Galectin-9 Induces Mitochondria-Mediated Apoptosis of Esophageal Cancer In Vitro and In Vivo in a Xenograft Mouse Model

 ,
,  , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results
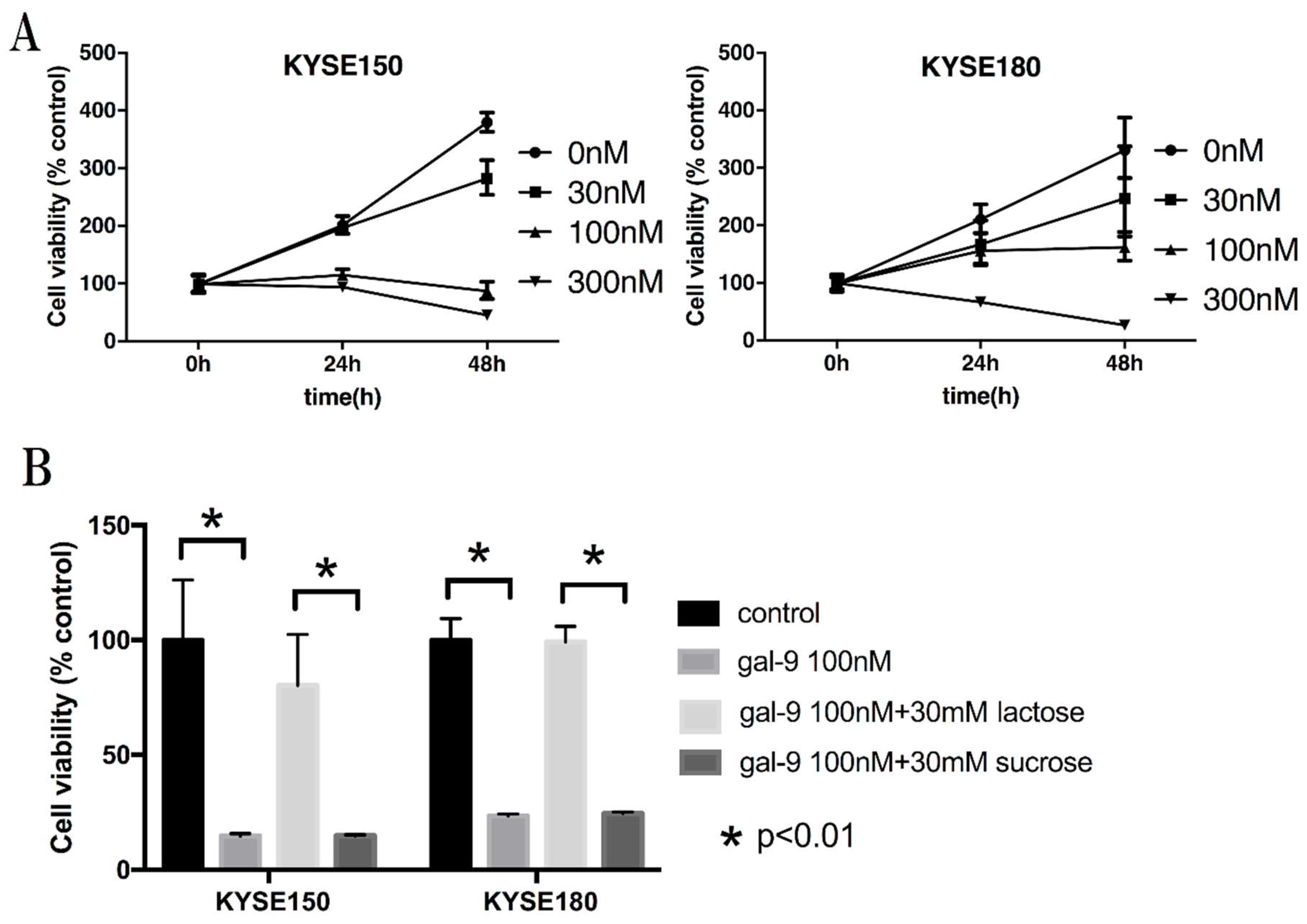
2.1. Galectin-9 (Gal-9) Inhibits the Proliferation of Human Esophageal Squamous Cell Carcinoma (ESCC) Cells In Vitro
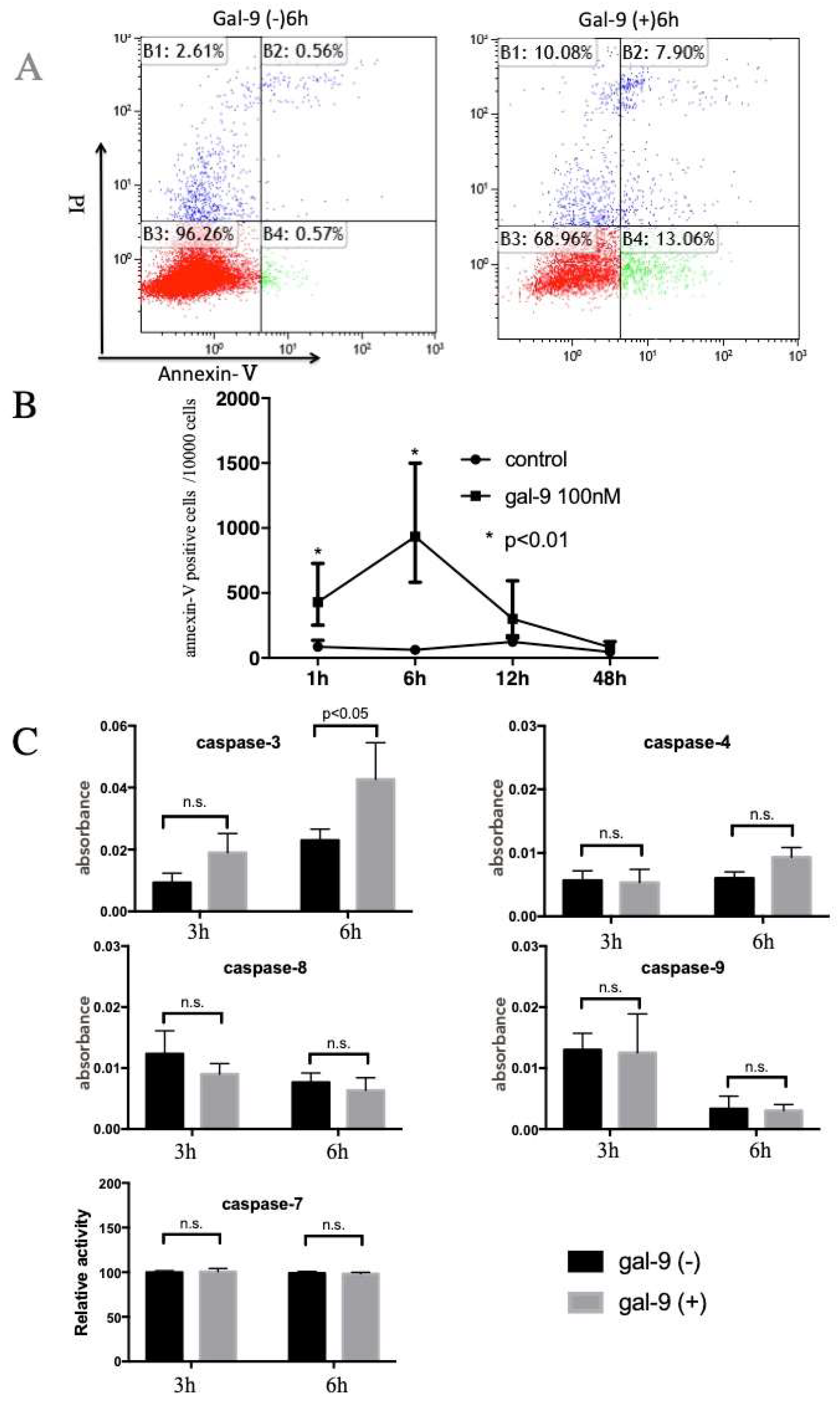
2.2. Gal-9 Induces Apoptosis of ESCC Cells
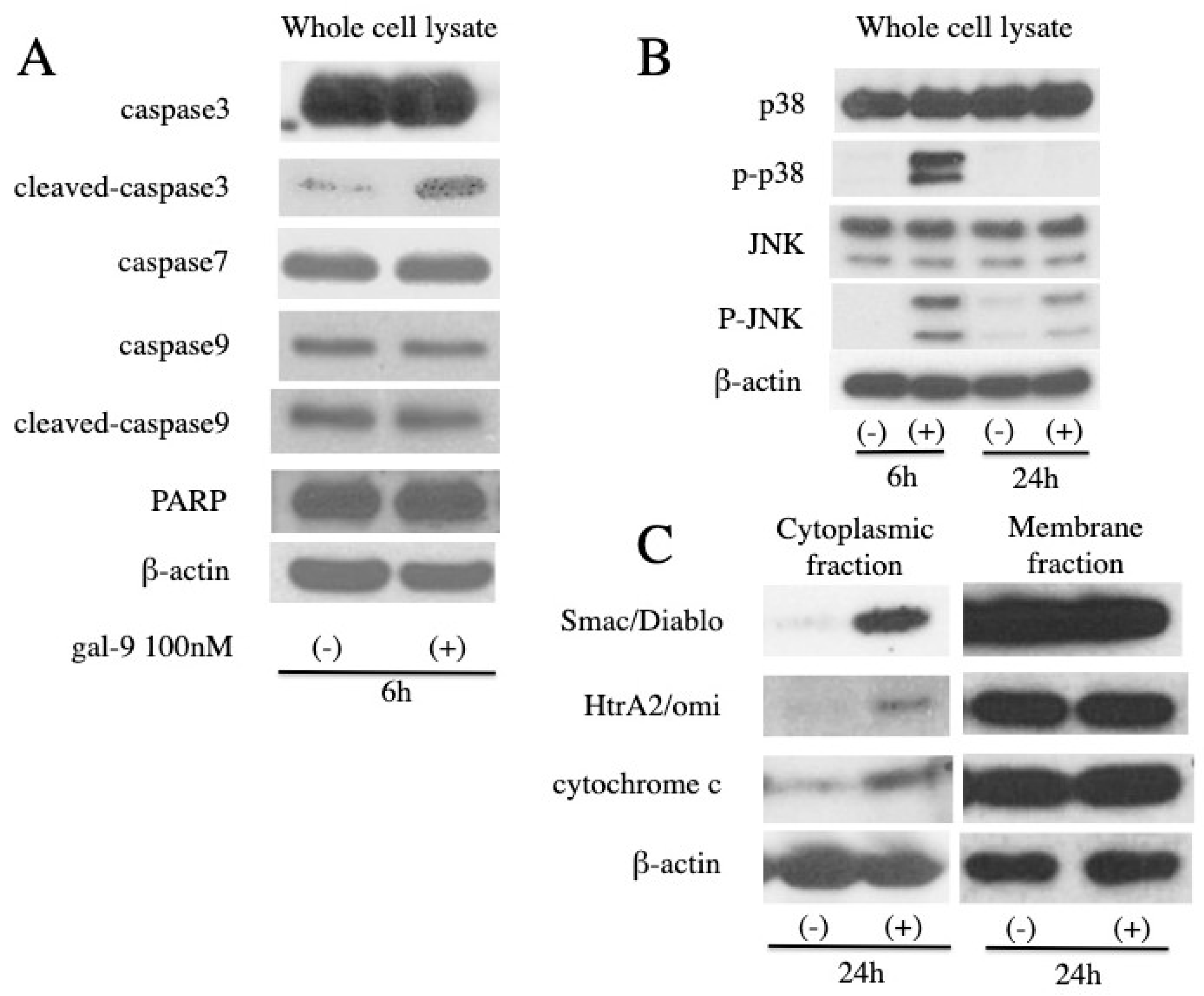
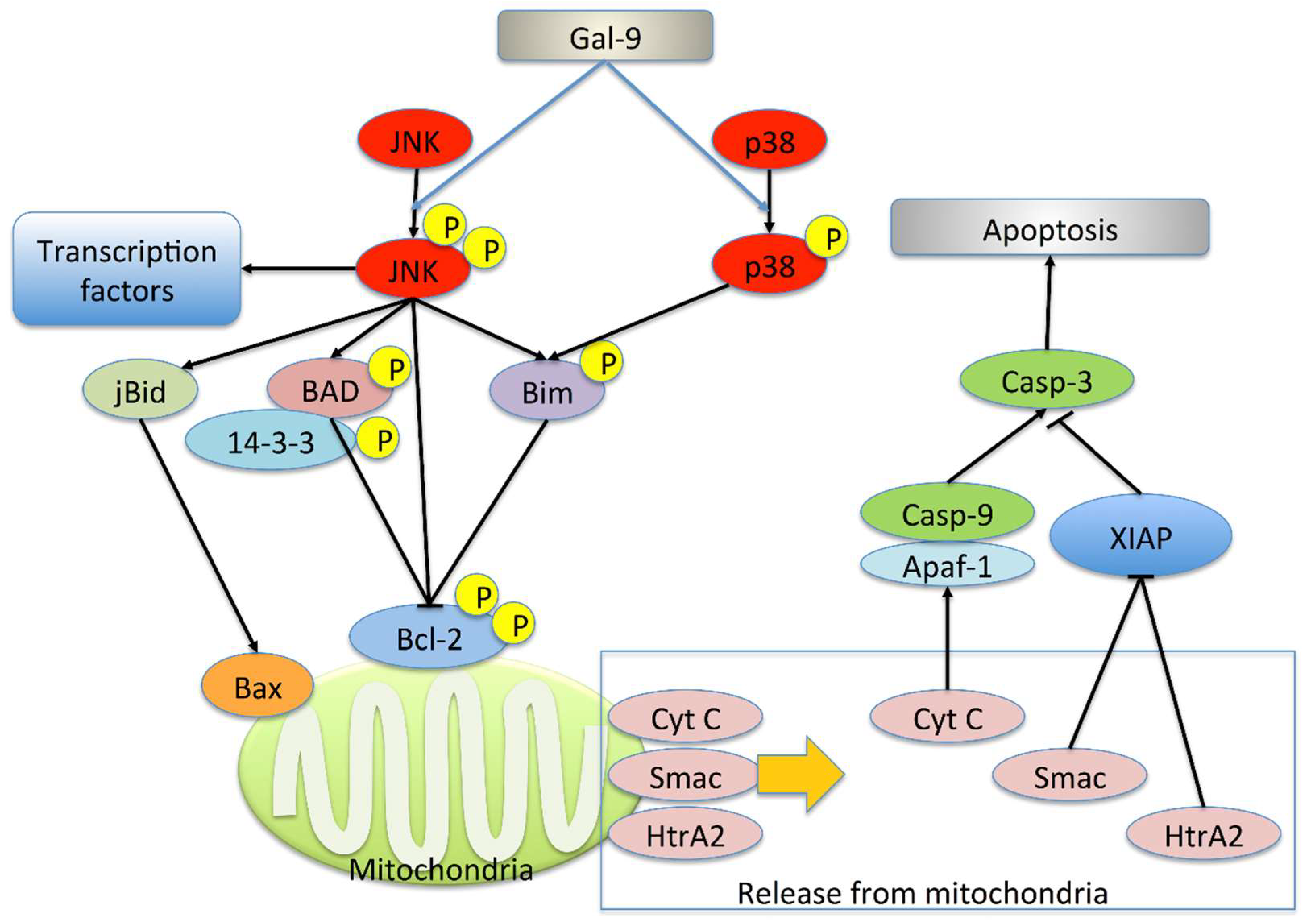
2.3. Gal-9 Changes the Level of Apoptosis-Related Protein
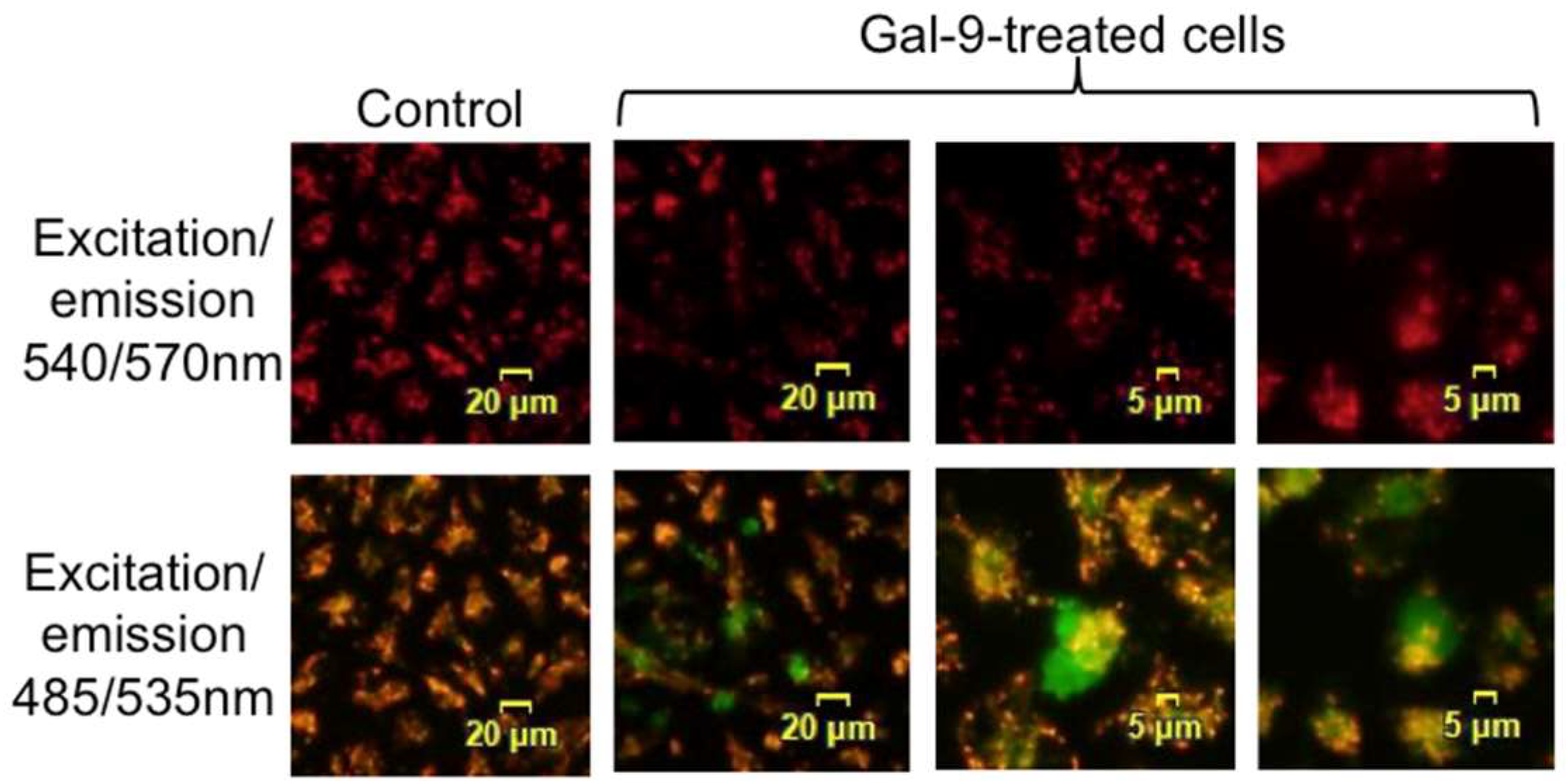
2.4. Gal-9 Induces the Collapse of Mitochondrial Potential
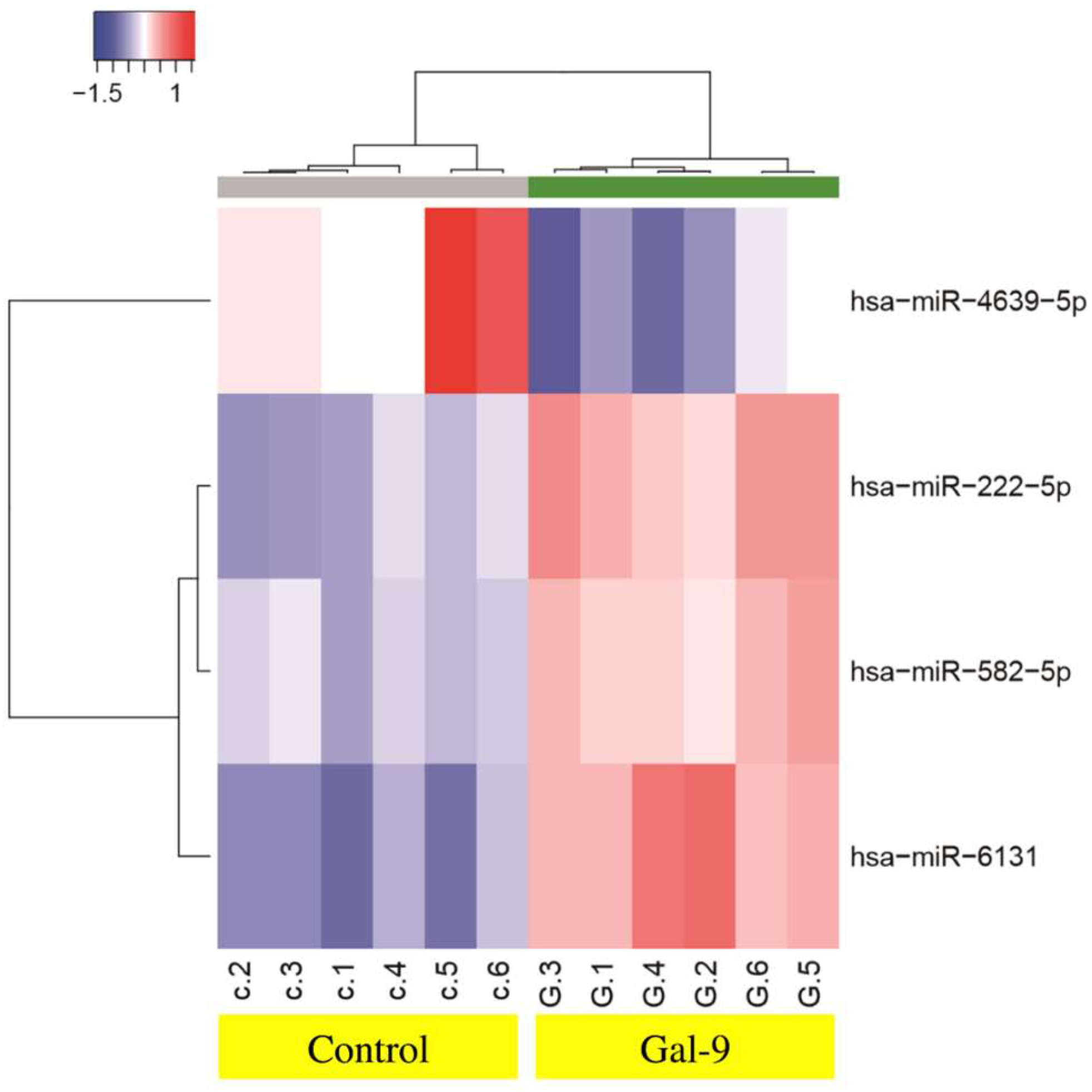
2.5. Effects of Gal-9 on the miRNA Expression
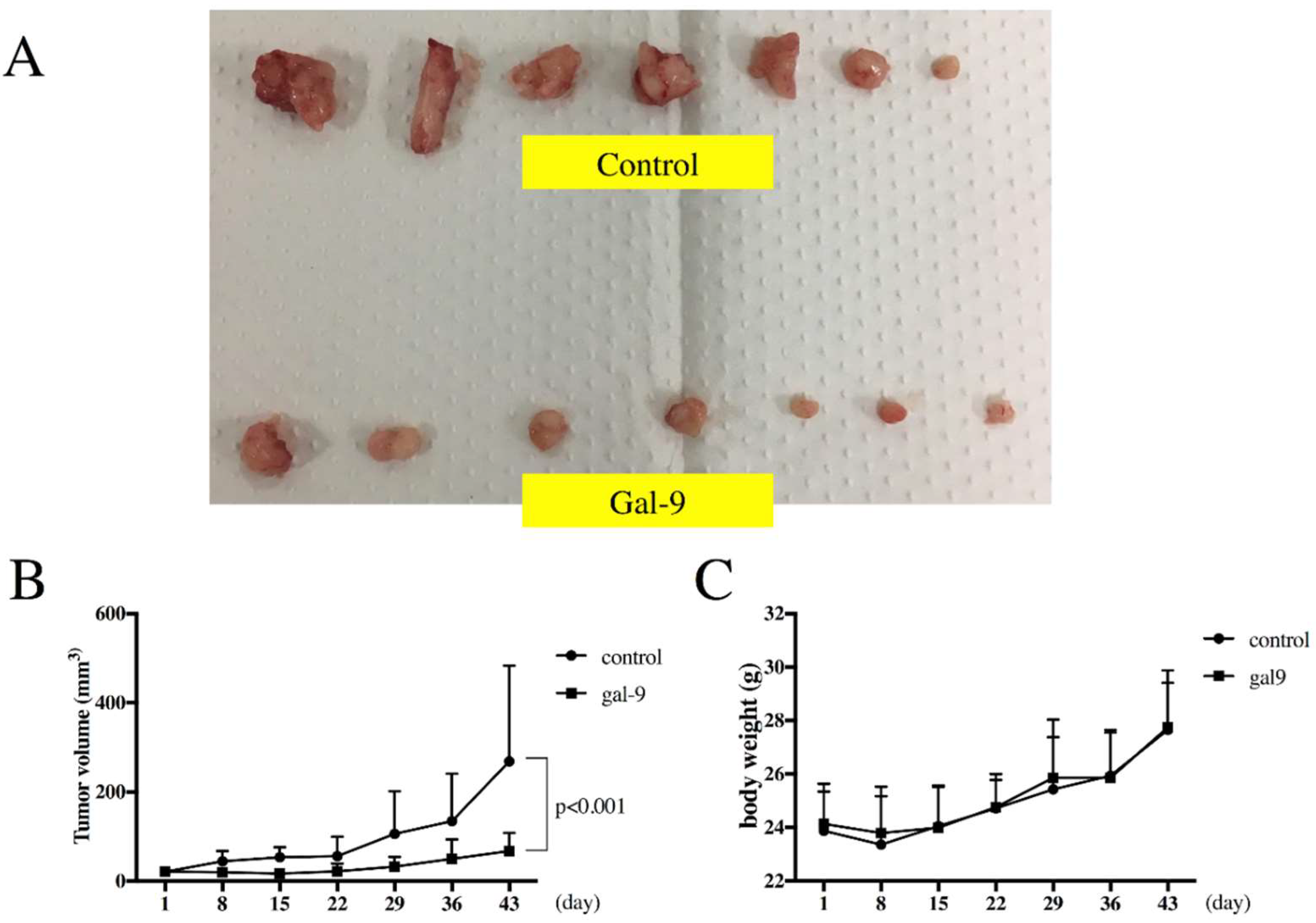
2.6. Gal-9 Inhibits the Growth of ESCC Cells in the Xenograft Mouse Model
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Lines and Reagents
4.3. Cell Proliferation Assay
4.4. Apoptosis Analysis
4.5. Colorimetric Assay for Caspase-3, -4, -7, -8 and -9 Activity
4.6. Gel Electrophoresis and Western Blot Analysis
4.7. Mitochondrial Membrane Potential Assay
4.8. Analysis of miRNA Microarray
4.9. Xenograft Model Analysis
4.10. Real-Time Polymerase Chain Reaction (PCR)
4.11. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Gal-9 | Galectin-9 |
| ESCC | esophageal squamous cell carcinoma |
| JNK | c-Jun NH2-terminal kinase |
| OXPHOS | mitochondrial oxidative phosphorylation system |
| p38 | p38 mitogen-activated protein kinase |
| EAC | esophageal adenocarcinoma |
References
- Kamangar, F.; Dores, G.M.; Anderson, W.F. Patterns of cancer incidence, mortality, and prevalence across five continents: Defining priorities to reduce cancer disparities in different geographic regions of the world. J. Clin. Oncol. 2006, 24, 2137–2150. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, H.; Luo, G.; Zhang, Y.; Wang, L.; Li, K. A systematic review and network meta-analysis of neoadjuvant therapy combined with surgery for patients with resectable esophageal squamous cell carcinoma. Int. J. Surg. 2017, 38, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Kato, K. Chemoradiotherapy for esophageal squamous cell cancer. Jpn. J. Clin. Oncol. 2016, 46, 805–810. [Google Scholar] [CrossRef] [Green Version]
- Wen, R.; Banik, B.; Pathak, R.K.; Kumar, A.; Kolishetti, N.; Dhar, S. Nanotechnology inspired tools for mitochondrial dysfunction related diseases. Adv. Drug Deliv. Rev. 2016, 99, 52–69. [Google Scholar] [CrossRef] [Green Version]
- Hirashima, M.; Kashio, Y.; Nishi, N.; Yamauchi, A.; Imaizumi, T.-A.; Kageshita, T.; Saita, N.; Nakamura, T. Galectin-9 in physiological and pathological conditions. Glycoconj. J. 2002, 19, 593–600. [Google Scholar] [CrossRef]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Oomizu, S.; Sakata, K.; Sakata, A.; Arikawa, T.; Watanabe, K.; Ito, K.; Takeshita, K.; Niki, T.; Saita, N.; et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin. Immunol. 2008, 127, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Madireddi, S.; Eun, S.-Y.; Lee, S.-W.; Nemčovičová, I.; Mehta, A.K.; Zajonc, D.M.; Nishi, N.; Niki, T.; Hirashima, M.; Croft, M. Galectin-9 controls the therapeutic activity of 4-1BB-targeting antibodies. J. Exp. Med. 2014, 211, 1433–1448. [Google Scholar] [CrossRef]
- Nagahara, K.; Arikawa, T.; Oomizu, S.; Kontani, K.; Nobumoto, A.; Tateno, H.; Watanabe, K.; Niki, T.; Katoh, S.; Miyake, M.; et al. Galectin-9 increases Tim-3+ dendritic cells and CD8 T cells and enhances antitumor immunity via Galectin-9-Tim-3 interactions. J. Immunol. 2008, 181, 7660–7669. [Google Scholar] [CrossRef]
- Thijssen, V.L.; Heusschen, R.; Caers, J.; Griffioen, A.W. Galectin expression in cancer diagnosis and prognosis: A systematic review. Biochim. Biophys. Acta 2015, 1855, 235–247. [Google Scholar] [CrossRef]
- Akashi, E.; Fujihara, S.; Morishita, A.; Tadokoro, T.; Chiyo, T.; Fujikawa, K.; Kobara, H.; Mori, H.; Iwama, H.; Okano, K.; et al. Effects of galectin-9 on apoptosis, cell cycle and autophagy in human esophageal adenocarcinoma cells. Oncol. Rep. 2017, 38, 506–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadokoro, T.; Morishita, A.; Fujihara, S.; Iwama, H.; Niki, T.; Fujita, K.; Akashi, E.; Mimura, S.; Oura, K.; Sakamoto, T.; et al. Galectin-9: An anticancer molecule for gallbladder carcinoma. Int. J. Oncol. 2016, 48, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Iwama, H.; Sakamoto, T.; Okura, R.; Kobayashi, K.; Takano, J.; Katsura, A.; Tatsuta, M.; Maeda, E.; Mimura, S.; et al. Galectin-9 suppresses the growth of hepatocellular carcinoma via apoptosis in vitro and in vivo. Int. J. Oncol. 2015, 46, 2419–2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Morishita, A.; Iwama, H.; Fujita, K.; Okura, R.; Fujihara, S.; Yamashita, T.; Fujimori, T.; Kato, K.; Kamada, H.; et al. Galectin-9 suppresses cholangiocarcinoma cell proliferation by inducing apoptosis but not cell cycle arrest. Oncol. Rep. 2015, 34, 1761–1770. [Google Scholar] [CrossRef]
- Zamore, P.D.; Haley, B. Ribo-gnome: The Big World of Small RNAs. Science 2005, 309, 1519–1524. [Google Scholar] [CrossRef] [Green Version]
- Morishita, A.; Masaki, T. miRNA in hepatocellular carcinoma. Hepatol. Res. 2015, 45, 128–141. [Google Scholar] [CrossRef]
- Wiersma, V.R.; de Bruyn, M.; van Ginkel, R.J.; Sigar, E.; Hirashima, M.; Niki, T.; Nishi, N.; Samplonius, D.F.; Helfrich, W.; Bremer, E. The glycan-binding protein galectin-9 has direct apoptotic activity toward melanoma cells. J. Investig. Dermatol. 2012, 132, 2302–2305. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Kuroda, J.; Ashihara, E.; Oomizu, S.; Terui, Y.; Taniyama, A.; Adachi, S.; Takagi, T.; Yamamoto, M.; Sasaki, N.; et al. Galectin-9 exhibits anti-myeloma activity through JNK and p38 MAP kinase pathways. Leukemia 2010, 24, 843–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, J.; Yamamoto, M.; Nagoshi, H.; Kobayashi, T.; Sasaki, N.; Shimura, Y.; Horiike, S.; Kimura, S.; Yamauchi, A.; Hirashima, M.; et al. Targeting activating transcription factor 3 by galectin-9 induces apoptosis and overcomes various types of treatment resistance in chronic myelogenous Leukemia. Mol. Cancer Res. 2010, 8, 994–1001. [Google Scholar] [CrossRef]
- Wang, K.; Johnson, A.; Ali, S.M.; Klempner, S.J.; Bekaii-Saab, T.; Vacirca, J.L.; Khaira, D.; Yelensky, R.; Chmielecki, J.; Elvin, J.A.; et al. Comprehensive genomic profiling of advanced esophageal squamous cell carcinomas and esophageal adenocarcinomas reveals similarities and differences. Oncologist 2015, 20, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.-J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.-C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.-Y.; et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): A double-blind, randomised phase 3 trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; dos Santos, L.V.; Aprile, G.; Ferry, D.R.; et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef]
- Brown, L.M.; Devesa, S.S.; Chow, W.-H. Incidence of adenocarcinoma of the esophagus among White Americans by sex, stage, and age. JNCI J. Natl. Cancer Inst. 2008, 100, 1184–1187. [Google Scholar] [CrossRef]
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global cancer incidence and mortality rates and trends—An update. Cancer Epidemiol. Prev. Biomark. 2016, 25, 16–27. [Google Scholar] [CrossRef]
- Miyanishi, N.; Nishi, N.; Abe, H.; Kashio, Y.; Shinonaga, R.; Nakakita, S.-i.; Sumiyoshi, W.; Yamauchi, A.; Nakamura, T.; Hirashima, M.; et al. Carbohydrate-recognition domains of galectin-9 are involved in intermolecular interaction with galectin-9 itself and other members of the galectin family. Glycobiology 2007, 17, 423–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, R.; Matsumoto, H.; Seki, M.; Hata, M.; Asano, Y.; Kanegasaki, S.; Stevens, R.L.; Hirashima, M. Human ecalectin, a variant of human galectin-9, is a novel eosinophil chemoattractant produced by T lymphocytes. J. Biol. Chem. 1998, 273, 16976–16984. [Google Scholar] [CrossRef]
- Moriyama, K.; Kukita, A.; Li, Y.-J.; Uehara, N.; Zhang, J.-Q.; Takahashi, I.; Kukita, T. Regulation of osteoclastogenesis through Tim-3: Possible involvement of the Tim-3/galectin-9 system in the modulation of inflammatory bone destruction. Lab. Investig. 2014, 94, 1200–1211. [Google Scholar] [CrossRef] [PubMed]
- Niki, T.; Tsutsui, S.; Hirose, S.; Aradono, S.; Sugimoto, Y.; Takeshita, K.; Nishi, N.; Hirashima, M. Galectin-9 is a high affinity IgE-binding lectin with anti-allergic effect by blocking IgE-antigen complex formation. J. Biol. Chem. 2009, 284, 32344–32352. [Google Scholar] [CrossRef]
- Muniz, J.M.; Bibiano Borges, C.R.; Beghini, M.; de Araújo, M.S.; Miranda Alves, P.; de Lima, L.M.B.; de Lima Pereira, S.A.; Nogueira, R.D.; Napimoga, M.H.; Rodrigues, D.B.R. Galectin-9 as an important marker in the differential diagnosis between oral squamous cell carcinoma, oral leukoplakia and oral lichen planus. Immunobiology 2015, 220, 1006–1011. [Google Scholar] [CrossRef]
- Chan, S.W.; Kallarakkal, T.G.; Abraham, M.T. Changed expression of E-cadherin and galectin-9 in oral squamous cell carcinomas but lack of potential as prognostic markers. Asian Pac. J. Cancer Prev. 2014, 15, 2145–2152. [Google Scholar] [CrossRef]
- Duray, A.; De Maesschalck, T.; Decaestecker, C.; Remmelink, M.; Chantrain, G.; Neiveyans, J.; Horoi, M.; Leroy, X.; Gabius, H.-J.; Saussez, S. Galectin fingerprinting in naso-sinusal diseases. Oncol. Rep. 2014, 32, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, M.; Ueno, M.; Oomizu, S.; Arikawa, T.; Shinonaga, R.; Zhang, S.; Yamauchi, A.; Hirashima, M. Galectin-9 expression links to malignant potential of cervical squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2008, 134, 899–907. [Google Scholar] [CrossRef]
- Van Loo, G.; van Gurp, M.; Depuydt, B.; Srinivasula, S.M.; Rodriguez, I.; Alnemri, E.S.; Gevaert, K.; Vandekerckhove, J.; Declercq, W.; Vandenabeele, P. The serine protease Omi/HtrA2 is released from mitochondria during apoptosis. Omi interacts with caspase-inhibitor XIAP and induces enhanced caspase activity. Cell Death Differ. 2002, 9, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, B.; Chang, S.H.; Becker, E.B.E.; Bonni, A.; Xia, Z. p38 MAP kinase mediates apoptosis through phosphorylation of Bim EL at Ser-65. J. Biol. Chem. 2006, 281, 25215–25222. [Google Scholar] [CrossRef]
- Wei, Y.; Sinha, S.; Levine, B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008, 4, 949–951. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, W.; Ran, Y.; Xiong, Y.; Zhong, Z.; Fan, X.; Wang, Z.; Ye, Q. miR-582-5p inhibits proliferation of hepatocellular carcinoma by targeting CDK1 and AKT3. Tumor Biol. 2015, 36, 8309–8316. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Yang, J.; Li, S.; Chen, J. Upregulation of miR-582-5p inhibits cell proliferation, cell cycle progression and invasion by targeting Rab27a in human colorectal carcinoma. Cancer Gene Ther. 2015, 22, 475–480. [Google Scholar] [CrossRef]
- Uchino, K.; Takeshita, F.; Takahashi, R.; Kosaka, N.; Fujiwara, K.; Naruoka, H.; Sonoke, S.; Yano, J.; Sasaki, H.; Nozawa, S.; et al. Therapeutic effects of microRNA-582-5p and -3p on the inhibition of bladder cancer progression. Mol. Ther. 2013, 21, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-W.; Chen, B.; Lei, C.-B.; Liu, G.-X.; Wang, Y.-G.; Yi, C.; Wang, Y.-Y.; Zhang, S.-Y. miR-582-5p inhibits invasion and migration of salivary adenoid cystic carcinoma cells by targeting FOXC1. Jpn. J. Clin. Oncol. 2017, 47, 690–698. [Google Scholar] [CrossRef]
- Mengshol, J.A.; Golden-Mason, L.; Arikawa, T.; Smith, M.; Niki, T.; McWilliams, R.; Randall, J.A.; McMahan, R.; Zimmerman, M.A.; Rangachari, M.; et al. A crucial role for Kupffer cell-derived galectin-9 in regulation of T cell immunity in hepatitis C infection. PLoS ONE 2010, 5, e9504. [Google Scholar] [CrossRef]
- Nishi, N.; Itoh, A.; Fujiyama, A.; Yoshida, N.; Araya, S.I.; Hirashima, M.; Shoji, H.; Nakamura, T. Development of highly stable galectins: Truncation of the linker peptide confers protease-resistance on tandem-repeat type galectins. FEBS Lett. 2005, 579, 2058–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashio, Y.; Nakamura, K.; Abedin, M.J.; Seki, M.; Nishi, N.; Yoshida, N.; Nakamura, T.; Hirashima, M. Galectin-9 induces apoptosis through the calcium-calpain-caspase-1 pathway. J. Immunol. 2003, 170, 3631–3636. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Downregulated | Fold (Treated/Untreated) | p Value | Chromosomal Localization |
|---|---|---|---|
| miR-4639-5p | 0.45 | 0.0022 | 6 |
| Upregulated | |||
| miR-222-5p | 1.81 | 0.0022 | X chromosome |
| miR-582-5p | 1.52 | 0.0022 | 5 |
| miR-6131 | 2.27 | 0.0022 | 5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiyo, T.; Fujita, K.; Iwama, H.; Fujihara, S.; Tadokoro, T.; Ohura, K.; Matsui, T.; Goda, Y.; Kobayashi, N.; Nishiyama, N.; et al. Galectin-9 Induces Mitochondria-Mediated Apoptosis of Esophageal Cancer In Vitro and In Vivo in a Xenograft Mouse Model. Int. J. Mol. Sci. 2019, 20, 2634. https://doi.org/10.3390/ijms20112634
Chiyo T, Fujita K, Iwama H, Fujihara S, Tadokoro T, Ohura K, Matsui T, Goda Y, Kobayashi N, Nishiyama N, et al. Galectin-9 Induces Mitochondria-Mediated Apoptosis of Esophageal Cancer In Vitro and In Vivo in a Xenograft Mouse Model. International Journal of Molecular Sciences. 2019; 20(11):2634. https://doi.org/10.3390/ijms20112634
Chicago/Turabian StyleChiyo, Taiga, Koji Fujita, Hisakazu Iwama, Shintaro Fujihara, Tomoko Tadokoro, Kyoko Ohura, Takanori Matsui, Yasuhiro Goda, Nobuya Kobayashi, Noriko Nishiyama, and et al. 2019. "Galectin-9 Induces Mitochondria-Mediated Apoptosis of Esophageal Cancer In Vitro and In Vivo in a Xenograft Mouse Model" International Journal of Molecular Sciences 20, no. 11: 2634. https://doi.org/10.3390/ijms20112634
APA StyleChiyo, T., Fujita, K., Iwama, H., Fujihara, S., Tadokoro, T., Ohura, K., Matsui, T., Goda, Y., Kobayashi, N., Nishiyama, N., Yachida, T., Morishita, A., Kobara, H., Mori, H., Niki, T., Hirashima, M., Himoto, T., & Masaki, T. (2019). Galectin-9 Induces Mitochondria-Mediated Apoptosis of Esophageal Cancer In Vitro and In Vivo in a Xenograft Mouse Model. International Journal of Molecular Sciences, 20(11), 2634. https://doi.org/10.3390/ijms20112634