Targeting Angiogenesis in Prostate Cancer



Abstract
:1. Introduction
2. Background
2.1. Prostate Cancer
2.2. Treatment Options in Prostate Cancer
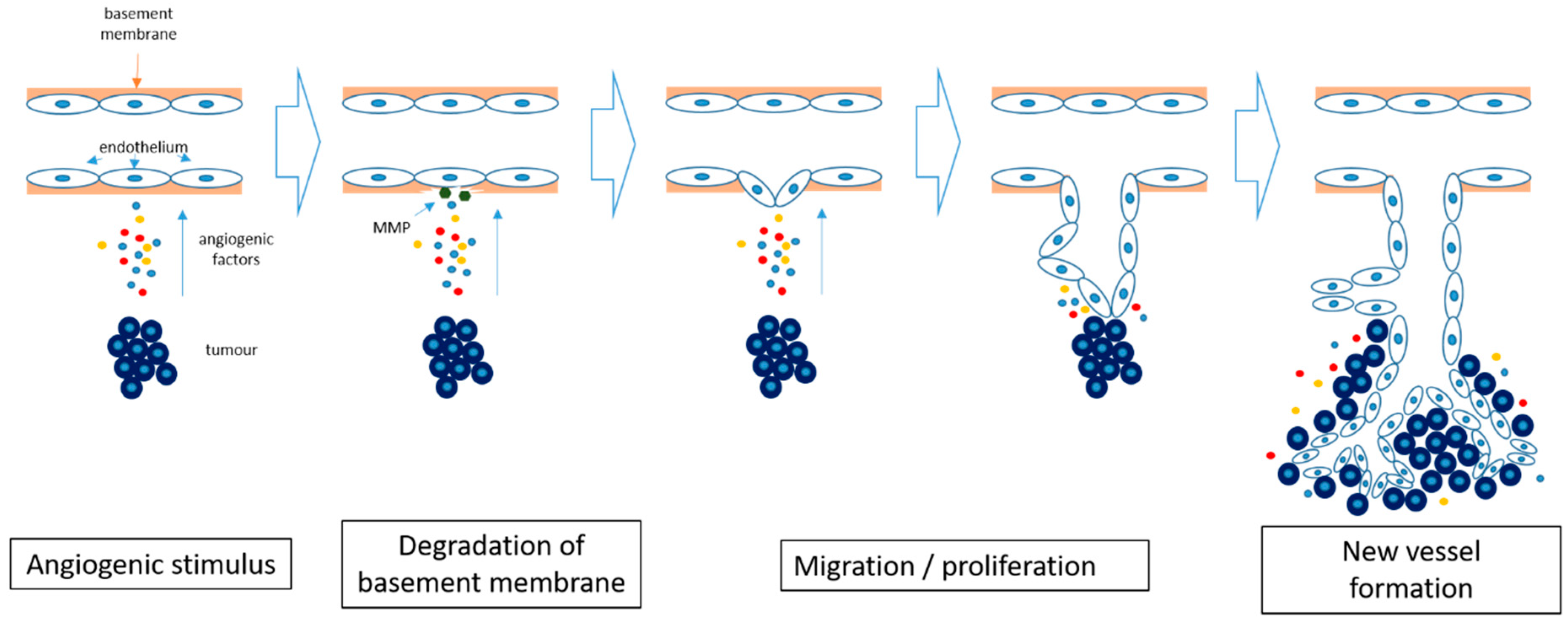
2.3. Angiogenesis in Cancer
2.4. Angiogenesis Inhibition in Cancer
3. Results
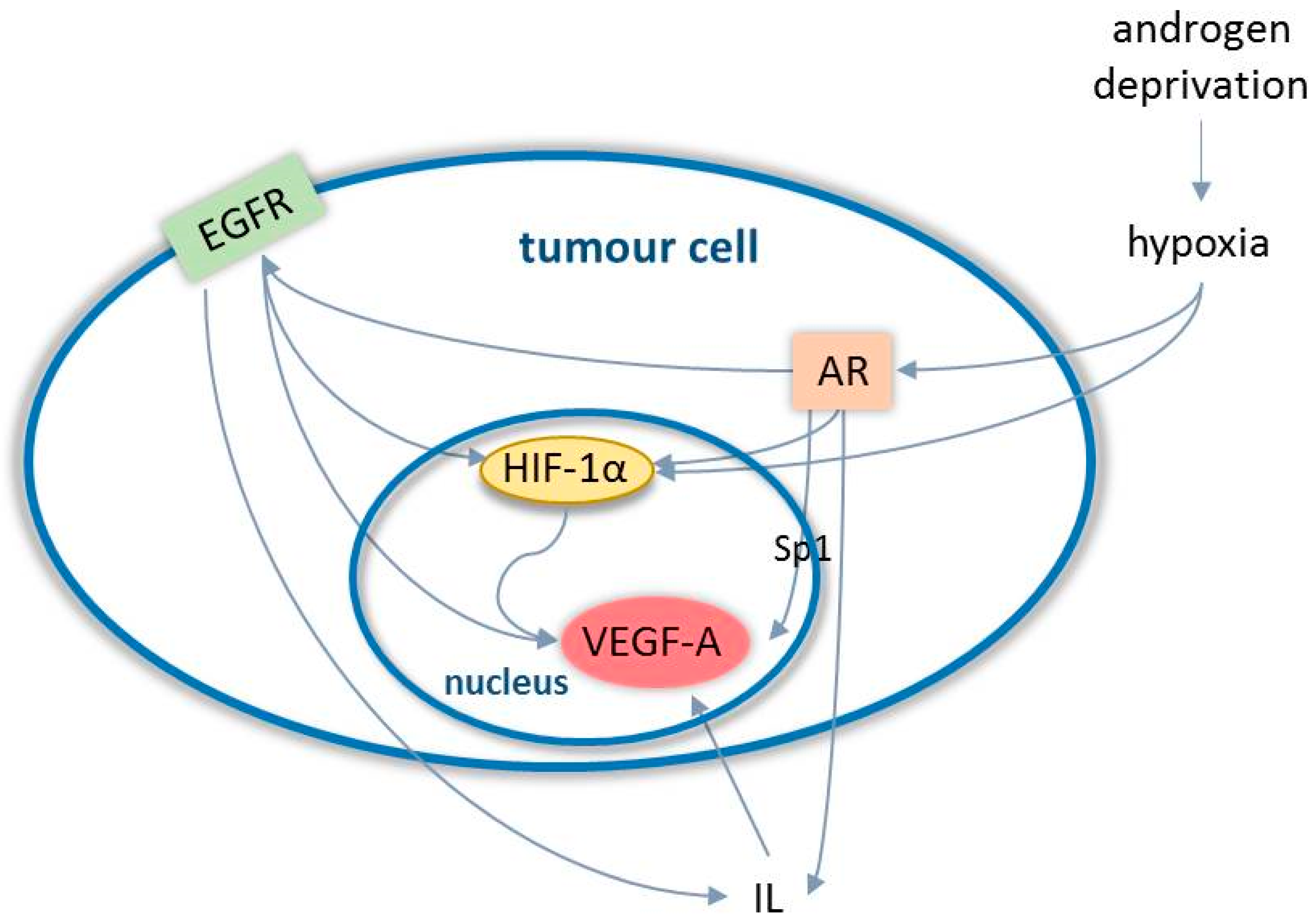
3.1. Angiogenesis in Prostate Cancer
3.2. Anti-Angiogenesis Clinical Studies in Prostate Cancer
4. Discussion
5. Materials and Methods
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- National Cancer Institute. SEER Stat Fact Sheets: Prostate; National Cancer Institute: Bethesda, MD, USA. Available online: https://seer.cancer.gov/statfacts/html/prost.html#prevalence (accessed on 10 March 2018).
- American Cancer Society. Cancer Facts and Figures; American Cancer Society: Atlanta, GA, USA, 2018; Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2018/cancer-facts-and-figures-2018.pdf (accessed on 10 March 2018).
- Cancer Research UK. Prostate Cancer Incidence Statistics [Internet]. 2014. Available online: http://www.cancerresearchuk.org/cancer-info/cancerstats/types/prostate/incidence/#age (accessed on 14 August 2018).
- Zlotta, A.R.; Egawa, S.; Pushkar, D.; Govorov, A.; Kimura, T.; Kido, M.; Takahashi, H.; Kuk, C.; Kovylina, M.; Aldaoud, N.; et al. Prevalence of prostate cancer on autopsy: Cross-sectional study on unscreened Caucasian and Asian men. J. Natl. Cancer Inst. 2013, 105, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Cancer Facts and Figures; American Cancer Society: Atlanta, GA, USA, 2012; Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2012/estimated-number-of-new-cancer-cases-and-deaths-by-sex-2012.pdf (accessed on 19 August 2018).
- The National Cancer Registration Service, Eastern Office [Internet]. Available online: http://www.ncras.nhs.uk/ncrs-east/ (accessed on 14 August 2018).
- Zelefsky, M.J.; Eastham, J.A.; Sartor, A.O. Cancer of the prostate. In Cancer: Principles and Practice of Oncology, 9th ed.; De Vita, V.T., Jr., Lawrence, T.S., Rosenberg, S.A., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2011; pp. 1220–7121. [Google Scholar]
- PDQ Adult Treatment Editorial Board. Prostate Cancer Treatment (PDQ®): Patient Version. 30 April 2018. In PDQ Cancer Information Summaries [Internet]; National Cancer Institute (US): Bethesda, MD, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK65915/ (accessed on 19 August 2018).
- Hamdy, F.C.; Donovan, J.L.; Lane, J.A.; Mason, M.; Metcalfe, C.; Holding, P.; Davis, M.; Peters, T.J.; Turner, E.L.; Martin, R.M.; et al. 10-Year Outcomes after Monitoring, Surgery, or Radiotherapy for Localized Prostate Cancer. N. Engl. J. Med. 2016, 375, 1415–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, J.; Kirkbride, P.; Cann, K.; Hasler, E.; Prettyjohns, M. Prostate cancer:summary of updated NICE guidance. BMJ 2014, 8, 348. [Google Scholar] [CrossRef]
- Ragde, H.; Blasko, J.C.; Grimm, P.D.; Kenny, G.M.; Sylvester, J.E.; Hoak, D.C.; Landin, K.; Cavanagh, W. Interstitial iodine-125 radiation without adjuvant therapy in the treatment of clinically localized prostate carcinoma. Cancer 1997, 80, 442–453. [Google Scholar] [CrossRef]
- The Medical Research Council Prostate Cancer Working Party Investigators Group. Immediate versus deferred treatment for advanced prostatic cancer: Initial results of the Medical Research Council Trial. Br. J. Urol. 1997, 79, 235–426. [Google Scholar]
- Dearnaley, D.P.; Mason, M.D.; Parmar, M.K.; Sanders, K.; Sydes, M.R. Adjuvant therapy with oral sodium clodronate in locally advanced and metastatic prostate cancer: Long-term overall survival results from the MRC PR04 and PR05 randomizedcontrolled trials. Lancet Oncol. 2009, 10, 872–876. [Google Scholar] [CrossRef]
- James, N.D.; de Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.; Amos, C.L.; Gilson, C.; Jones, R.J. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef]
- De Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L.; et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomizedopen-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- Fizazi, K.; Carducci, M.; Smith, M.; Damião, R.; Brown, J.; Karsh, L.; Milecki, P.; Shore, N.; Rader, M.; Wang, H.; et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: A randomized, double-blind study. Lancet 2011, 377, 813–822. [Google Scholar] [CrossRef]
- Oosterhof, G.O.N.; Roberts, J.T.; de Reijke, T.M.; Engelholm, S.A.; Horenblas, S.; von der Maase, H.; Neymark, N.; Debois, M. ColletteL. Strontium (89) chloride versus palliative local field radiotherapy in patients with hormonal escaped prostate cancer: A phase III study of the European Organisation for Research and Treatment of Cancer, Genitourinary Group. Eur. Urol. 2003, 44, 519–526. [Google Scholar] [CrossRef]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef]
- Rajabi, M.; Mousa, S.A. The Role of Angiogenesis in Cancer Treatment. Biomedicines 2017, 21, 34. [Google Scholar] [CrossRef]
- Winkler, F. Hostile takeover: How tumors hijack pre-existing vascular environments to thrive. J. Pathol. 2017, 242, 267–272. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Pavlakovic, H.; Havers, W.; Schweigerer, L. Multiple angiogenesis stimulators in a single malignancy: Implications for anti-angiogenic tumor therapy. Angiogenesis 2001, 4, 259–262. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef]
- Gressett, M.; Shah, S.R. Intricacies of bevacizumab-induced toxicities and their management. Ann. Pharmacother. 2009, 43, 490–501. [Google Scholar] [CrossRef]
- Kamba, T.; McDonald, D.M. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br. J. Cancer 2007, 96, 1788–1795. [Google Scholar] [CrossRef]
- Ferrara, N. VEGF as a therapeutic target in cancer. Oncology 2005, 69 (Suppl. 3), 11–16. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [Green Version]
- El-Kenawi, A.E.; El-Remessy, A.B. Angiogenesis inhibitors in cancer therapy: Mechanistic perspective on classification and treatment rationales. Br. J. Pharmacol. 2013, 170, 712–729. [Google Scholar] [CrossRef]
- Mundel, T.M.; Kalluri, R. Type IV collagen-derived angiogenesis inhibitors. Microvasc. Res. 2007, 74, 85–89. [Google Scholar] [CrossRef] [Green Version]
- Kurozumi, K.; Ichikawa, T.; Onishi, M.; Fujii, K.; Date, I. Cilengitide treatment for malignant glioma: Current status and future direction. Neurol. Med. Chir. 2012, 52, 539–547. [Google Scholar] [CrossRef]
- Su, J.; Cai, M.; Li, W.; Hou, B.; He, H.; Ling, C.; Huang, T.; Liu, H.; Guo, Y. Molecularly Targeted Drugs Plus Radiotherapy and Temozolomide Treatment for Newly Diagnosed Glioblastoma: A Meta-Analysis and Systematic Review. Oncol. Res. 2016, 24, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Herbert, S.P.; Stainier, D.Y. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat. Rev. Mol. Cell. Biol. 2011, 12, 551–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolin, K.; Gordon, M.S.; Holmgren, E.; Gaudreault, J.; Novotny, W.; Fyfe, G.; Adelman, D.; Stalter, S.; Breed, J. Phase Ib trial of intravenous recombinant humanized monoclonal antibody to vascular endothelial growth factor in combination with chemotherapy in patients with advanced cancer: Pharmacologic and long-term safety data. J. Clin. Oncol. 2011, 19, 851–856. [Google Scholar] [CrossRef]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Kroetz, D.L. Bevacizumab-induced hypertension: Clinical presentation and molecular understanding. Pharmacol. Ther. 2018, 182, 152–160. [Google Scholar] [CrossRef]
- Minder, P.; Zajac, E.; Quigley, J.P.; Deryugina, E.I. EGFR Regulates the Development and Microarchitecture of Intratumoral Angiogenic Vasculature Capable of Sustaining Cancer Cell Intravasation. Neoplasia 2015, 17, 634–649. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Sharma, M.C.; Sarkar, C. Morphology of angiogenesis in human cancer: A conceptual overview, histoprognostic perspective and significance of neoangiogenesis. Histopathology 2005, 46, 481–489. [Google Scholar] [CrossRef]
- Bono, A.V.; Celato, N.; Cova, V.; Salvadore, M.; Chinetti, S.; Novario, R. Microvessel density in prostate carcinoma. Prostate Cancer Prostatic Dis. 2002, 5, 123–127. [Google Scholar] [CrossRef]
- Borre, M.; Offersen, B.V.; Nerstrom, B.; Overgaard, J. Microvessel density predicts survival in prostate cancer patients subjected to watchful waiting. Br. J. Cancer 1998, 78, 940–944. [Google Scholar] [CrossRef]
- Jiang, J.; Chen, Y.; Zhu, Y.; Yao, X.; Qi, J. Contrast-enhanced ultrasonography for the detection and characterization of prostate cancer: Correlation with microvessel density and Gleason score. Clin. Radiol. 2011, 66, 732–737. [Google Scholar] [CrossRef]
- Tretiakova, M.; Antic, T.; Binder, D.; Kocherginsky, M.; Liao, C.; Taxy, J.B.; Oto, A. Microvessel density is not increased in prostate cancer: Digital imaging of routine sections and tissue microarrays. Hum. Pathol. 2013, 44, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Saka, H. Reconsideration of the clinical and histopathological significance of angiogenesis in prostate cancer: Usefulness and limitations of microvessel density measurement. Int. J. Urol. 2015, 22, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Taverna, G.; Grizzi, F.; Colombo, P.; Seveso, M.; Giusti, G.; Proietti, S.; Fiorini, G.; Lughezzani, G.; Casale, P.; Buffi, N.; et al. Two-dimensional neovascular complexity is significantly higher in nontumor prostate tissue than in low-risk prostate cancer. Korean J. Urol. 2015, 56, 435–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taverna, G.; Grizzi, F.; Colombo, P.; Graziotti, P. Is angiogenesis a hallmark of prostate cancer? Front. Oncol. 2013, 3, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Brot, S.; Ntekim, A.; Cardenas, R.; James, V.; Allegrucci, C.; Heery, D.M.; Bates, D.O.; Ødum, N.; Persson, J.L.; Mongan, N.P. Regulation of vascular endothelial growth factor in prostate cancer. Endocr. Relat. Cancer 2015, 22, 107–123. [Google Scholar] [CrossRef]
- Wong, S.Y.; Haack, H.; Crowley, D.; Barry, M.; Bronson, R.T.; Hynes, R.O. Tumor-secreted vascular endothelial growth factor-C is necessary for prostate cancer lymphangiogenesis, but lymphangiogenesis is unnecessary for lymph node metastasis. Cancer Res. 2005, 65, 9789–9798. [Google Scholar] [CrossRef]
- Wegiel, B.; Bjartell, A.; Ekberg, J.; Gadaleanu, V.; Brunhoff, C.; Persson, J.L. A role for cyclin A1 in mediating the autocrine expression of vascular endothelial growth factor in prostate cancer. Oncogene 2005, 24, 6385–6393. [Google Scholar] [CrossRef] [Green Version]
- Green, M.M.; Hiley, C.T.; Shanks, J.H.; Bottomley, I.C.; West, C.M.; Cowan, R.A.; Stratford, I.J. Expression of vascular endothelial growth factor (VEGF) in locally invasive prostate cancer is prognostic for radiotherapy outcome. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 84–90. [Google Scholar] [CrossRef]
- Duque, J.L.; Loughlin, K.R.; Adam, R.M.; Kantoff, P.W.; Zurakowski, D.; Freeman, M.R. Plasma levels of vascular endothelial growth factor are increased in patients with metastatic prostate cancer. Urology 1999, 54, 523–527. [Google Scholar] [CrossRef]
- Hrouda, D.; Nicol, D.L.; Gardiner, R.A. The role of angiogenesis in prostate development and the pathogenesis of prostate cancer. Urol. Res. 2003, 30, 347–355. [Google Scholar]
- McKay, R.R.; Zurita, A.J.; Werner, L.; Bruce, J.Y.; Carducci, M.A.; Stein, M.N.; Heath, E.I.; Hussain, A.; Tran, H.T.; Sweeney, C.J.; et al. Randomized Phase II Trial of Short-Course Androgen Deprivation Therapy With or Without Bevacizumab for Patients With Recurrent Prostate Cancer After Definitive Local Therapy. J. Clin. Oncol. 2016, 34, 1913–1920. [Google Scholar] [CrossRef]
- Kelly, W.K.; Halabi, S.; Carducci, M.; George, D.; Mahoney, J.F.; Stadler, W.M.; Morris, M.; Kantoff, P.; Monk, J.P.; Kaplan, E.; et al. Randomized, double-blind, placebo-controlled phase III trial comparing docetaxel and prednisone with or without bevacizumab in men with metastatic castration-resistant prostate cancer: CALGB 90401. J. Clin. Oncol. 2012, 30, 1534–1540. [Google Scholar] [CrossRef]
- Tannock, I.F.; Fizazi, K.; Ivanov, S.; Karlsson, C.T.; Fléchon, A.; Skoneczna, I.; Orlandi, F.; Gravis, G.; Matveev, V.; Bavbek, S.; et al. Aflibercept versus placebo in combination with docetaxel and prednisone for treatment of men with metastatic castration-resistant prostate cancer (VENICE): A phase 3, double-blind randomizedtrial. Lancet Oncol. 2013, 14, 760–768. [Google Scholar] [CrossRef]
- Michaelson, M.D.; Oudard, S.; Ou, Y.C.; Sengeløv, L.; Saad, F.; Houede, N.; Ostler, P.; Stenzl, A.; Daugaard, G.; Jones, R.; et al. Randomized, placebo-controlled, phase III trial of sunitinib plus prednisone versus prednisone alone in progressive, metastatic, castration-resistant prostate cancer. J. Clin. Oncol. 2014, 32, 76–82. [Google Scholar] [CrossRef]
- Keizman, D.; Zahurak, M.; Sinibaldi, V.; Carducci, M.; Denmeade, S.; Drake, C.; Pili, R.; Antonarakis, E.S.; Hudock, S.; Eisenberger, M. Lenalidomide in nonmetastatic biochemically relapsed prostate cancer: Results of a phase I/II double-blinded, randomized study. Clin. Cancer Res. 2010, 16, 5269–5276. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Vogelzang, N.J.; Budnik, N.; Wiechno, P.J.; Sternberg, C.N.; Doner, K.; Bellmunt, J.; Burke, J.M.; de Olza, M.O.; Choudhury, A.; et al. Docetaxel and prednisone with or without lenalidomide in chemotherapy-naivepatients with metastatic castration-resistant prostate cancer (MAINSAIL): Arandomized, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2015, 16, 417–425. [Google Scholar] [CrossRef]
- Mangoni, M.; Vozenin, M.C.; Biti, G.; Deutsch, E. Normal tissues toxicities triggered by combined anti-angiogenic and radiation therapies: Hurdles might be ahead. Br. J. Cancer 2012, 107, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Ogita, S.; Tejwani, S.; Heilbrun, L.; Fontana, J.; Heath, E.; Freeman, S.; Smith, D.; Baranowski, K.; Vaishampayan, U. Pilot Phase II Trial of Bevacizumab Monotherapy in Nonmetastatic Castrate-Resistant Prostate Cancer. ISRN Oncol. 2012, 2012, 242850. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Vacca, A. New Insights in Anti-Angiogenesis in Multiple Myeloma. Int. J. Mol. Sci. 2018, 19, 2031. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-Q.; Fang, J.-M.; Xiao, Y.-Y.; Zhao, Y.; Cui, R.; Hu, F.; Xu, Q. Prognostic role of vascular endothelial growth factor in prostate cancer: A systematic review and meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 2289–2298. [Google Scholar] [PubMed]
- Wang, K.; Peng, H.L.; Li, L.K. Prognostic value of vascular endothelial growth factorexpression in patients with prostate cancer: A systematic review withmeta-analysis. Asian Pac. J. Cancer Prev. 2012, 13, 5665–5669. [Google Scholar] [CrossRef] [PubMed]
- Scholz, A.; Harter, P.N.; Cremer, S.; Yalcin, B.H.; Gurnik, S.; Yamaji, M.; Di Tacchio, M.; Sommer, K.; Baumgarten, P.; Bähr, O.; et al. Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol. Med. 2016, 8, 39–57. [Google Scholar] [CrossRef]
- Lindholm, E.M.; Krohn, M.; Iadevaia, S.; Kristian, A.; Mills, G.B.; Mælandsmo, G.M.; Engebraaten, O. Proteomic characterization of breast cancer xenografts identifies early and late bevacizumab-induced responses and predicts effective drug combinations. Clin. Cancer Res. 2014, 20, 404–412. [Google Scholar] [CrossRef]
- Madan, R.A.; Karzai, F.H.; Ning, Y.-M.; Adesunloye, B.A.; Huang, X.; Harold, N.; Couvillon, A.; Chun, G.; Cordes, L.; Sissung, T.; et al. Phase II trial of docetaxel, bevacizumab, lenalidomide and prednisone in patients with metastatic castration-resistant prostate cancer. BJU Int. 2016, 118, 590–597. [Google Scholar] [CrossRef]
- Brauer, M.J.; Zhuang, G.; Schmidt, M.; Yao, J.; Wu, X.; Kaminker, J.S.; Jurinka, S.S.; Kolumam, G.; Chung, A.S.; Jubb, A.; et al. Identification and analysis of in vivo VEGF downstream markers link VEGF pathway activity with efficacy of anti-VEGF therapies. Clin. Cancer Res. 2013, 19, 3681–3692. [Google Scholar] [CrossRef]
- De Haas, S.; Delmar, P.; Bansal, A.T.; Moisse, M.; Miles, D.W.; Leighl, N.; Escudier, B.; Van Cutsem, E.; Carmeliet, P.; Scherer, S.J.; et al. Genetic variability of VEGF pathway genes in six randomized Phase III trials assessing the addition of bevacizumab to standard therapy. Angiogenesis 2014, 17, 909–920. [Google Scholar] [CrossRef]
- Golovine, K.; Kutikov, A.; Teper, E.; Simhan, J.; Makhov, P.B.; Canter, D.J.; Uzzo, R.G.; Kolenko, V.M. Modulation of Akt/mTOR signalling overcomes sunitinib resistance in renal and prostate cancer cells. Mol. Cancer Ther. 2012, 11, 1510–1517. [Google Scholar]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef]
- Wang, Y.; Kreisberg, J.I.; Ghosh, P.M. Cross-talk between the androgen receptor and the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer. Curr. Cancer Drug Targets 2007, 7, 591–604. [Google Scholar] [CrossRef]
- Yamamoto, Y.; A De Velasco, M.; Kura, Y.; Nozawa, M.; Hatanaka, Y.; Oki, T.; Ozeki, T.; Shimizu, N.; Minami, T.; Yoshimura, K.; et al. Evaluation of in vivo responses of sorafenib therapy in a preclinical mouse model of PTEN-deficient of prostate cancer. J. Transl. Med. 2015, 13, 150. [Google Scholar] [CrossRef]
- De Velasco, M.A.; Kura, Y.; Yoshikawa, K.; Nishio, K.; Davies, B.R.; Uemura, H. Efficacy of targeted AKT inhibition in genetically engineered mouse models of PTEN-deficient prostate cancer. Oncotarget 2016, 7, 15959–15976. [Google Scholar] [CrossRef] [Green Version]
- Sordello, S.; Bertrand, N.; Plouet, J. Vascular endothelial growth factor is up-regulated in vitro and in vivo by androgens. Biochem. Biophys. Res. Commun. 1998, 251, 287–290. [Google Scholar] [CrossRef]
- Eisermann, K.; Fraizer, G. The Androgen Receptor and VEGF: Mechanisms of Androgen-Regulated Angiogenesis in Prostate Cancer. Cancers 2017, 9, 32. [Google Scholar] [CrossRef]
- Kashyap, V.; Ahmad, S.; Nilsson, E.M.; Helczynski, L.; Kenna, S.; Persson, J.L.; Gudas, L.J.; Mongan, N.P. The lysine specific demethylase-1 (LSD1/KDM1A) regulates VEGF-A expression in prostate cancer. Mol. Oncol. 2013, 7, 555–566. [Google Scholar] [CrossRef]
- Deng, X.; Shao, G.; Zhang, H.; Li, C.; Zhang, D.; Cheng, L.; Elzey, B.; Pili, R.; Ratliff, T.; Huang, J. Proteinarginine methyltransferase 5 functions as an epigenetic activator of the androgen receptor to promote prostate cancer cell growth. Oncogene 2016, 36, 1223–1231. [Google Scholar] [CrossRef]
- Eisermann, K.; Broderick, C.J.; Bazarov, A.; Moazam, M.M.; Fraizer, G.C. Androgen up-regulates vascular endothelial growth factor expression in prostate cancer cells via an Sp1 binding site. Mol. Cancer 2013, 12, 7. [Google Scholar] [CrossRef]
- Antonarakis, E.; Armstrong, A.; Dehm, S.; Luo, J. Androgen receptor variant-driven prostate cancer: Clinical implications and therapeutic targeting. Prostate Cancer Prostatic Dis. 2016, 19, 231–241. [Google Scholar] [CrossRef]
- Fernandez, E.V.; Reece, K.M.; Ley, A.M.; Troutman, S.M.; Sissung, T.M.; Price, D.K.; Chau, C.H.; Figg, W.D. Dual targeting of the androgen receptor and hypoxia-inducible factor 1α pathways synergistically inhibits castration-resistant prostate cancer cells. Mol. Pharmacol. 2015, 87, 1006–1012. [Google Scholar] [CrossRef]
- Pignon, J.C.; Koopmansch, B.; Nolens, G.; Delacroix, L.; Waltregny, D.; Winkler, R. Androgen receptor controls EGFR and ERBB2 gene expression at different levels in prostate cancer cell lines. Cancer Res. 2009, 69, 2941–2949. [Google Scholar] [CrossRef]
- Zheng, Y.; Izumi, K.; Yao, J.L.; Miyamoto, H. Dihydrotestosterone upregulates the expression of epidermal growth factor receptor and ERBB2 in androgen receptor-positive bladder cancer cells. Endocr. Relat. Cancer 2011, 18, 451–464. [Google Scholar] [CrossRef]
- Tabernero, J. The role of VEGF and EGFR inhibition: Implications for combining anti-VEGF and anti-EGFR agents. Mol. Cancer Res. 2007, 5, 203–220. [Google Scholar] [CrossRef]
- Mabjeesh, N.J.; Willard, M.T.; Frederickson, C.E.; Zhong, H.; Simons, J.W. Androgens stimulate hypoxia-inducible factor 1 activation via autocrine loop of tyrosine kinase receptor/phosphatidylinositol 3′-kinase/protein kinase B in prostate cancer cells. Clin. Cancer Res. 2003, 9, 2416–2425. [Google Scholar]
- Cereda, V.; Formica, V.; Roselli, M. Issues and promises of bevacizumab in prostate cancer treatment. Exp. Opin. Biol. Ther. 2018, 18, 707–717. [Google Scholar] [CrossRef]
- Shabsigh, A.; Ghafar, M.A.; De La Taille, A.; Burchardt, M.; Kaplan, S.A.; Anastasiadis, A.G.; Buttyan, R. Biomarker analysis demonstrates a hypoxic environment in the castrated rat ventral prostate gland. J. Cell Biochem. 2001, 81, 437–444. [Google Scholar] [CrossRef]
- Halin, S.; Hammarsten, P.; Wikström, P.; Bergh, A. Androgen-insensitive prostate cancer cells transiently respond to castration treatment when growing in an androgen-dependent prostate environment. Prostate 2007, 67, 370–377. [Google Scholar] [CrossRef]
- Mitani, T.; Harada, N.; Nakano, Y.; Inui, H.; Yamaji, R. Coordinated action of hypoxia-inducible factor-1α and β-catenin in androgen receptor signaling. J. Biol. Chem. 2012, 287, 33594–33606. [Google Scholar] [CrossRef]
- Horii, K.; Suzuki, Y.; Kondo, Y.; Akimoto, M.; Nishimura, T.; Yamabe, Y.; Sakaue, M.; Sano, T.; Kitagawa, T.; Himeno, S.; et al. Androgen-dependent gene expression of prostate-specific antigen is enhanced synergistically by hypoxia in human prostate cancer cells. Mol. Cancer Res. 2007, 5, 383–391. [Google Scholar] [CrossRef]
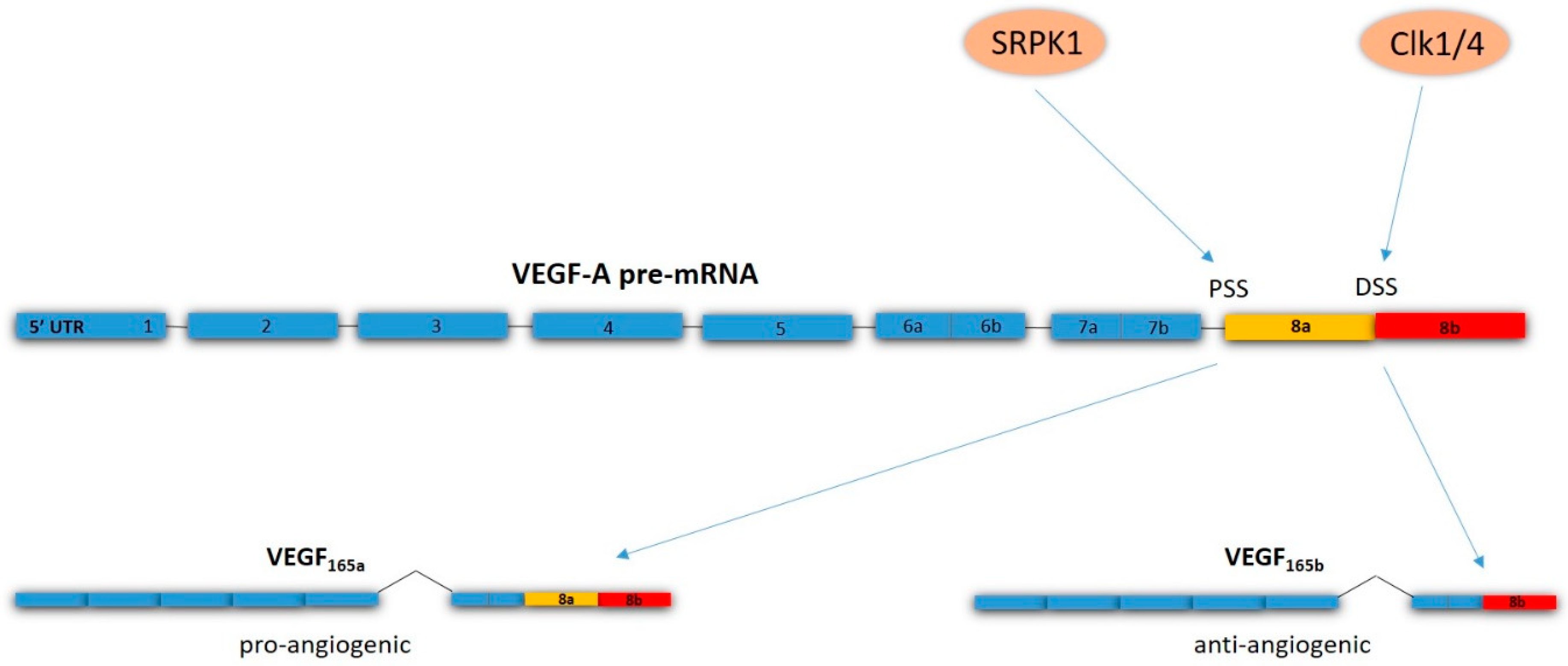
- Bates, D.; Cui, T.-G.; Doughty, J.M.; Winkler, M.; Sugiono, M.; Shields, J.D.; Peat, D.; Gillatt, D.; Harper, S.J. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002, 62, 4123–4131. [Google Scholar]
- Woolard, J.; Wang, W.Y.; Bevan, H.S.; Qiu, Y.; Morbidelli, L.; Pritchard-Jones, R.O.; Cui, T.G.; Sugiono, M.; Waine, E.; Perrin, R.; et al. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: Mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004, 64, 7822–7835. [Google Scholar] [CrossRef]
- Oltean, S.; Gammons, M.; Hulse, R.; Hamdollah-Zadeh, M.; Mavrou, A.; Donaldson, L.; Salmon, A.H.; Harper, S.J.; Ladomery, M.R.; Bates, D.O. SRPK1 inhibition in vivo: Modulation of VEGF splicing and potential treatment for multiple diseases. Biochem. Soc. Trans. 2012, 40, 831–835. [Google Scholar] [CrossRef]
- Auboeuf, D.; Dowhan, D.H.; Kang, Y.K.; Larkin, K.; Lee, J.W.; Berget, S.M.; O’Malley, B.W. Differential recruitment of nuclear receptor coactivators may determine alternative RNA splice site choice in target genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2270–2274. [Google Scholar] [CrossRef] [Green Version]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef]
- Amin, E.M.; Oltean, S.; Hua, J.; Gammons, M.V.; Hamdollah-Zadeh, M.; Welsh, G.I.; Cheung, M.-K.; Ni, L.; Kase, S.; Rennel, E.S.; et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 2011, 20, 768–780. [Google Scholar] [CrossRef]
- Nowak, D.G.; Woolard, J.; Amin, E.M.; Konopatskaya, O.; Saleem, M.A.; Churchill, A.J.; Ladomery, M.R.; Harper, S.J.; Bates, D.O. Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J. Cell Sci. 2008, 121, 3487–3495. [Google Scholar] [CrossRef] [Green Version]
- Nowak, D.G.; Amin, E.M.; Rennel, E.S.; Hoareau-Aveilla, C.; Gammons, M.; Damodoran, G.; Hagiwara, M.; Harper, S.J.; Woolard, J.; Ladomery, M.R.; et al. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: A novel therapeutic strategy for angiogenesis. J. Biol. Chem. 2010, 285, 5532–5540. [Google Scholar] [CrossRef]
- Mavrou, A.; Brakspear, K.; Hamdollah-Zadeh, M.; Damodaran, G.; Babaei-Jadidi, R.; Oxley, J.; Gillatt, D.A.; Ladomery, M.R.; Harper, S.J.; Bates, D.O.; Oltean, S. Serine-arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene 2015, 34, 4311–4319. [Google Scholar] [CrossRef] [PubMed]
- Mavrou, A.; Oltean, S. SRPK1 inhibition in prostate cancer: A novel anti-angiogenic treatment through modulation of VEGF alternative splicing. Pharmacol. Res. 2016, 107, 276–281. [Google Scholar] [CrossRef] [Green Version]
- Van den Brûle, F.A.; Waltregny, D.; Castronovo, V. Increased expression of galectin-1 in carcinoma-associated stroma predicts poor outcome in prostate carcinoma patients. J. Pathol. 2001, 193, 80–87. [Google Scholar] [CrossRef]
- Stanley, P. Galectin-1 Pulls the Strings on VEGFR2. Cell 2014, 156, 625–626. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, F.M.; Gentilini, L.D.; Gueron, G.; Meiss, R.P.; Ortiz, E.G.; Berguer, P.M.; Ahmed, A.; Navone, N.; Rabinovich, G.A.; Compagno, D.; et al. In Vivo Hemin Conditioning Targets the Vascular and Immunologic Compartments and Restrains Prostate Tumor Development. Clin. Cancer Res. 2017, 23, 5135–5148. [Google Scholar] [CrossRef] [PubMed]
- Laderach, D.J.; Gentilini, L.D.; Giribaldi, L.; Delgado, V.C.; Nugnes, L.; Croci, D.O.; Al Nakouzi, N.; Sacca, P.; Casas, G.; Mazza, O.; et al. A Unique Galectin Signature in Human Prostate Cancer Progression Suggests Galectin-1 as a Key Target for Treatment of Advanced Disease. Cancer Res. 2013, 73, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Goud, N.S.; Soukya, P.S.L.; Ghouse, M.; Komal, D.; Alvala, R.; Alvala, M. Human Galectin-1 and its inhibitors: Privileged target for cancer and HIV. Mini Rev. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Level of Risk | PSA Level (ng/mL) | Gleason Score | Clinical Stage | ||
|---|---|---|---|---|---|
| Low risk | <10 | and | ≤6 | and | T1–T2a |
| Intermediate risk | 10–20 | or | 7 | or | T2b |
| High risk | >20 | or | 8–10 | or | ≥T2c |
| Drug | Mechanism of Action | Phase of the Clinical Trial | Number of Patients | Outcome |
|---|---|---|---|---|
| Bevacizumab | Recombinant humanized monoclonal antibody that blocks VEGF-A | II | 99 | Improved relapse-free survival [54] |
| III | 1050 | No improvement in overall survival [55] | ||
| Aflibercept | Binds to circulating VEGF-A | III | 1224 | No improvement in overall survival [56] |
| Sunitinib | Receptor tyrosine kinase inhibitor | III | 873 | No improvement in overall survival [57] |
| Lenalidomide | Multiple mechanisms, including inhibition of VEGF-induced PI3K-Akt pathway signalling | I/II | 60 | Disease stabilisation, decrease in PSA [58] |
| III | 1059 | Worse overall survival [59] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melegh, Z.; Oltean, S. Targeting Angiogenesis in Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 2676. https://doi.org/10.3390/ijms20112676
Melegh Z, Oltean S. Targeting Angiogenesis in Prostate Cancer. International Journal of Molecular Sciences. 2019; 20(11):2676. https://doi.org/10.3390/ijms20112676
Chicago/Turabian StyleMelegh, Zsombor, and Sebastian Oltean. 2019. "Targeting Angiogenesis in Prostate Cancer" International Journal of Molecular Sciences 20, no. 11: 2676. https://doi.org/10.3390/ijms20112676
APA StyleMelegh, Z., & Oltean, S. (2019). Targeting Angiogenesis in Prostate Cancer. International Journal of Molecular Sciences, 20(11), 2676. https://doi.org/10.3390/ijms20112676