(E)-2-methoxy-4-(3-(4-methoxyphenyl) prop-1-en-1-yl) Phenol Ameliorates MPTP-Induced Dopaminergic Neurodegeneration by Inhibiting the STAT3 Pathway



Abstract
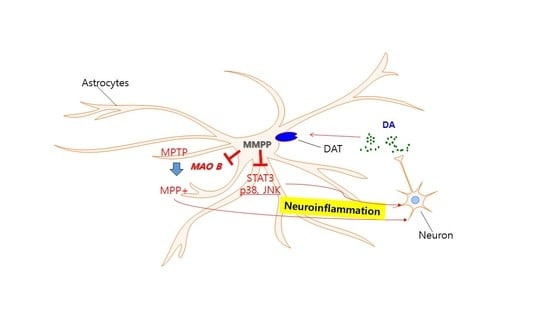
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Effect of MMPP in MPTP-Induced Behavioral Impairments
2.2. Effect of MMPP on the Expression of Astrogliosis and Microgliosis-Related Proteins
2.3. Effect of MMPP on Dopaminergic Neurodegeneration
2.4. Effect of MMPP on MAO-B Expression and Activation of STAT3
2.5. Effect of MMPP on MPP+-Induced Inflammation in Primary Cultured Cells
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Materials
4.3. MPTP Injections
4.4. Rotarod Test
4.5. Pole Test
4.6. Gait Test
4.7. Immunohistochemical Staining
4.8. HPLC Analysis of Dopamine and DOPAC
4.9. Nuclear Extraction and Gel Mobility Shift Assay
4.10. Primary Cell Culture
4.11. Western Blotting
4.12. Cell Viability Assay
4.13. MAO-B Activity Assay
4.14. Luciferase Assay
4.15. Statistical Analysis
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| MMPP | (E)-2-methoxy-4-(3-(4-methoxyphenyl) prop-1-en-1-yl) phenol |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MPP+ | 1-methyl-4-phenylpyridinium |
| STAT3 | signal transducer and activator of transcription 3 |
| Iba1 | including ionized calcium binding adaptor molecule 1 |
| iNOS | inducible nitric oxide synthase |
| GFAP | glial fibrillary acidic protein |
| MAPK | mitogen-activated protein kinase |
| PD | Parkinson’s Disease |
| DA | Dopamine |
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis |
| JNK | c-Jun N-terminal kinases |
| BHPB | (E)-2,4-bis(p-hydroxyphenyl)-2-butenal |
| DOPAC | 3,4-Dihydroxyphenylacetic acid |
| MAO-B | monoamine oxidase B |
| WDI | World Drug Index |
| CMC | chemistry, manufacturing, and control |
References
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D. The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2014, 29, 1583–1590. [Google Scholar] [CrossRef]
- Binukumar, B.K.; Pant, H.C. TFP5/TP5 peptide provides neuroprotection in the MPTP model of Parkinson’s disease. Neural Regen. Res. 2016, 11, 698–701. [Google Scholar]
- Javed, H.; Kamal, M.A.; Ojha, S. An overview on the role of alpha-synuclein in experimental models of Parkinson’s disease from pathogenesis to therapeutics. Cns Neurol. Disord. Drug Targets 2016, 15, 1240–1252. [Google Scholar] [CrossRef]
- Klemann, C.J.; Martens, G.J.; Poelmans, G.; Visser, J.E. Validity of the MPTP-Treated Mouse as a Model for Parkinson’s Disease. Mol. Neurobiol. 2016, 53, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Meredith, G.E.; Rademacher, D.J. MPTP mouse models of Parkinson’s disease: An update. J. Parkinson’s Dis. 2011, 1, 19–33. [Google Scholar]
- Bahat-Stroomza, M.; Gilgun-Sherki, Y.; Offen, D.; Panet, H.; Saada, A.; Krool-Galron, N.; Barzilai, A.; Atlas, D.; Melamed, E. A novel thiol antioxidant that crosses the blood brain barrier protects dopaminergic neurons in experimental models of Parkinson’s disease. Eur. J. Neurosci. 2005, 21, 637–646. [Google Scholar] [CrossRef]
- Inazu, M.; Kubota, N.; Takeda, H.; Oguchi, K.; Koizumi, M.; Kimura, S.; Matsumiya, T. Methyl-4-phenylpyridinium (MPP(+))-evoked dopamine release from rat striatal slices: Possible roles of voltage-dependent calcium channels and reverse dopamine transport. Neurochem. Int. 2001, 39, 253–260. [Google Scholar] [CrossRef]
- Smeyne, R.J.; Jackson-Lewis, V. The MPTP model of Parkinson’s disease. Brain Res. Mol. Brain Res. 2005, 134, 57–66. [Google Scholar] [CrossRef]
- Boger, H.A.; Middaugh, L.D.; Zaman, V.; Hoffer, B.; Granholm, A.C. Differential effects of the dopamine neurotoxin MPTP in animals with a partial deletion of the GDNF receptor, GFR alpha1, gene. Brain Res. 2008, 1241, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Sedelis, M.; Schwarting, R.K.; Huston, J.P. Behavioral phenotyping of the MPTP mouse model of Parkinson’s disease. Behav. Brain Res. 2001, 125, 109–125. [Google Scholar] [CrossRef]
- O’Callaghan, J.P.; Kelly, K.A.; VanGilder, R.L.; Sofroniew, M.V.; Miller, D.B. Early activation of STAT3 regulates reactive astrogliosis induced by diverse forms of neurotoxicity. PLoS ONE 2014, 9, e102003. [Google Scholar] [CrossRef]
- Sriram, K.; Benkovic, S.A.; Hebert, M.A.; Miller, D.B.; O’Callaghan, J.P. Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration: Key signaling pathway for astrogliosis in vivo? J. Biol. Chem. 2004, 279, 19936–19947. [Google Scholar]
- Di Domenico, F.; Casalena, G.; Sultana, R.; Cai, J.; Pierce, W.M.; Perluigi, M.; Cini, C.; Baracca, A.; Solaini, G.; Lenaz, G.; et al. Involvement of Stat3 in mouse brain development and sexual dimorphism: A proteomics approach. Brain Res. 2010, 1362, 1–12. [Google Scholar] [CrossRef]
- Sonawane, P.J.; Gupta, V.; Sasi, B.K.; Kalyani, A.; Natarajan, B.; Khan, A.A.; Sahu, B.S.; Mahapatra, N.R. Transcriptional regulation of the novel monoamine oxidase renalase: Crucial roles of transcription factors Sp1, STAT3, and ZBP89. Biochemistry 2014, 53, 6878–6892. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Shukla, M.; Yakubenko, V.P.; Mulya, A.; Kundu, S.; Cathcart, M.K. IL-4 and IL-13 employ discrete signaling pathways for target gene expression in alternatively activated monocytes/macrophages. Free Radic. Biol. Med. 2013, 54, 1–16. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhu, F.; Pu, Y.; Wang, D.; Huang, A.; Hu, X.; Qin, S.; Sun, X.; Su, Z.; He, C. Neuroprotective effects of nitidine against traumatic CNS injury via inhibiting microglia activation. BrainBehav. Immun. 2015, 48, 287–300. [Google Scholar] [CrossRef]
- Peng, J.; Andersen, J.K. The role of c-Jun N-terminal kinase (JNK) in Parkinson’s disease. Iubmb Life 2003, 55, 267–271. [Google Scholar] [CrossRef]
- Correa, S.A.; Eales, K.L. The Role of p38 MAPK and Its Substrates in Neuronal Plasticity and Neurodegenerative Disease. J. Signal Transduct. 2012, 2012, 649079. [Google Scholar] [CrossRef]
- Jha, S.K.; Jha, N.K.; Kar, R.; Ambasta, R.K.; Kumar, P. p38 MAPK and PI3K/AKT Signalling Cascades inParkinson’s Disease. Int. J. Mol. Cell. Med. 2015, 4, 67–86. [Google Scholar]
- Takeda, K.; Ichijo, H. Neuronal p38 MAPK signalling: An emerging regulator of cell fate and function in the nervous system. Genes Cells 2002, 7, 1099–1111. [Google Scholar] [CrossRef]
- Toulouse, A.; Nolan, Y.M. A role for mitogen-activated protein kinase phosphatase 1 (MKP1) in neural cell development and survival. Neural Regen. Res. 2015, 10, 1748–1749. [Google Scholar] [CrossRef]
- Hunot, S.; Vila, M.; Teismann, P.; Davis, R.J.; Hirsch, E.C.; Przedborski, S.; Rakic, P.; Flavell, R.A. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 665–670. [Google Scholar] [CrossRef]
- Wong, W.K.; Ou, X.M.; Chen, K.; Shih, J.C. Activation of human monoamine oxidase B gene expression by a protein kinase C MAPK signal transduction pathway involves c-Jun and Egr-1. J. Biol. Chem. 2002, 277, 22222–22230. [Google Scholar] [CrossRef]
- Xiong, M.; Wang, L.; Yu, H.L.; Han, H.; Mao, D.; Chen, J.; Zeng, Y.; He, N.; Liu, Z.G.; Wang, Z.Y.; et al. Ginkgetin exerts growth inhibitory and apoptotic effects on osteosarcoma cells through inhibition of STAT3 and activation of caspase-3/9. Oncol. Rep. 2016, 35, 1034–1040. [Google Scholar] [CrossRef]
- Si, Y.; Zhang, Y.; Han, L.; Chen, L.; Xu, Y.; Sun, F.; Ji, M.; Yang, J.; Bao, H. Dexmedetomidine Acts via the JAK2/STAT3 Pathway to Attenuate Isoflurane-Induced Neurocognitive Deficits in Senile Mice. PLoS ONE 2016, 11, e0164763. [Google Scholar] [CrossRef]
- Ban, J.O.; Kim, D.H.; Lee, H.P.; Hwang, C.J.; Shim, J.H.; Kim, D.J.; Kim, T.M.; Jeong, H.S.; Nah, S.S.; Chen, H.; et al. Anti-arthritis effects of (E)-2,4-bis(p-hydroxyphenyl)-2-butenal are mediated by inhibition of the STAT3 pathway. Br. J. Pharmacol. 2014, 171, 2900–2912. [Google Scholar] [CrossRef]
- Cho, S.H.; Park, M.H.; Lee, H.P.; Back, M.K.; Sung, H.C.; Chang, H.W.; Kim, J.H.; Jeong, H.S.; Han, S.B.; Hong, J.T. (E)-2,4-Bis(p-hydroxyphenyl)-2-butenal enhanced TRAIL-induced apoptosis in ovarian cancer cells through downregulation of NF-kappaB/STAT3 pathway. Arch. Pharmacal Res. 2014, 37, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Kim, J.A.; Choi, D.Y.; Lee, Y.J.; Jung, H.S.; Hong, J.T. Anti-inflammatory and anti-amyloidogenic effects of a small molecule, 2,4-bis(p-hydroxyphenyl)-2-butenal in Tg2576 Alzheimer’s disease mice model. J. Neuroinflammation 2013, 10, 2. [Google Scholar] [CrossRef]
- Son, D.J.; Zheng, J.; Jung, Y.Y.; Hwang, C.J.; Lee, H.P.; Woo, J.R.; Baek, S.Y.; Ham, Y.W.; Kang, M.W.; Shong, M.; et al. MMPP Attenuates Non-Small Cell Lung Cancer Growth by Inhibiting the STAT3 DNA-Binding Activity via Direct Binding to the STAT3 DNA-Binding Domain. Theranostics 2017, 7, 4632–4642. [Google Scholar] [CrossRef]
- de Pablos, R.M.; Herrera, A.J.; Espinosa-Oliva, A.M.; Sarmiento, M.; Munoz, M.F.; Machado, A.; Venero, J.L. Chronic stress enhances microglia activation and exacerbates death of nigral dopaminergic neurons under conditions of inflammation. J. Neuroinflammation 2014, 11, 34. [Google Scholar] [CrossRef]
- Gao, H.M.; Liu, B.; Zhang, W.; Hong, J.S. Novel anti-inflammatory therapy for Parkinson’s disease. Trends Pharmacol. Sci. 2003, 24, 395–401. [Google Scholar] [CrossRef]
- Liu, B.; Du, L.; Hong, J.S. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J. Pharmacol. Exp. Ther. 2000, 293, 607–617. [Google Scholar]
- Delgado, M.; Ganea, D. Neuroprotective effect of vasoactive intestinal peptide (VIP) in a mouse model of Parkinson’s disease by blocking microglial activation. Faseb J. 2003, 17, 944–946. [Google Scholar] [CrossRef]
- Hwang, C.J.; Lee, H.P.; Choi, D.Y.; Jeong, H.S.; Kim, T.H.; Lee, T.H.; Kim, Y.M.; Moon, D.B.; Park, S.S.; Kim, S.Y.; et al. Inhibitory effect of thiacremonone on MPTP-induced dopaminergic neurodegeneration through inhibition of p38 activation. Oncotarget 2016, 7, 46943–46958. [Google Scholar] [CrossRef] [Green Version]
- Fricke, I.B.; Viel, T.; Worlitzer, M.M.; Collmann, F.M.; Vrachimis, A.; Faust, A.; Wachsmuth, L.; Faber, C.; Dolle, F.; Kuhlmann, M.T.; et al. 6-hydroxydopamine-induced Parkinson’s disease-like degeneration generates acute microgliosis and astrogliosis in the nigrostriatal system but no bioluminescence imaging-detectable alteration in adult neurogenesis. Eur. J. Neurosci. 2016, 43, 1352–1365. [Google Scholar] [CrossRef]
- Vollbrecht, P.J.; Simmler, L.D.; Blakely, R.D.; Deutch, A.Y. Dopamine denervation of the prefrontal cortex increases expression of the astrocytic glutamate transporter GLT-1. J. Neurochem. 2014, 130, 109–114. [Google Scholar] [CrossRef]
- Jackson-Lewis, V.; Przedborski, S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2007, 2, 141–151. [Google Scholar] [CrossRef]
- Onofrj, M.; Ghilardi, M.F. MPTP induced parkinsonian syndrome: Long term follow-up and neurophysiological study. Ital. J. Neurol. Sci. 1990, 11, 445–458. [Google Scholar] [CrossRef]
- Teo, K.C.; Ho, S.L. Monoamine oxidase-B (MAO-B) inhibitors: Implications for disease-modification in Parkinson’s disease. Transl. Neurodegener 2013, 2, 19. [Google Scholar] [CrossRef]
- Fowler, J.S.; Logan, J.; Volkow, N.D.; Shumay, E.; McCall-Perez, F.; Jayne, M.; Wang, G.J.; Alexoff, D.L.; Apelskog-Torres, K.; Hubbard, B.; et al. Evidence that formulations of the selective MAO-B inhibitor, selegiline, which bypass first-pass metabolism, also inhibit MAO-A in the human brain. Neuropsychopharmacol 2015, 40, 650–657. [Google Scholar] [CrossRef]
- Lecht, S.; Haroutiunian, S.; Hoffman, A.; Lazarovici, P. Rasagiline—A novel MAO B inhibitor in Parkinson’s disease therapy. Ther. Clin. Risk Manag. 2007, 3, 467–474. [Google Scholar]
- Wang, L.W.; Tu, Y.F.; Huang, C.C.; Ho, C.J. JNK signaling is the shared pathway linking neuroinflammation, blood-brain barrier disruption, and oligodendroglial apoptosis in the white matter injury of the immature brain. J. Neuroinflammation 2012, 9, 175. [Google Scholar] [CrossRef]
- De Zutter, G.S.; Davis, R.J. Pro-apoptotic gene expression mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Proc. Natl. Acad. Sci. USA 2001, 98, 6168–6173. [Google Scholar] [CrossRef] [Green Version]
- Ben Haim, L.; Carrillo-de Sauvage, M.A.; Ceyzeriat, K.; Escartin, C. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 278. [Google Scholar] [CrossRef] [Green Version]
- Brzozowski, M.J.; Jenner, P.; Rose, S. Inhibition of i-NOS but not n-NOS protects rat primary cell cultures against MPP(+)-induced neuronal toxicity. J. Neural Transm. 2015, 122, 779–788. [Google Scholar] [CrossRef]
- Turkson, J.; Bowman, T.; Adnane, J.; Zhang, Y.; Djeu, J.Y.; Sekharam, M.; Frank, D.A.; Holzman, L.B.; Wu, J.; Sebti, S.; et al. Requirement for Ras/Rac1-mediated p38 and c-Jun N-terminal kinase signaling in Stat3 transcriptional activity induced by the Src oncoprotein. Mol. Cell. Biol. 1999, 19, 7519–7528. [Google Scholar] [CrossRef]
- Tang, J.; Li, Z.H.; Ge, S.N.; Wang, W.; Mei, X.P.; Wang, W.; Zhang, T.; Xu, L.X.; Li, J.L. The Inhibition of Spinal Astrocytic JAK2-STAT3 Pathway Activation Correlates with the Analgesic Effects of Triptolide in the Rat Neuropathic Pain Model. Evid. -Based Complementary Altern. Med. 2012, 2012, 185167. [Google Scholar] [CrossRef]
- Meng, A.; Zhang, X.; Shi, Y. Role of p38 MAPK and STAT3 in lipopolysaccharide-stimulated mouse alveolar macrophages. Exp. Ther. Med. 2014, 8, 1772–1776. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Hwang, C.J.; Lee, D.Y.; Gu, S.M.; Lee, H.P.; Choi, D.Y.; Oh, K.W.; Han, S.B.; Hong, J.T. (E)-2-Methoxy-4-(3-(4-methoxyphenyl) prop-1-en-1-yl) Phenol Ameliorates LPS-Mediated Memory Impairment by Inhibition of STAT3 Pathway. Neuromolecular Med. 2017, 19, 555–570. [Google Scholar] [CrossRef] [Green Version]
- Son, D.J.; Kim, D.H.; Nah, S.S.; Park, M.H.; Lee, H.P.; Han, S.B.; Venkatareddy, U.; Gann, B.; Rodriguez, K.; Burt, S.R.; et al. Novel synthetic (E)-2-methoxy-4-(3-(4-methoxyphenyl) prop-1-en-1-yl) phenol inhibits arthritis by targeting signal transducer and activator of transcription 3. Sci. Rep. 2016, 6, 36852. [Google Scholar] [CrossRef] [Green Version]
- Hwang, C.J.; Kim, Y.E.; Son, D.J.; Park, M.H.; Choi, D.Y.; Park, P.H.; Hellstrom, M.; Han, S.B.; Oh, K.W.; Park, E.K.; et al. Parkin deficiency exacerbate ethanol-induced dopaminergic neurodegeneration by P38 pathway dependent inhibition of autophagy and mitochondrial function. Redox Biol. 2017, 11, 456–468. [Google Scholar] [CrossRef]
- Lee, Y.J.; Choi, I.S.; Park, M.H.; Lee, Y.M.; Song, J.K.; Kim, Y.H.; Kim, K.H.; Hwang, D.Y.; Jeong, J.H.; Yun, Y.P.; et al. 4-O-Methylhonokiol attenuates memory impairment in presenilin 2 mutant mice through reduction of oxidative damage and inactivation of astrocytes and the ERK pathway. Free Radic. Biol. Med. 2011, 50, 66–77. [Google Scholar] [CrossRef]
- Menzfeld, C.; John, M.; van Rossum, D.; Regen, T.; Scheffel, J.; Janova, H.; Gotz, A.; Ribes, S.; Nau, R.; Borisch, A.; et al. Tyrphostin AG126 exerts neuroprotection in CNS inflammation by a dual mechanism. Glia 2015, 63, 1083–1099. [Google Scholar] [CrossRef] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.Y.; Yun, J.; Hwang, C.J.; Lee, H.P.; Kim, H.D.; Chun, H.; Park, P.-H.; Choi, D.Y.; Han, S.-B.; Hong, J.T. (E)-2-methoxy-4-(3-(4-methoxyphenyl) prop-1-en-1-yl) Phenol Ameliorates MPTP-Induced Dopaminergic Neurodegeneration by Inhibiting the STAT3 Pathway. Int. J. Mol. Sci. 2019, 20, 2632. https://doi.org/10.3390/ijms20112632
Choi JY, Yun J, Hwang CJ, Lee HP, Kim HD, Chun H, Park P-H, Choi DY, Han S-B, Hong JT. (E)-2-methoxy-4-(3-(4-methoxyphenyl) prop-1-en-1-yl) Phenol Ameliorates MPTP-Induced Dopaminergic Neurodegeneration by Inhibiting the STAT3 Pathway. International Journal of Molecular Sciences. 2019; 20(11):2632. https://doi.org/10.3390/ijms20112632
Chicago/Turabian StyleChoi, Ji Yeon, Jaesuk Yun, Chul Ju Hwang, Hee Pom Lee, Hae Deun Kim, Hyungok Chun, Pil-Hoon Park, Dong Young Choi, Sang-Bae Han, and Jin Tae Hong. 2019. "(E)-2-methoxy-4-(3-(4-methoxyphenyl) prop-1-en-1-yl) Phenol Ameliorates MPTP-Induced Dopaminergic Neurodegeneration by Inhibiting the STAT3 Pathway" International Journal of Molecular Sciences 20, no. 11: 2632. https://doi.org/10.3390/ijms20112632
APA StyleChoi, J. Y., Yun, J., Hwang, C. J., Lee, H. P., Kim, H. D., Chun, H., Park, P. -H., Choi, D. Y., Han, S. -B., & Hong, J. T. (2019). (E)-2-methoxy-4-(3-(4-methoxyphenyl) prop-1-en-1-yl) Phenol Ameliorates MPTP-Induced Dopaminergic Neurodegeneration by Inhibiting the STAT3 Pathway. International Journal of Molecular Sciences, 20(11), 2632. https://doi.org/10.3390/ijms20112632