar] [CrossRef] [Green Version]
- Insua-Rodriguez, J.; Pein, M.; Hongu, T.; Meier, J.; Descot, A.; Lowy, C.M.; De Braekeleer, E.; Sinn, H.P.; Spaich, S.; Sutterlin, M.; et al. Stress signaling in breast cancer cells induces matrix components that promote chemoresistant metastasis. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Thakur, R.; Mishra, D.P. Matrix reloaded: CCN, tenascin and SIBLING group of matricellular proteins in orchestrating cancer hallmark capabilities. Pharmacol. Ther. 2016, 168, 61–74. [Google Scholar] [CrossRef]
- Shao, H.; Kirkwood, J.M.; Wells, A. Tenascin-C Signaling in melanoma. Cell Adh. Migr. 2015, 9, 125–130. [Google Scholar] [CrossRef]
- Abdeen, A.A.; Weiss, J.B.; Lee, J.; Kilian, K.A. Matrix composition and mechanics direct proangiogenic signaling from mesenchymal stem cells. Tissue Eng. Part A 2014, 20, 2737–2745. [Google Scholar] [CrossRef] [PubMed]
- Boink, M.A.; van den Broek, L.J.; Roffel, S.; Nazmi, K.; Bolscher, J.G.; Gefen, A.; Veerman, E.C.; Gibbs, S. Different wound healing properties of dermis, adipose, and gingiva mesenchymal stromal cells. Wound Repair Regen. 2016, 24, 100–109. [Google Scholar] [CrossRef]
- Ragelle, H.; Naba, A.; Larson, B.L.; Zhou, F.; Prijic, M.; Whittaker, C.A.; Del Rosario, A.; Langer, R.; Hynes, R.O.; Anderson, D.G. Comprehensive proteomic characterization of stem cell-derived extracellular matrices. Biomaterials 2017, 128, 147–159. [Google Scholar] [CrossRef]
- Melzer, C.; von der Ohe, J.; Hass, R. MSC stimulate ovarian tumor growth during intercellular communication but reduce tumorigenicity after fusion with ovarian cancer cells. Cell Commun. Signal 2018, 16, 67. [Google Scholar] [CrossRef]
- Xue, J.; Zhu, Y.; Sun, Z.; Ji, R.; Zhang, X.; Xu, W.; Yuan, X.; Zhang, B.; Yan, Y.; Yin, L.; et al. Tumorigenic hybrids between mesenchymal stem cells and gastric cancer cells enhanced cancer proliferation, migration and stemness. BMC Cancer 2015, 15, 793. [Google Scholar] [CrossRef]
- Zhang, L.N.; Kong, C.F.; Zhao, D.; Cong, X.L.; Wang, S.S.; Ma, L.; Huang, Y.H. Fusion with mesenchymal stem cells differentially affects tumorigenic and metastatic abilities of lung cancer cells. J. Cell Physiol. 2019, 234, 3570–3582. [Google Scholar] [CrossRef]
- Wei, H.J.; Nickoloff, J.A.; Chen, W.H.; Liu, H.Y.; Lo, W.C.; Chang, Y.T.; Yang, P.C.; Wu, C.W.; Williams, D.F.; Gelovani, J.G.; et al. FOXF1 mediates mesenchymal stem cell fusion-induced reprogramming of lung cancer cells. Oncotarget 2014, 5, 9514–9529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Yuan, Y.; Zhang, L.; Min, Z.; Zhou, D.; Yu, S.; Wang, P.; Ju, S.; Jun, L.; Fu, J. Impact of cell fusion in myeloma marrow microenvironment on tumor progression. Oncotarget 2018, 9, 30997–31006. [Google Scholar] [CrossRef]
- Wang, Y.; Chu, Y.J.; Yue, B.; Ma, X.X.; Zhang, G.Q.; Xiang, H.F.; Liu, Y.; Wang, T.R.; Wu, X.L.; Chen, B.H. Adipose-derived mesenchymal stem cells promote osteosarcoma proliferation and metastasis by activating the STAT3 pathway. Oncotarget 2017, 8, 23803–23816. [Google Scholar] [CrossRef]
- Zhang, T.; Lee, Y.W.; Rui, Y.F.; Cheng, T.Y.; Jiang, X.H.; Li, G. Bone marrow-derived mesenchymal stem cells promote growth and angiogenesis of breast and prostate tumors. Stem Cell Res. Ther. 2013, 4, 15. [Google Scholar] [CrossRef]
- Huang, W.H.; Chang, M.C.; Tsai, K.S.; Hung, M.C.; Chen, H.L.; Hung, S.C. Mesenchymal stem cells promote growth and angiogenesis of tumors in mice. Oncogene 2013, 32, 4343–4354. [Google Scholar] [CrossRef]
- Suzuki, K.; Sun, R.W.; Origuchi, M.; Kanehira, M.; Takahata, T.; Itoh, J.; Umezawa, A.; Kijima, H.; Fukuda, S.; Saijo, Y. Mesenchymal Stromal Cells Promote Tumor Growth through the Enhancement of Neovascularization. Mol. Med. 2011, 17, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Wang, Y.N.; Wang, S.C.; Cai, J.Y.; Shi, J.Q.; Sui, X.; Cao, Y.; Huang, W.J.; Chen, X.Y.; Cai, Z.J.; et al. Bone marrow-derived mesenchymal stem cell-secreted IL-8 promotes the angiogenesis and growth of colorectal cancer. Oncotarget 2015, 6, 42825–42837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelagalli, A.; Nardelli, A.; Fontanella, R.; Zannetti, A. Inhibition of AQP1 Hampers Osteosarcoma and Hepatocellular Carcinoma Progression Mediated by Bone Marrow-Derived Mesenchymal Stem Cells. Int. J. Mol. Sci. 2016, 17, 1102. [Google Scholar] [CrossRef] [PubMed]
- Tu, B.; Zhu, J.; Liu, S.; Wang, L.; Fan, Q.M.; Hao, Y.Q.; Fan, C.Y.; Tang, T.T. Mesenchymal stem cells promote osteosarcoma cell survival and drug resistance through activation of STAT3. Oncotarget 2016, 7, 48296–48308. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Zhou, Y.; Li, W.; Zhang, B.; Zhang, H.; Zhao, S.; Zheng, P.; Wu, H.; Yang, J. Tumor-derived mesenchymal-stem-cell-secreted IL-6 enhances resistance to cisplatin via the STAT3 pathway in breast cancer. Oncol. Lett. 2018, 15, 9142–9150. [Google Scholar] [CrossRef]
- Skolekova, S.; Matuskova, M.; Bohac, M.; Toro, L.; Demkova, L.; Gursky, J.; Kucerova, L. Cisplatin-induced mesenchymal stromal cells-mediated mechanism contributing to decreased antitumor effect in breast cancer cells. Cell Commun. Signal. 2016, 14, 13. [Google Scholar] [CrossRef]
- Wang, W.W.; Zhong, W.; Yuan, J.H.; Yan, C.C.; Hu, S.P.; Tong, Y.P.; Mao, Y.B.; Hu, T.H.; Zhang, B.; Song, G. Involvement of Wnt/beta-catenin signaling in the mesenchymal stem cells promote metastatic growth and chemoresistance of cholangiocarcinoma. Oncotarget 2015, 6, 42276–42289. [Google Scholar] [CrossRef]
- Grahovac, J.; Becker, D.; Wells, A. Melanoma cell invasiveness is promoted at least in part by the epidermal growth factor-like repeats of tenascin-C. J. Invest. Dermatol. 2013, 133, 210–220. [Google Scholar] [CrossRef]
- Rodrigues, M.; Yates, C.C.; Nuschke, A.; Griffith, L.; Wells, A. The matrikine tenascin-C protects multipotential stromal cells/mesenchymal stem cells from death cytokines such as FasL. Tissue Eng. Part A 2013, 19, 1972–1983. [Google Scholar] [CrossRef]
- Mantovani, A. MSCs, macrophages, and cancer: A dangerous menage-a-trois. Cell Stem Cell 2012, 11, 730–732. [Google Scholar] [CrossRef]
- Ren, G.; Zhao, X.; Wang, Y.; Zhang, X.; Chen, X.; Xu, C.; Yuan, Z.R.; Roberts, A.I.; Zhang, L.; Zheng, B.; et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFalpha. Cell Stem Cell 2012, 11, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Zhang, X.; Wang, M.; Zhang, J.; Huang, F.; Cai, J.; Zhang, Q.; Mao, F.; Zhu, W.; Qian, H.; et al. Activation of mesenchymal stem cells by macrophages prompts human gastric cancer growth through NF-kappaB pathway. PLoS ONE 2014, 9, e97569. [Google Scholar] [CrossRef]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef]
- Yang, M.; Ma, B.; Shao, H.; Clark, A.M.; Wells, A. Macrophage phenotypic subtypes diametrically regulate epithelial-mesenchymal plasticity in breast cancer cells. BMC Cancer 2016, 16, 419. [Google Scholar] [CrossRef]
- Jia, X.H.; Feng, G.W.; Wang, Z.L.; Du, Y.; Shen, C.; Hui, H.; Peng, D.; Li, Z.J.; Kong, D.L.; Tian, J. Activation of mesenchymal stem cells by macrophages promotes tumor progression through immune suppressive effects. Oncotarget 2016, 7, 20934–20944. [Google Scholar] [CrossRef]
- Mathew, E.; Brannon, A.L.; Del Vecchio, A.; Garcia, P.E.; Penny, M.K.; Kane, K.T.; Vinta, A.; Buckanovich, R.J.; di Magliano, M.P. Mesenchymal Stem Cells Promote Pancreatic Tumor Growth by Inducing Alternative Polarization of Macrophages. Neoplasia 2016, 18, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.Y.; Du, L.M.; Cao, K.; Huang, Y.; Yu, P.F.; Zhang, L.Y.; Li, F.Y.; Wang, Y.; Shi, Y.F. Tumour cell-derived exosomes endow mesenchymal stromal cells with tumour-promotion capabilities. Oncogene 2016, 35, 6038–6042. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.F.; Huang, Y.; Han, Y.Y.; Lin, L.Y.; Sun, W.H.; Rabson, A.B.; Wang, Y.; Shi, Y.F. TNFalpha-activated mesenchymal stromal cells promote breast cancer metastasis by recruiting CXCR2(+) neutrophils. Oncogene 2017, 36, 482–490. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhang, X.; Zhang, L.; Li, W.; Wu, H.; Yuan, X.; Mao, F.; Wang, M.; Zhu, W.; Qian, H.; et al. The IL-6-STAT3 axis mediates a reciprocal crosstalk between cancer-derived mesenchymal stem cells and neutrophils to synergistically prompt gastric cancer progression. Cell Death Dis. 2014, 5, e1295. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.H.; Yoon, N.; Reneau, J.C.; Prockop, D.J. Preactivation of human MSCs with TNF-alpha enhances tumor-suppressive activity. Cell Stem Cell 2012, 11, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Waterman, R.S.; Henkle, S.L.; Betancourt, A.M. Mesenchymal Stem Cell 1 (MSC1)-Based Therapy Attenuates Tumor Growth Whereas MSC2-Treatment Promotes Tumor Growth and Metastasis. PLoS ONE 2012, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Waterman, R.S.; Tomchuck, S.L.; Henkle, S.L.; Betancourt, A.M. A New Mesenchymal Stem Cell (MSC) Paradigm: Polarization into a Pro-Inflammatory MSC1 or an Immunosuppressive MSC2 Phenotype. PLoS ONE 2010, 5, 14. [Google Scholar] [CrossRef]
- Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Research 2008, 68, 4331–4339. [Google Scholar] [CrossRef] [PubMed]
- Paunescu, V.; Bojin, F.M.; Tatu, C.A.; Gavriliuc, O.I.; Rosca, A.; Gruia, A.T.; Tanasie, G.; Bunu, C.; Crisnic, D.; Gherghiceanu, M.; et al. Tumour-associated fibroblasts and mesenchymal stem cells: More similarities than differences. J. Cell Mol. Med. 2011, 15, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018, 215, 3075–3093. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Y.; Wells, A.; Sylakowski, K.; Clark, A.M.; Ma, B. Adult Stem Cell Functioning in the Tumor Micro-Environment. Int. J. Mol. Sci. 2019, 20, 2566. https://doi.org/10.3390/ijms20102566
Jiang Y, Wells A, Sylakowski K, Clark AM, Ma B. Adult Stem Cell Functioning in the Tumor Micro-Environment. International Journal of Molecular Sciences. 2019; 20(10):2566. https://doi.org/10.3390/ijms20102566
Chicago/Turabian StyleJiang, Yuhan, Alan Wells, Kyle Sylakowski, Amanda M. Clark, and Bo Ma. 2019. "Adult Stem Cell Functioning in the Tumor Micro-Environment" International Journal of Molecular Sciences 20, no. 10: 2566. https://doi.org/10.3390/ijms20102566
APA StyleJiang, Y., Wells, A., Sylakowski, K., Clark, A. M., & Ma, B. (2019). Adult Stem Cell Functioning in the Tumor Micro-Environment. International Journal of Molecular Sciences, 20(10), 2566. https://doi.org/10.3390/ijms20102566