Graptopetalum paraguayense Inhibits Liver Fibrosis by Blocking TGF-β Signaling In Vivo and In Vitro

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
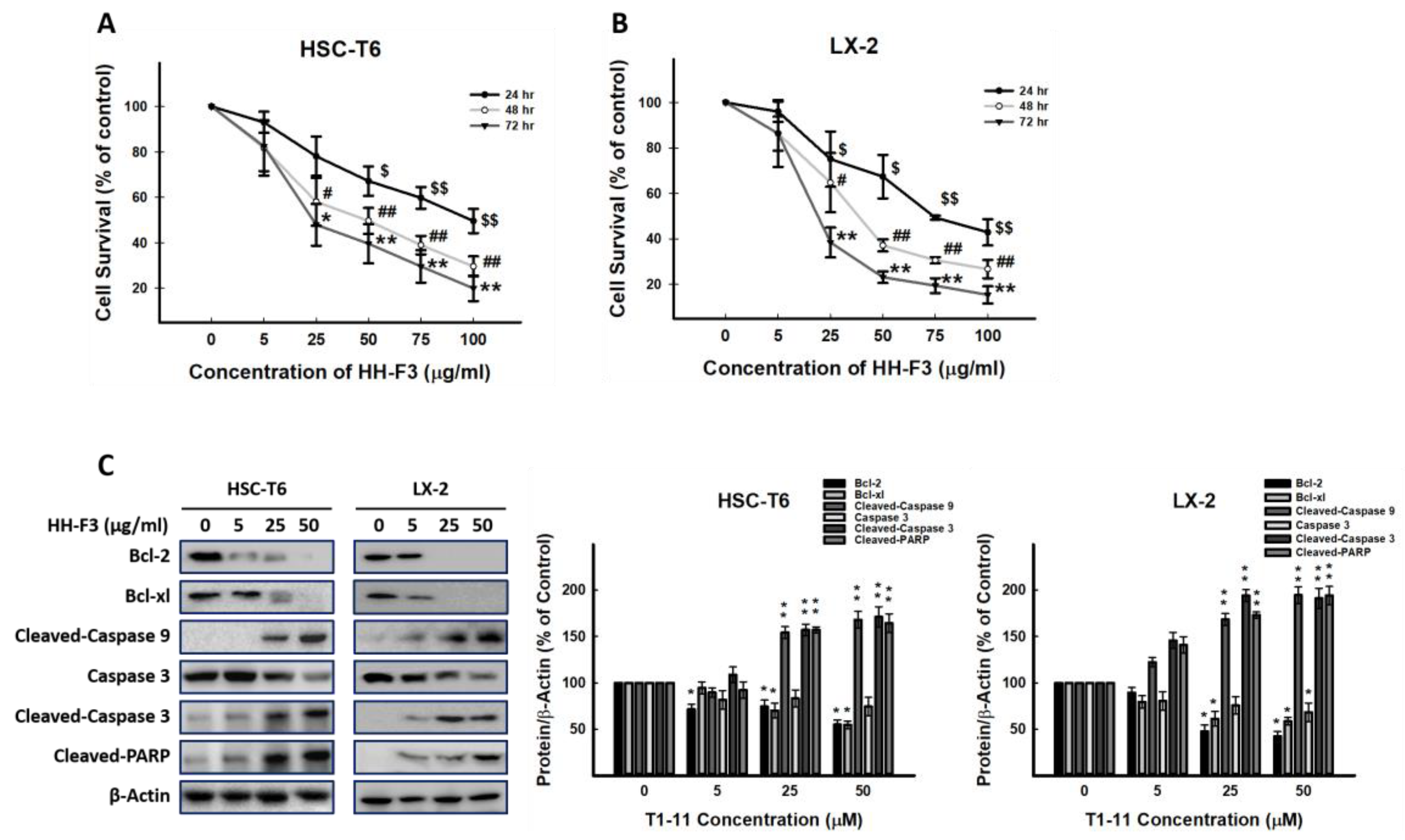
2.1. The Effects of Different GP Preparations on HSC-T6 Cells
2.2. HH-F3 Reduces the Viability of Hepatic Stellate Cells and Has No Cytotoxic Effect on Hepatocytes
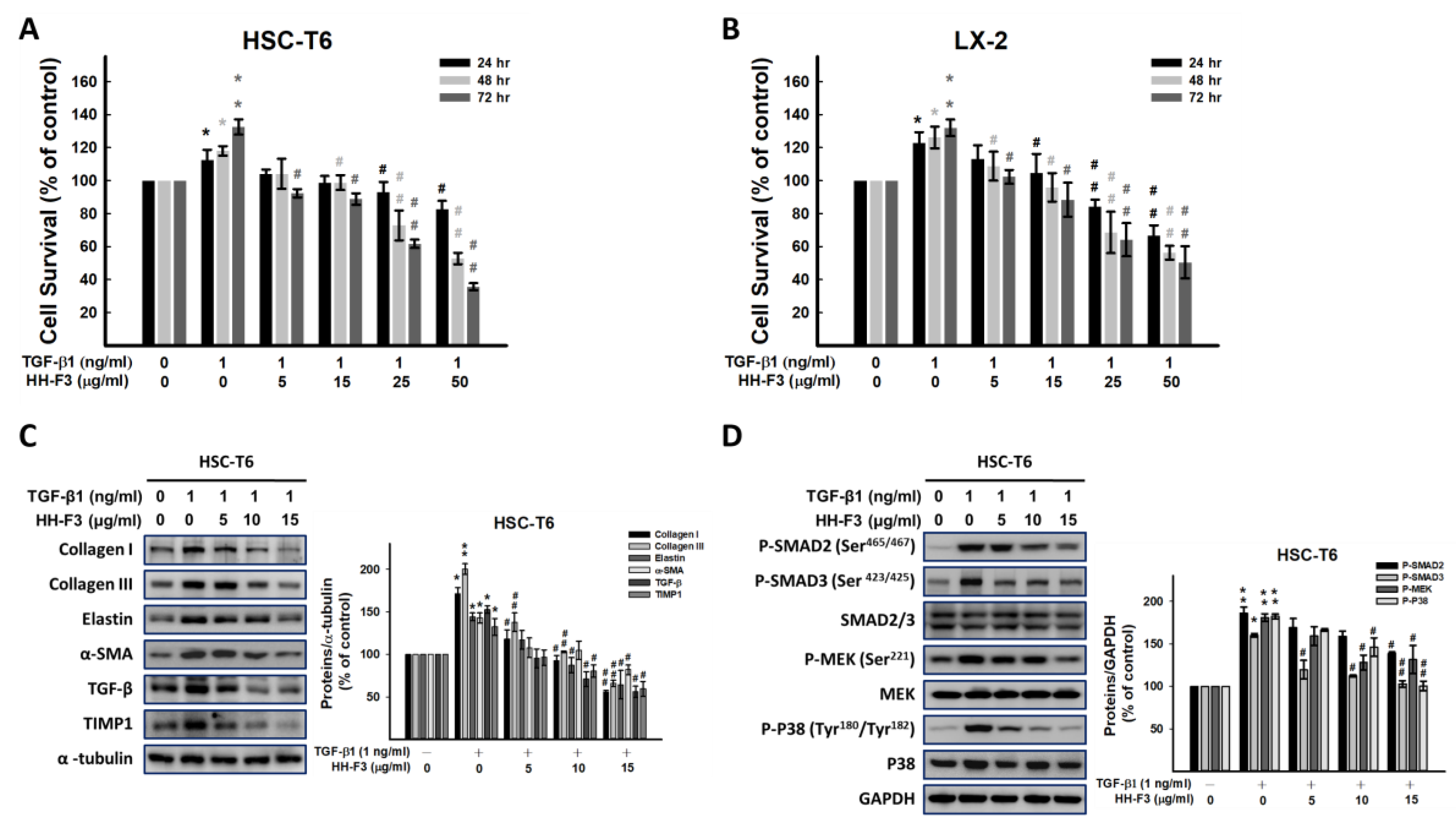
2.3. GP/HH-F3 Inhibits TGF-β1-Induced HSC-T6 Cell Proliferation
2.4. GP/HH-F3 Inhibit the TGF-β1-Induced HSC Activation of Collagen Matrix Expression and the TGF-β Signaling Pathway
2.5. HH-F3 Inhibits TGF-β1-Induced Cell Migration/Invasion
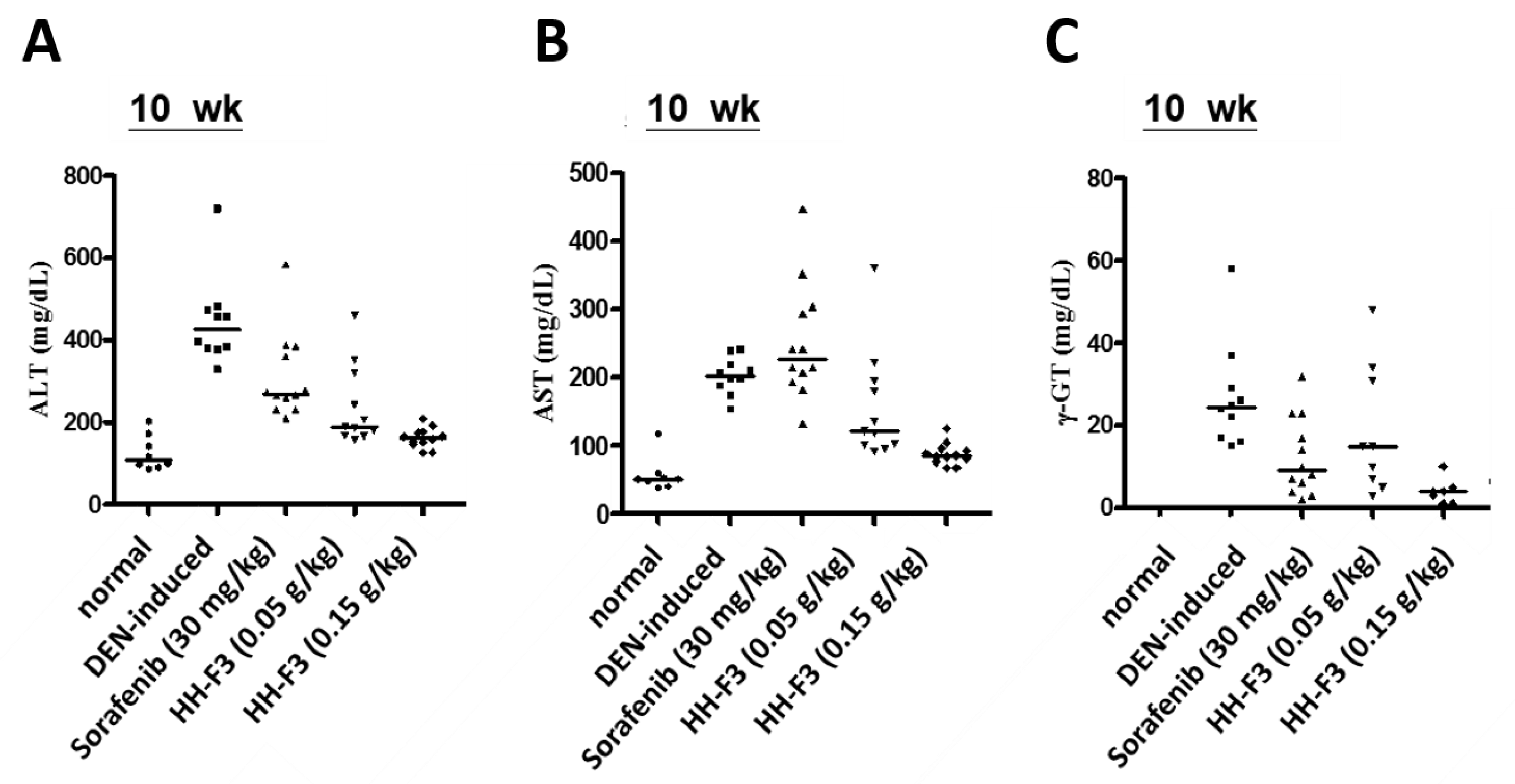
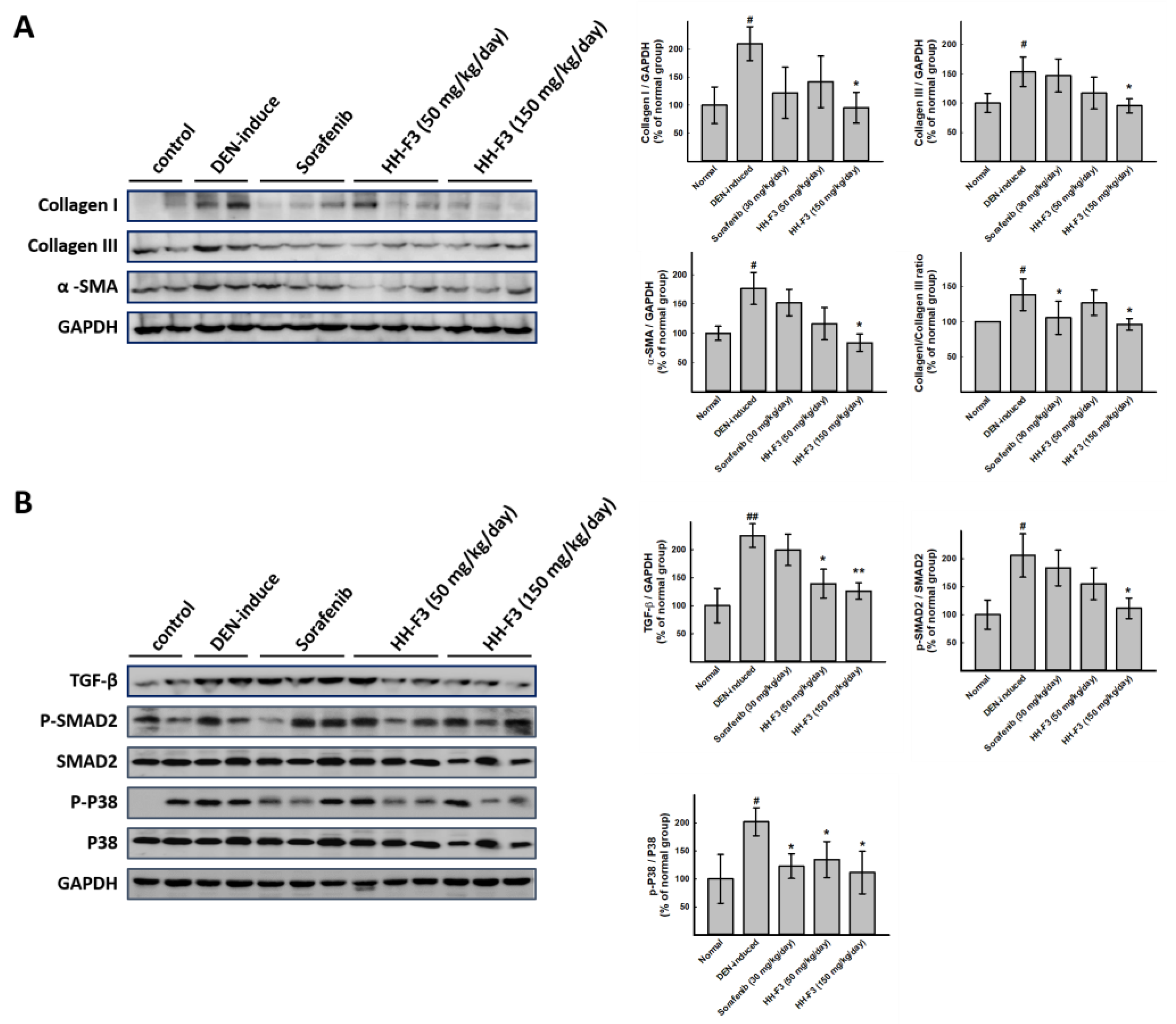
2.6. HH-F3 Effectively Alleviates DEN-Mediated Liver Injury and Fibrosis In Vivo
3. Discussion
4. Methods
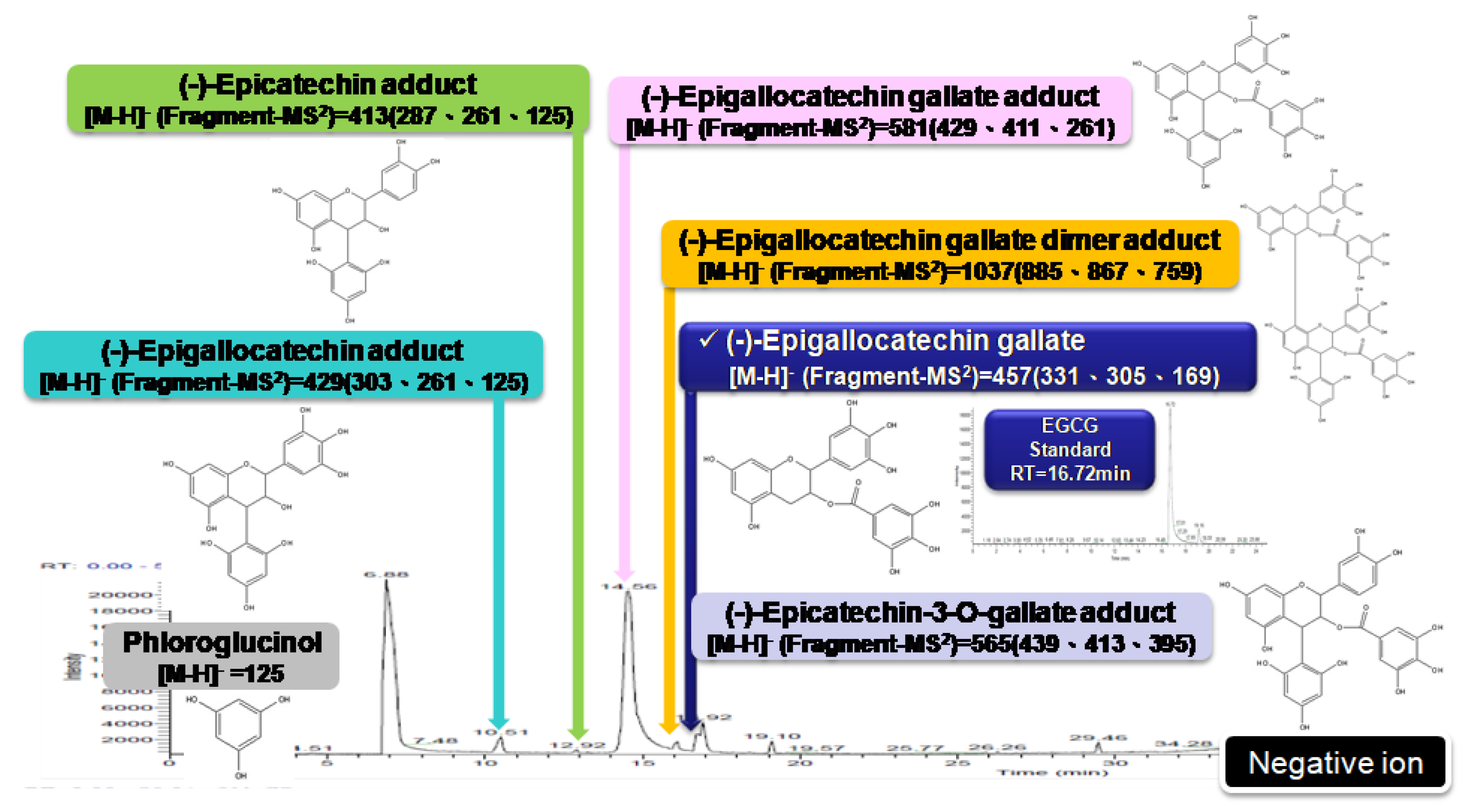
4.1. GP Extraction and HH-F3 Purification
4.2. Cell Culture
4.3. MTT Assay
4.4. Cell Counting
4.5. Sulforhodamine B (SRB) Assay
4.6. Western Blot
4.7. Transwell Cell Migration/Invasion Assay
4.8. Wound-Healing Assay
4.9. Animal Model
4.10. Ethics Statement
4.11. Hepatic Enzyme Measurement
4.12. Histological Analysis
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Alanine aminotransferase | ALT |
| Alcoholic steatohepatitis | ASH |
| Alpha-smooth muscle actin | α-SMA |
| Aspartate aminotransferase | AST |
| Aurora kinase B | AURKB |
| Diethylnitrosamine | DEN |
| Dimethyl sulfoxide | DMSO |
| Dimethylnitrosamine | DMN |
| Dulbecco’s Modified Eagle medium | DMEM |
| Epidermal growth factor receptor | EGFR |
| Extracellular matrix | ECM |
| Graptopetalum paraguayense | GP |
| Hepatic stellate cell | HSC |
| Hepatitis B virus | HBV |
| Hepatitis C virus | HCV |
| Hepatocellular carcinoma | HCC |
| Horseradish peroxidase | HRP |
| Lipid peroxidation | LPO |
| Matrix-assisted laser desorption Ionization-time of flight | MALDI-TOF |
| Matrix metalloproteinase | MMP |
| Methanolic extract of GP | MGP |
| Mitogen-activated protein kinase | MAPK |
| Mitogen-activated protein kinase kinase | MEK |
| Nonalcoholic fatty liver disease | NAFLD |
| Nonalcoholic steatohepatitis | NASH |
| Optical density | OD |
| Peroxisome proliferator-activated receptor Coactivator 1-alpha | PGC-1α |
| Platelet derived growth factor | PDGF |
| Polyvinylidene difluoride | PVDF |
| Reactive oxygen species | ROS |
| Sulforhodamine B | SRB |
| Trichloroacetic acid | TCA |
| Tissue inhibitor of metalloproteinase | TIMP |
| Transforming growth factor beta | TGF-β |
| Trichloroacetic acid | TCA |
| γ-Glutamyltransferase | γ-GT |
References
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Invest. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D. Structure of the extracellular matrix in normal and fibrotic liver: Collagens and glycoproteins. Semin. Liver Dis. 1990, 10, 1–10. [Google Scholar] [CrossRef]
- Wanless, I.R. Pathogenesis of cirrhosis. J. Gastroenterol. Hepatol. 2004, 19, S369–S371. [Google Scholar] [CrossRef]
- Maher, J.J.; McGuire, R.F. Extracellular matrix gene expression increases preferentially in rat lipocytes and sinusoidal endothelial cells during hepatic fibrosis in vivo. J. Clin. Invest. 1990, 86, 1641–1648. [Google Scholar] [CrossRef]
- Milani, S.; Herbst, H.; Schuppan, D.; Kim, K.Y.; Riecken, E.O.; Stein, H. Procollagen expression by nonparenchymal rat liver cells in experimental biliary fibrosis. Gastroenterology 1990, 98, 175–184. [Google Scholar] [CrossRef]
- Moreira, R.K. Hepatic stellate cells and liver fibrosis. Arch. Pathol. Lab. Med. 2007, 131, 1728–1734. [Google Scholar]
- Shek, F.W.; Benyon, R.C. How can transforming growth factor beta be targeted usefully to combat liver fibrosis? Eur. J. Gastroenterol. Hepatol. 2004, 16, 123–126. [Google Scholar] [CrossRef]
- Wiercinska, E.; Wickert, L.; Denecke, B.; Said, H.M.; Hamzavi, J.; Gressner, A.M.; Thorikay, M.; ten Dijke, P.; Mertens, P.R.; Breitkopf, K.; et al. Id1 is a critical mediator in TGF-beta-induced transdifferentiation of rat hepatic stellate cells. Hepatology 2006, 43, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Schnabl, B.; Kweon, Y.O.; Frederick, J.P.; Wang, X.F.; Rippe, R.A.; Brenner, D.A. The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology 2001, 34, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Dooley, S.; Hamzavi, J.; Breitkopf, K.; Wiercinska, E.; Said, H.M.; Lorenzen, J.; Ten Dijke, P.; Gressner, A.M. Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology 2003, 125, 178–191. [Google Scholar] [CrossRef]
- Huang, K.F.; Chen, Y.W.; Chang, C.T.; Chou, S.T. Studies on the inhibitory effect of Graptopetalum paraguayense E. Walther extracts on mushroom tyrosinase. Food Chem. 2005, 89, 583–587. [Google Scholar] [CrossRef]
- Chung, Y.C.; Chen, S.J.; Hsu, C.K.; Chang, C.T.; Chou, S.T. Studies on the antioxidative activity of Graptopetalum paraguayense E. Walther. Food Chem. 2005, 91, 419–424. [Google Scholar] [CrossRef]
- Chen, S.J.; Chang, C.T.; Chung, Y.C.; Chou, S.T. Studies on the inhibitory effect of Graptopetalum paraguayense E. Walther extracts on the angiotensin converting enzyme. Food Chem. 2007, 100, 1032–1036. [Google Scholar] [CrossRef]
- Chung, Y.C.; Chen, S.J.; Peng, H.Y.; Chou, S.T. Antihypertensive and antioxidant effects of the Graptopetalum paraguayense E. Walther extract in spontaneously hypertensive rats. J. Sci. Food Agric. 2009, 89, 2678–2686. [Google Scholar] [CrossRef]
- Duh, P.D.; Lin, S.L.; Wu, S.C. Hepatoprotection of Graptopetalum paraguayense E. Walther on CCl4-induced liver damage and inflammation. J. Ethnopharmacol. 2011, 134, 379–385. [Google Scholar] [CrossRef]
- Chen, S.J.; Chung, J.G.; Chung, Y.C.; Chou, S.T. In vitro antioxidant and antiproliferative activity of the stem extracts from Graptopetalum paraguayense. Am. J. Chin. Med. 2008, 36, 369–383. [Google Scholar] [CrossRef]
- Hsu, W.H.; Chang, C.C.; Huang, K.W.; Chen, Y.C.; Hsu, S.L.; Wu, L.C.; Tsou, A.P.; Lai, J.M.; Huang, C.Y. Evaluation of the medicinal herb Graptopetalum paraguayense as a treatment for liver cancer. PloS ONE 2015, 10, e0121298. [Google Scholar] [CrossRef]
- Jhuang, H.J.; Hsu, W.H.; Lin, K.T.; Hsu, S.L.; Wang, F.S.; Chou, C.K.; Lee, K.H.; Tsou, A.P.; Lai, J.M.; Yeh, S.F.; et al. Gluconeogenesis, lipogenesis, and HBV replication are commonly regulated by PGC-1alpha-dependent pathway. Oncotarget 2015, 6, 7788–7803. [Google Scholar] [CrossRef]
- Su, L.J.; Chang, C.C.; Yang, C.H.; Hsieh, S.J.; Wu, Y.C.; Lai, J.M.; Tseng, T.L.; Huang, C.Y.; Hsu, S.L. Graptopetalum paraguayense ameliorates chemical-induced rat hepatic fibrosis in vivo and inactivates stellate cells and Kupffer cells in vitro. PloS ONE 2013, 8, e53988. [Google Scholar] [CrossRef] [PubMed]
- Su, L.J.; Yang, C.H.; Huang, S.F.; Yuo, Y.L.; Hsieh, H.C.; Tseng, T.L.; Chen, C.H.; Hsu, S.L.; Huang, C.Y. Evaluation of the Chinese Medicinal Herb, Graptopetalum paraguayense, as a Therapeutic Treatment for Liver Damage in Rat Models. Evid. Based Complement. Altern. Med. 2012, 2012, 256561. [Google Scholar] [CrossRef] [PubMed]
- Meindl-Beinker, N.M.; Dooley, S. Transforming growth factor-beta and hepatocyte transdifferentiation in liver fibrogenesis. J. Gastroenterol. Hepatol. 2008, 23, S122–S127. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.X.; He, R.H.; Yang, G.; Tan, J.J.; Zhou, L.; Meng, X.M.; Huang, X.R.; Lan, H.Y. Asiatic acid inhibits liver fibrosis by blocking TGF-beta/Smad signaling in vivo and in vitro. PloS ONE 2012, 7, e31350. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, J.H.; Joo, C.K. TGF-beta1 regulates cell fate during epithelial-mesenchymal transition by upregulating survivin. Cell Death Dis. 2013, 4, e714. [Google Scholar] [CrossRef] [PubMed]
- Latella, G.; Vetuschi, A.; Sferra, R.; Catitti, V.; D’Angelo, A.; Zanninelli, G.; Flanders, K.C.; Gaudio, E. Targeted disruption of Smad3 confers resistance to the development of dimethylnitrosamine-induced hepatic fibrosis in mice. Liver Int. 2009, 29, 997–1009. [Google Scholar] [CrossRef]
- Dooley, S.; Hamzavi, J.; Ciuclan, L.; Godoy, P.; Ilkavets, I.; Ehnert, S.; Ueberham, E.; Gebhardt, R.; Kanzler, S.; Geier, A.; et al. Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediated fibrogenesis and protects against liver damage. Gastroenterology 2008, 135, 642–659. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Jacobson, K.; Schaller, M.D. MAP kinases and cell migration. J. Cell Sci. 2004, 117, 4619–4628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saika, S.; Okada, Y.; Miyamoto, T.; Yamanaka, O.; Ohnishi, Y.; Ooshima, A.; Liu, C.Y.; Weng, D.; Kao, W.W. Role of p38 MAP kinase in regulation of cell migration and proliferation in healing corneal epithelium. Invest. Ophthalmol. Vis. Sci. 2004, 45, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.W.; Huang, Y.C.; Tai, K.F.; Chen, B.H.; Lee, P.H.; Hwang, L.H. Dual Therapeutic Effects of Interferon-alpha Gene Therapy in a Rat Hepatocellular Carcinoma Model With Liver Cirrhosis. Mol. Ther. 2008, 16, 1681–1687. [Google Scholar] [CrossRef]
- Ma, R.; Chen, J.; Liang, Y.; Lin, S.; Zhu, L.; Liang, X.; Cai, X. Sorafenib: A potential therapeutic drug for hepatic fibrosis and its outcomes. Biomed. Pharm. 2017, 88, 459–468. [Google Scholar] [CrossRef]
- Stefano, J.T.; Pereira, I.V.; Torres, M.M.; Bida, P.M.; Coelho, A.M.; Xerfan, M.P.; Cogliati, B.; Barbeiro, D.F.; Mazo, D.F.; Kubrusly, M.S.; et al. Sorafenib prevents liver fibrosis in a non-alcoholic steatohepatitis (NASH) rodent model. Braz. J. Med. Biol Res. 2015, 48, 408–414. [Google Scholar] [CrossRef] [Green Version]
- Prosser, C.C.; Yen, R.D.; Wu, J. Molecular therapy for hepatic injury and fibrosis: Where are we? World J. Gastroenterol. 2006, 12, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Gressner, O.A.; Weiskirchen, R.; Gressner, A.M. Biomarkers of liver fibrosis: Clinical translation of molecular pathogenesis or based on liver-dependent malfunction tests. Clin. Chim. Acta 2007, 381, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Iwaisako, K.; Brenner, D.A.; Kisseleva, T. What’s new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. J. Gastroenterol. Hepatol. 2012, 27, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Huseini, H.F.; Alavian, S.M.; Heshmat, R.; Heydari, M.R.; Abolmaali, K. The efficacy of Liv-52 on liver cirrhotic patients: A randomized, double-blind, placebo-controlled first approach. Phytomedicine 2005, 12, 619–624. [Google Scholar] [CrossRef]
- Tsai, J.H.; Liu, J.Y.; Wu, T.T.; Ho, P.C.; Huang, C.Y.; Shyu, J.C.; Hsieh, Y.S.; Tsai, C.C.; Liu, Y.C. Effects of silymarin on the resolution of liver fibrosis induced by carbon tetrachloride in rats. J. Viral. Hepat. 2008, 15, 508–514. [Google Scholar] [CrossRef]
- Lee, J.K.; Kim, J.H.; Shin, H.K. Therapeutic effects of the oriental herbal medicine Sho-saiko-to on liver cirrhosis and carcinoma. Hepatol. Res. 2011, 41, 825–837. [Google Scholar] [CrossRef]
- Takahashi, Y.; Soejima, Y.; Kumagai, A.; Watanabe, M.; Uozaki, H.; Fukusato, T. Inhibitory effects of Japanese herbal medicines sho-saiko-to and juzen-taiho-to on nonalcoholic steatohepatitis in mice. PloS ONE 2014, 9, e87279. [Google Scholar] [CrossRef]
- Lin, Y.L.; Wu, C.H.; Luo, M.H.; Huang, Y.J.; Wang, C.N.; Shiao, M.S.; Huang, Y.T. In vitro protective effects of salvianolic acid B on primary hepatocytes and hepatic stellate cells. J. Ethnopharmacol. 2006, 105, 215–222. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Vallee, A.; Lecarpentier, Y.; Vallee, J.N. Thermodynamic Aspects and Reprogramming Cellular Energy Metabolism during the Fibrosis Process. Int. J. Mol. Sci. 2017, 18, 2537. [Google Scholar] [CrossRef]
- Leask, A.; Abraham, D.J. TGF-beta signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, Y.; Okazaki, I. Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut 2007, 56, 284–292. [Google Scholar] [CrossRef]
- Rippe, R.A.; Brenner, D.A. From quiescence to activation: Gene regulation in hepatic stellate cells. Gastroenterology 2004, 127, 1260–1262. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.M.; Gaston Pravia, K.A. Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radic. Biol. Med. 2010, 48, 1–15. [Google Scholar] [CrossRef]
- Liu, X.; Hu, H.; Yin, J.Q. Therapeutic strategies against TGF-beta signaling pathway in hepatic fibrosis. Liver Int. 2006, 26, 8–22. [Google Scholar] [CrossRef]
- Lindert, S.; Wickert, L.; Sawitza, I.; Wiercinska, E.; Gressner, A.M.; Dooley, S.; Breitkopf, K. Transdifferentiation-dependent expression of alpha-SMA in hepatic stellate cells does not involve TGF-beta pathways leading to coinduction of collagen type I and thrombospondin-2. Matrix Biol. 2005, 24, 198–207. [Google Scholar] [CrossRef]
- Yang, C.; Zeisberg, M.; Mosterman, B.; Sudhakar, A.; Yerramalla, U.; Holthaus, K.; Xu, L.; Eng, F.; Afdhal, N.; Kalluri, R. Liver fibrosis: Insights into migration of hepatic stellate cells in response to extracellular matrix and growth factors. Gastroenterology 2003, 124, 147–159. [Google Scholar] [CrossRef]
- Tolba, R.; Kraus, T.; Liedtke, C.; Schwarz, M.; Weiskirchen, R. Diethylnitrosamine (DEN)-induced carcinogenic liver injury in mice. Lab. Anim. 2015, 49, 59–69. [Google Scholar] [CrossRef] [Green Version]
- de Lima, V.M.; Oliveira, C.P.; Alves, V.A.; Chammas, M.C.; Oliveira, E.P.; Stefano, J.T.; de Mello, E.S.; Cerri, G.G.; Carrilho, F.J.; Caldwell, S.H. A rodent model of NASH with cirrhosis, oval cell proliferation and hepatocellular carcinoma. J. Hepatol. 2008, 49, 1055–1061. [Google Scholar] [CrossRef]
- Berasain, C.; Castillo, J.; Prieto, J.; Avila, M.A. New molecular targets for hepatocellular carcinoma: The ErbB1 signaling system. Liver Int. 2007, 27, 174–185. [Google Scholar] [CrossRef]
- Xu, Y.; Li, N.; Xiang, R.; Sun, P. Emerging roles of the p38 MAPK and PI3K/AKT/mTOR pathways in oncogene-induced senescence. Trends Biochem. Sci. 2014, 39, 268–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.L.; Wu, C.F.; Huang, Y.T. Effects of rhubarb on migration of rat hepatic stellate cells. J. Gastroenterol. Hepatol 2009, 24, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.G.; Wu, X.; Guan, J.L. Wound-healing assay. Methods Mol. Biol. 2005, 294, 23–29. [Google Scholar]
- Li, B.; Chen, D.; Li, W.; Xiao, D. 20(S)-Protopanaxadiol saponins inhibit SKOV3 cell migration. Oncol. Lett. 2016, 11, 1693–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, W.-H.; Liao, S.-C.; Chyan, Y.-J.; Huang, K.-W.; Hsu, S.-L.; Chen, Y.-C.; Siu, M.-L.; Chang, C.-C.; Chung, Y.-S.; Huang, C.-Y.F. Graptopetalum paraguayense Inhibits Liver Fibrosis by Blocking TGF-β Signaling In Vivo and In Vitro. Int. J. Mol. Sci. 2019, 20, 2592. https://doi.org/10.3390/ijms20102592
Hsu W-H, Liao S-C, Chyan Y-J, Huang K-W, Hsu S-L, Chen Y-C, Siu M-L, Chang C-C, Chung Y-S, Huang C-YF. Graptopetalum paraguayense Inhibits Liver Fibrosis by Blocking TGF-β Signaling In Vivo and In Vitro. International Journal of Molecular Sciences. 2019; 20(10):2592. https://doi.org/10.3390/ijms20102592
Chicago/Turabian StyleHsu, Wei-Hsiang, Se-Chun Liao, Yau-Jan Chyan, Kai-Wen Huang, Shih-Lan Hsu, Yi-Chen Chen, Ma-Li Siu, Chia-Chuan Chang, Yuh-Shan Chung, and Chi-Ying F. Huang. 2019. "Graptopetalum paraguayense Inhibits Liver Fibrosis by Blocking TGF-β Signaling In Vivo and In Vitro" International Journal of Molecular Sciences 20, no. 10: 2592. https://doi.org/10.3390/ijms20102592
APA StyleHsu, W.-H., Liao, S.-C., Chyan, Y.-J., Huang, K.-W., Hsu, S.-L., Chen, Y.-C., Siu, M.-L., Chang, C.-C., Chung, Y.-S., & Huang, C.-Y. F. (2019). Graptopetalum paraguayense Inhibits Liver Fibrosis by Blocking TGF-β Signaling In Vivo and In Vitro. International Journal of Molecular Sciences, 20(10), 2592. https://doi.org/10.3390/ijms20102592