2.4. Identification and Expression Analysis of DEGs
A RNA-Seq experiment was conducted using samples applied by GA
3 for 0, 2, and 4 h of LA hybrid lily and mapped the resulting reads to our reference transcriptome to determine the DEGs. The following DEGs were filtered with an RPKM≥ 0, FDR ≤ 0.05 and |log
2(ratio)| ≥2. The 2 h and 0 h libraries were compared and 7782 genes were found, a total of 3965 upregulated and 3817 downregulated genes were detected between the two libraries. The number of the upregulated genes and the downregulated genes are almost the same. There were 5475 upregulated and 3955 downregulated genes between the 4 h and 0 h libraries, 3657 upregulated and 2157 downregulated genes between the 4 h and 2 h libraries (
Figure 6). This suggests that the number of DEGs between 4 h and 0 h is larger than that between the 2 h and 0 h libraries, while the difference between the 4 h and 2 h libraries is the smallest of the three. We also found that the numbers of upregulated genes were higher than the numbers of downregulated genes after GA
3 treatment in LA lily.
Filtering with an RPKM≥ 2, FDR ≤ 0.05 and |log2(ratio)| ≥ 2, 2148 genes were differentially expressed among the three stages based on the transcriptome profiling results. A total of 1002 upregulated and 173 downregulated genes were detected between the 2 h and 0 h libraries. The upregulated genes in this group accounted for 46.6% of the total DEGs, almost the half of the total DEGs. The downregulated genes in this group accounted for 8% of the total DEGs. The total number DEGs between the 4 h and 0 h libraries accounted for 70.5% of the total DEGs, this is greater than the group of the 2 h and 0 h libraries which accounted for 54.6% of the total DEGs. However, the number of DEGs between the 2 h and 0 h libraries is more than half of the total DEGs which suggests that genes expressed rapidly in 2 h in response to GA
3 treatment. A total of 1252 upregulated and 264 downregulated genes were detected between the 4 h and 0 h libraries, accounting for 58.3% and 12.3% of the total DEGs, respectively. A total of 381 upregulated and 53 downregulated genes were detected between the 4 h and 2 h libraries, accounting for 17.7% and 2.5% of the total DEGs, respectively. Moreover, the number of the upregulated genes and the downregulated genes has a huge gap in each comparison group. As well as the above analysis of DEGs filtered with an RPKM ≥ 0, FDR ≤ 0.05 and |log2(ratio)| ≥ 2, the number of DEGs (RPKM ≥ 2, FDR ≤ 0.05 and |log2(ratio)| ≥ 2) between 4 h and 0 h libraries is the largest, followed by the 2 h and 0 h comparison group, the number of DEGs in 4 h and 2 h comparison group is the smallest of all. That means, in LA hybrid lily, transcript abundance changed dramatically from the stage without gibberellins treatment to the gibberellins treatment stages in which the GA response genes could be induced and expressed strongly. Meanwhile, the differentially expressions of these key genes determine the rapid response to the exogenous GA
3 in an instant in the lily (
Figure 6).
Through the Venn diagram analysis of the 2148 DEGs, three genes(contig_19485, Contig3551, contig_43047) were identified in three pairwise transcriptome comparisons, one of the genes is
LEA4 (
Late embryogenesis abundant group 4, contig_43047), the other two genes are uncharacterized, all three genes were constantly and dramatically up/downregulated between 0 h and 4 h, suggesting these genes may always play an important role in the whole process of the GA3 treatment. Furthermore, 414, 586, and 174 DEGs were found only in 2 h vs. 0 h, 4 h vs. 0 h, and 4 h vs. 2 h transcriptomes, accounting for 19.3%, 27.3%, and 8.1% of the total DEGs, respectively. 714 DEGs, accounting for 33.2%, were found both in 2 h vs. 0 h and 4 h vs. 0 h transcriptomes, in which 106 DEGs were characterized, such as
LEA (Late embryogenesis abundant protein, contig_123089),
NAC1 (NAC protein 1, Contig12404, Contig5556),
MYB TF 2 (
Myb transcription factor 2, Contig 5472),
WRKY TF 40, 33, 27, 21, 7 (
Transcription factor WRKY 40, 33, 27, 21, 7, Contig16070, Contig10474, contig_112644, contig_100827, contig_112645, contig_36867, Contig6963, Contig10395, contig_91975, contig_58912),
AP2/EREBP TF (
AP2/EREBP transcription factor superfamily protein, contig_15479), gene encoding the
xyloglucan endotransglucosylase/hydrolase protein (Contig 11584), and
P34 kinase (Contig 2523). 213 DEGs, accounting for 9.9%, were found both in 4 h vs. 0 h and 4 h vs. 2 h transcriptomes, in which 18 DEGs were characterized, one of which is the gene encoding the
diacylglycerol kinase protein (Contig 4850, contig_40020). 44 DEGs, accounting for 2%, were found both in 2 h vs. 0 h and 4 h vs. 2 h transcriptomes, in which only two genes were characterized, one of which is the gene encoding the
PEPC (
Phosphoenolpyruvate carboxylase, contig_98336) (
Figure 7).
With an algorithm developed from expression trend profiles of DEGs (RPKM ≥ 2, FDR ≤ 0.05 and |log
2(ratio)| ≥2), the 2148 DEGs were classified into eight expression-trend profiles. The results showed that the profile 6, of which the genes’ expression increased sharply from 0 h to 2 h and then kept stable from 2 h to 4 h accounted for the largest proportion, containing 1041 genes. It was closely followed by the profile 4, including 271genes, of which the genes’ expression remained temporarily almost the same from 0 h to 2 h and subsequently rose from 2 h to 4 h. The third is profile 7, which presented that the genes’ expression rose constantly from 0 h to 4 h, containing 242 genes. The next was the profile 2, including 195 genes, which showed the expression pattern of genes decreasing initially from 0 h to 2 h and then increasing from 2 h to 4 h. The following was the profile 0 which showed continuous downtrend in expression, including 150 genes. The sixth was the profile 1, including 107 genes, which showed a downward trend from 0 h to 2 h and kept a stable trend from 2 h to 4 h. The seventh was the profile 3, including 74 genes. The last was the profile 5, which contained 68 genes. Their expression trends were shown in
Figure 8 below. The heatmap of all the DEGs showed the consistent expression trends. On one side, some genes were downregulated from the 0 h to 2 h stages, but upregulated obviously from 2 h to 4 h stages. On the other side, it was showed that some genes increased expression at the 2 h stage, but decreased transcript abundance at the 4 h stage. Some genes were immediately expressed at the initial stage of 0 h, while others were upregulated subsequently (
Figure 9).
2.6. Differential Expression Analysis of Key Genes
Transcription factors: A large number of transcription factors were found, including
WRKY 85 (Contig 5897),
WRKY 40 (Contig 16070),
WRKY 33 (contig_100827, Contig10474, Contig6963, contig_36867),
WRKY 27 (contig_58912),
WRKY 21 (contig_112644, contig_112645),
WRKY 7 (contig_91975, Contig13143),
WRKY transcription factor (Contig10395, contig_74229), heat shock transcription factor
Heat shock A2 (contig_147190, contig_68192),
Heat Shock A-2b (Contig10086), transcription factor
AP2/EREBP (contig_15479, contig_64211, contig_6641),
AP2-like (contig_31023),
AP2-domain containing transcription factor (Contig16130), transcription factor
NAC1 (Contig12) 404, Contig5556),
NAC11 (contig_47533),
NAC-domain containing transcription factor (contig_47532, Contig2543, contig_122890, contig_5277), transcription factor
MYB (Contig10978, contig_107841, Contig2955),
R2R3 MYB (Contig806contig_135561, Contig10033, contig_112030),
MYB2 (Contig5427),
MYB5 (Contig9242),
MYB98 (contig_75829), MADS-box transcription factor
AGL20 (contig_14431), transcription factor
CBF (Contig9007), transcription factor
EREB1 (Contig9291), transcription factor
zinc referring to Zinc finger CCCH domain-containing protein 32 (contig_19526), ethylene response transcription factor
Ethylene responsive transcription factor 2b (contig_65834), and so on (
Figure S3).
Floral transition related genes: flowering integrator
AGL20/SOC1 (contig_14431, contig_16411, contig_70281, Contig1815, contig_44220, Contig12365, Contig1888, contig_10322, contig_10670), contributing flower factor
CO (Contig5082), transcription factor
bHLH (contig_14876, contig_55931, contig_66658, contig_80665, contig_91633), and transcription factor
bHLH25 (contig_95131) that may regulate the CO gene (
Figure S4).
Hormone-related genes:
cytochrome P450 (contig_495, Contig11622),
cytochrome P450 CYP81 (contig_57244, Contig81),
CYP8 (Contig1222),
cytochrome P450 89A2 (Contig15092),
DELLA protein
(DWARF8) (Contig3159),
GASA-like protein (contig_1561, contig_1562),
cold-regulated LTCOR12 (contig_67925), cold-regulated gibberellin regulatory protein
1LTCOR12 (contig_133755),
gibberellin 20 oxidase (contig_58931),
gibberellin 3-oxidase (Contig7586, Contig15483),
gibberellin 3-oxidase 2 (contig_30501),
gibberellin 2-oxidase 4 (contig_14262),
gibberellin 2-β-double oxygenase GA2ox (contig_79537),
9-cis epoxy carotenoid dioxygenase NCED3 (contig_127138, contig_82115),
tryptophan biosynthesis 1 (Contig 3534),
cytokinin oxidase/dehydrogenase 6 isoform 1 (Contig 14170),
SAUR-like auxin-responsive family protein (contig_105313, contig_96011, contig_96020),
auxin response GH3 family protein (contig_120322),
1-aminocyclopropane-1-carboxylic acid oxidase (Contig7564), E
3-ubiquitin-protein ligase UPL6 (Contig13942),
E3 ubiquitin E3 ubiquitin-protein ligase BRE1-like protein (Contig13942),
ring finger protein (contig_84742),
heat shock protein (Contig1525),
heat shock protein 70 (contig_8855),
cytoplasmic class I small heat shock protein 3B (contig_2205 4),
chaperone protein (contig_87062, Contig15475),
calmodulin-binding protein (Contig15102),
calmodulin-like protein (contig_127735),
60S ribosomal protein L31 (contig_121296), 4
-coumaric acid-CoA ligase 9 (contig_37094), p
hotosystem I P700 chlorophyll a apolipoprotein A1 (Second_Contig 1938), and so on (
Figure S5).
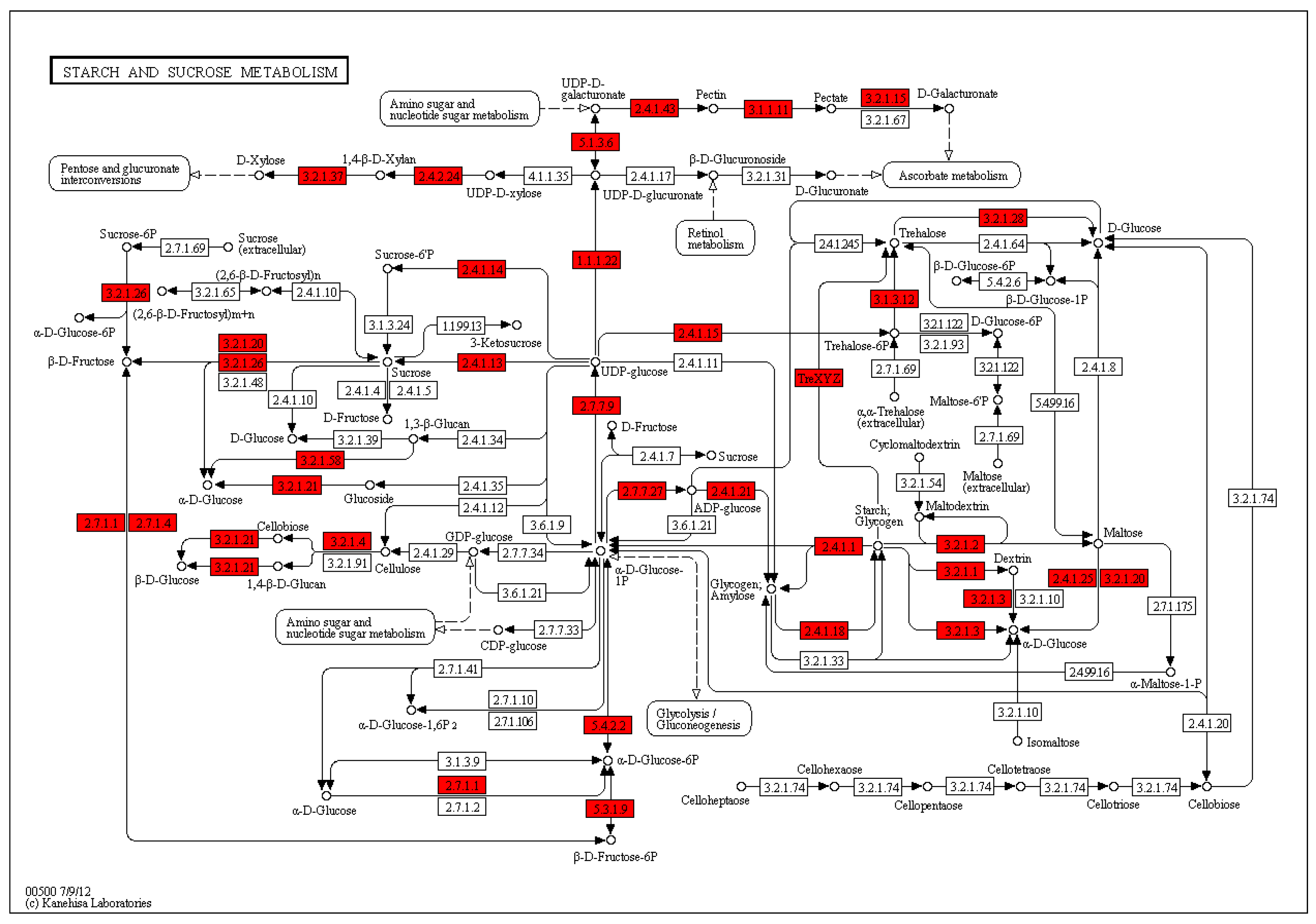
Carbohydrate metabolism-related genes:
glucose 6-phosphate dehydrogenase (Contig6729),
sucrose synthase (contig_22392, Contig4527),
fructose diphosphate aldolase (contig_6925),
Xylose isomerase (contig_21412),
endoglucanase (Second_Contig1564),
xyloglucan endo-glucokinase/hydrolase (Contig15472, Contig11584, contig_62659),
glycosyltransferase (contig_2821, contig_33603, contig_61457, contig_83432, contig_93519, contig_98102, Contig12157, Contig12635, Contig13119, Contig13294, Contig15238, Contig9400, Contig9565, Second_Contig294),
β-hexosaminidase (contig_46355),
hexyl glycosyltransferase (contig_27832, contig_8764, Contig5280),
anthocyanin 5,3-o-glucosyltransferase (Contig 13404),
soluble acid invertase (contig_68521), and so on (
Figure S6).
Stress-resistant related genes:
glutathione transferase (contig_44762, Contig8504, contig_101889, contig_53305, contig_53306, contig_58173, contig_79316, Contig11293, Contig13715),
thioredoxin (contig_3971, contig_3972, contig_98142),
actin depolymerization factor (contig_43718),
disease-resistant family protein LRR family protein (Contig602, contig_3570),
late embryogenesis-rich protein 1 (LEA1) (Contig15947, contig_24694),
late embryogenesis-rich protein 2 (contig_99782),
late embryogenesis-rich protein 4 (contig_43047),
serine/threonine protein kinase (Contig5432, Contig17002, Contig13127, Contig16418, Contig16448, contig_30231, contig_75279, contig_99357),
LRR receptor-like serine/threonine protein kinase (Contig16507),
receptor-like protein kinase (contig_56469),
P34 kinase (Contig2523),
histidine kinase 3 (Contig1188), and
mitogen-activated protein kinase (contig_53060) (
Figure S7).
Histone modification related genes: methylation-related gene
methyltransferase (Contig13028),
adenosine homocysteine (contig_61885, contig_101207),
Orcinol O-Methyltransferase methyltransferase (contig_37751, contig_53117),
ubiquitination-modification-related genomic protein H2A (contig_1031, contig_2045),
histone H3 (contig_19960, contig_425), ADP-ribosylation-related gene
poly[ADP-ribose] polymerase gene (contig_59490, Contig16608),
NADPH enzyme gene (contig_33047), and so on (
Figure S8).
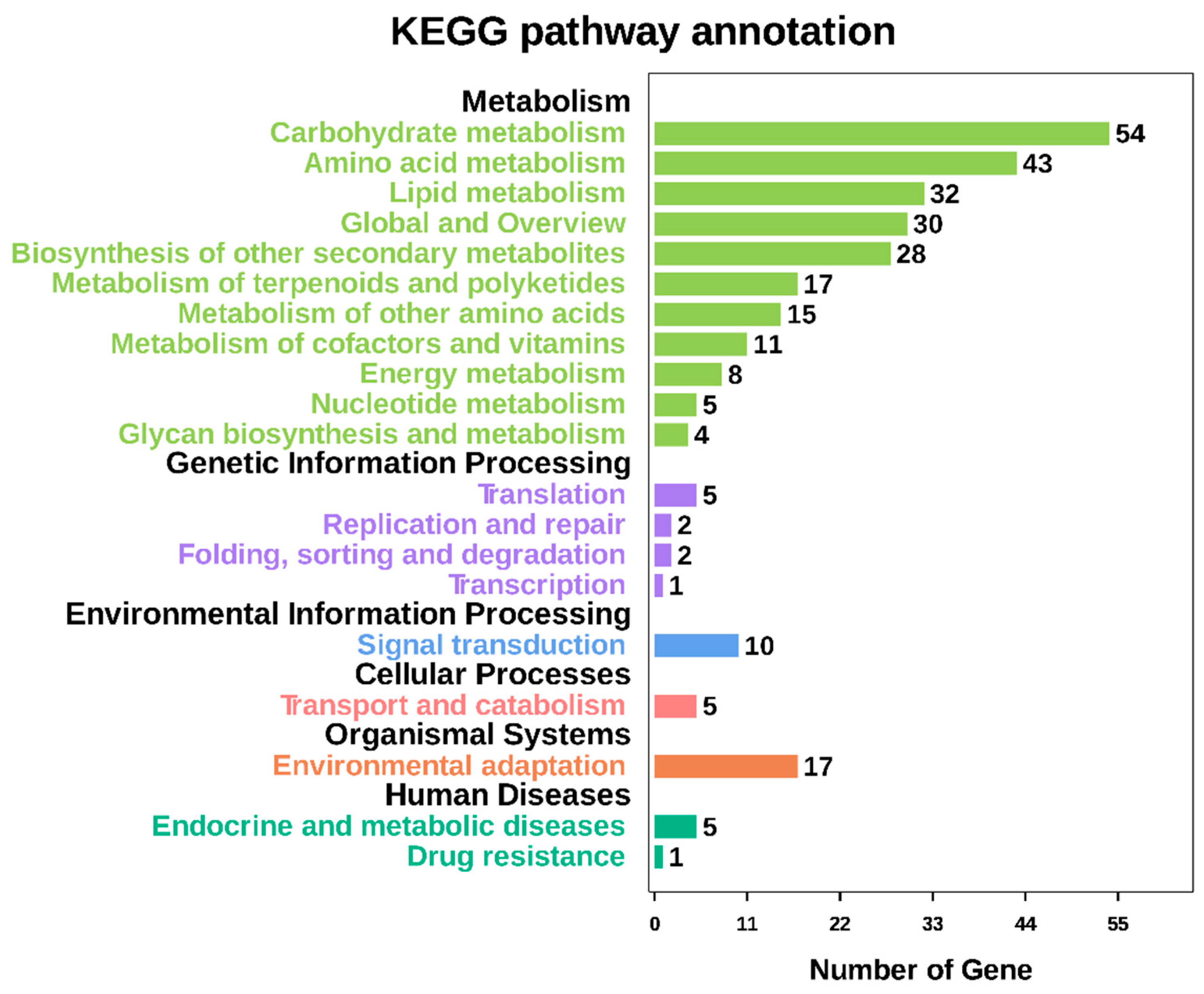
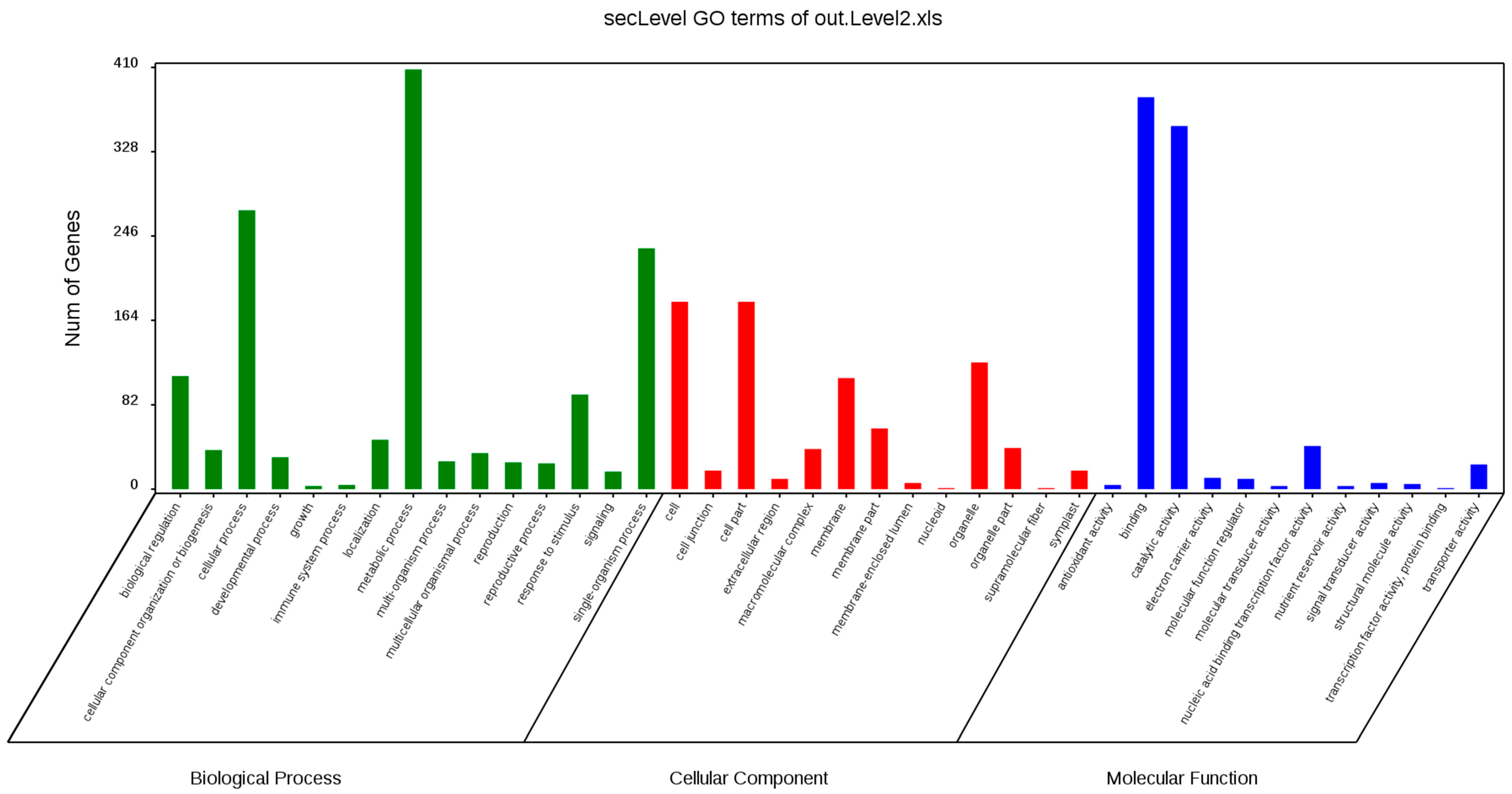
2.8. Functional Enrichment Analysis, Key Genes Mining, and Expression of MCL Modules
GO annotation is one of important steps in the GCN analysis to understand the biological functions of each detected module [
26]. First, we carried out a GO classification of the GCN (
Figure 13). Then, we carried out a GO enrichment analysis using AgriGO analysis tool, which revealed that modules were mainly involved in metabolic processes (GO:0008152), cellular process (GO:0009987), single organism process (GO:0044699), biological regulation (GO:0065007), response to stimulus (GO:0050896), and response to gibberellin (GO:0009739). The identification of over-represented and/or statistically significant biological processes can further reveal the functional features of each detected module. In this study, almost all modules were enriched to metabolic process, response to stimulus, biological regulation, response to gibberellin, and six of them (Module 1, 2, 3, 5, 12, and 14) were statistically significant, indicating that Module 1, Module 2, Module 3, Module 5, Module 12, and Module 14 are the GAs response related function modules.
In gene co-expression analysis, functional consistency among the highly inter connected genes present in the same modules at a given cut-off is very important for gene mining. In this study, functional enrichment analysis has resulted in significant enrichment of 6 modules (Module 1, 2, 3, 5, 12, and 14) containing genes principally involved in various kinds of stimulus response, biological regulation, and metabolic processes.
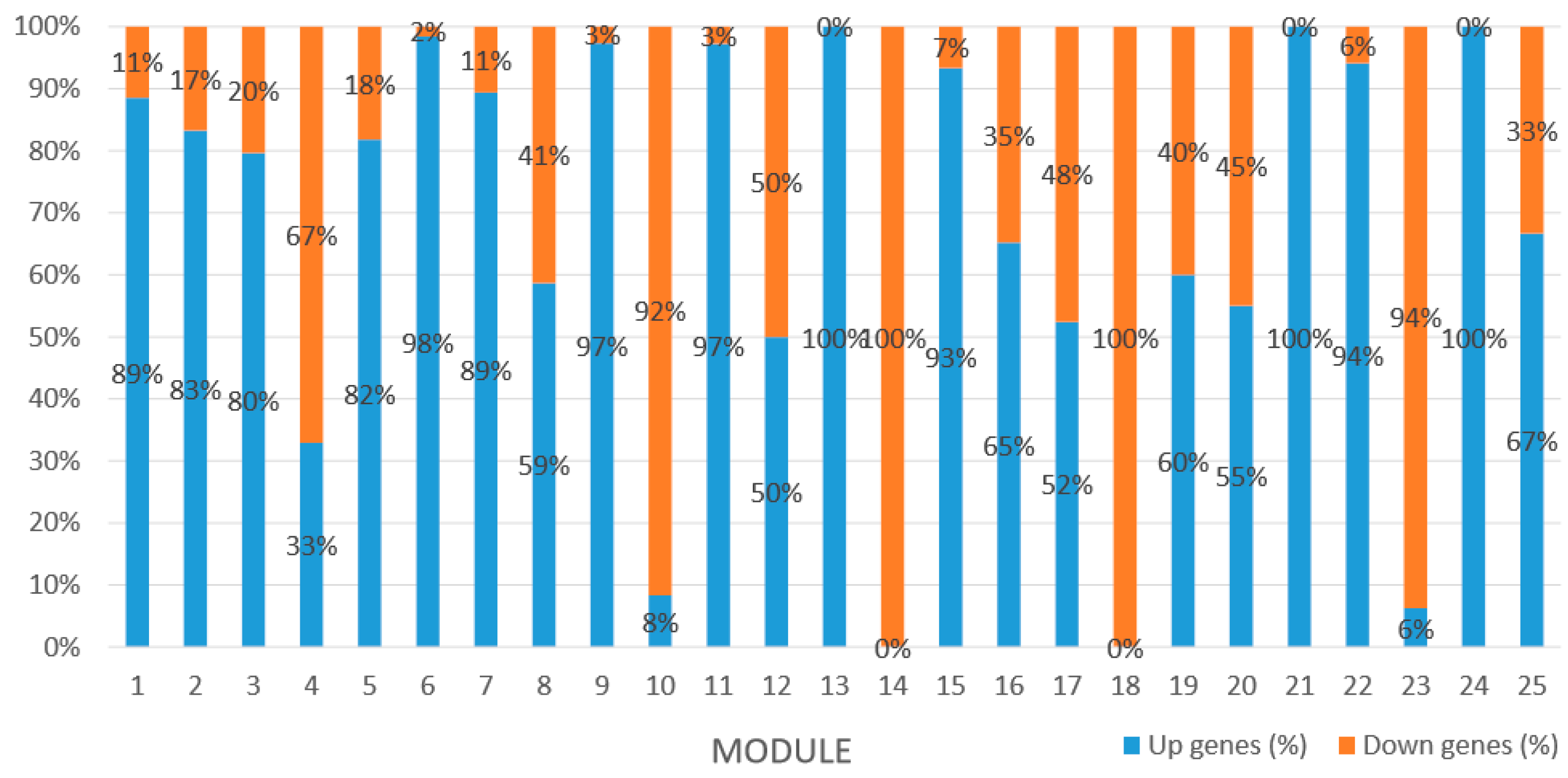
Module 1 consisted of 201 gene nodes and is highly enriched with regulation of transcription (GO:0006355, FDR = 0.00137), biosynthetic process (GO:0044283, FDR = 0.01), peptidyl-tyrosine modification (GO:0018212, FDR = 0.028), catabolic process (GO:0019320, FDR = 0.029), abscisic acid metabolic process (GO:0009687, FDR = 0.045). 89% of genes in the module are upregulated by GA
3, while rest of the genes is downregulated (
Figure 12). A total of 26 genes associated with various kinds of biological process (FDR ≤ 0.05), half of which were annotated genes in this module. The genes are labelled in different colors and co-express the network to show the interaction between genes, as shown in the figure below (
Figure 14). The expression of genes in Module 1 was presented through the heatmap (
Figure S9).
Red represents genes enriched to transcriptional regulation (contig_104756, contig_136393, contig_58865, contig_75642, contig_84469, contig_88830, Congtig11200 genes mentioned above are not annotated in the transcriptome contig_47532 NAC2, contig_47533 NAC11, contig_68192 Heat shock transcription factor A2, contig_91975 transcription factor WRKY7, Congtig10395 transcription factor WRKY, Congtig9007 transcription factor CBF).
Yellow represents enrichment to organisms Genes in anabolic processes (contig_112053, contig_14091, Congtig5132, Congtig9730 genes mentioned above were not annotated in transcriptome analysis, contig_12731 Myristylglycerol phosphate dehydratase, contig_49990 3-phosphonic acid 1-carboxylate Vinyl transferase, contig_58629 acyl-CoA oxidase, Congtig1084 phenylalanine ammonia lyase).
Blue represents a gene enriched for phosphorylation (contig_30231 serine/threonine protein kinase, Congtig16507 LRR receptor-like serine/threonine protein kinase).
Pink represents a gene involved in glucose metabolism (Congtig6131 2,3-independent bisphosphoglycerate). Purple represents the related gene enriched in the process of abscisic acid metabolism (contig_46737 phosphoglycerate mutase PGAM glycase).
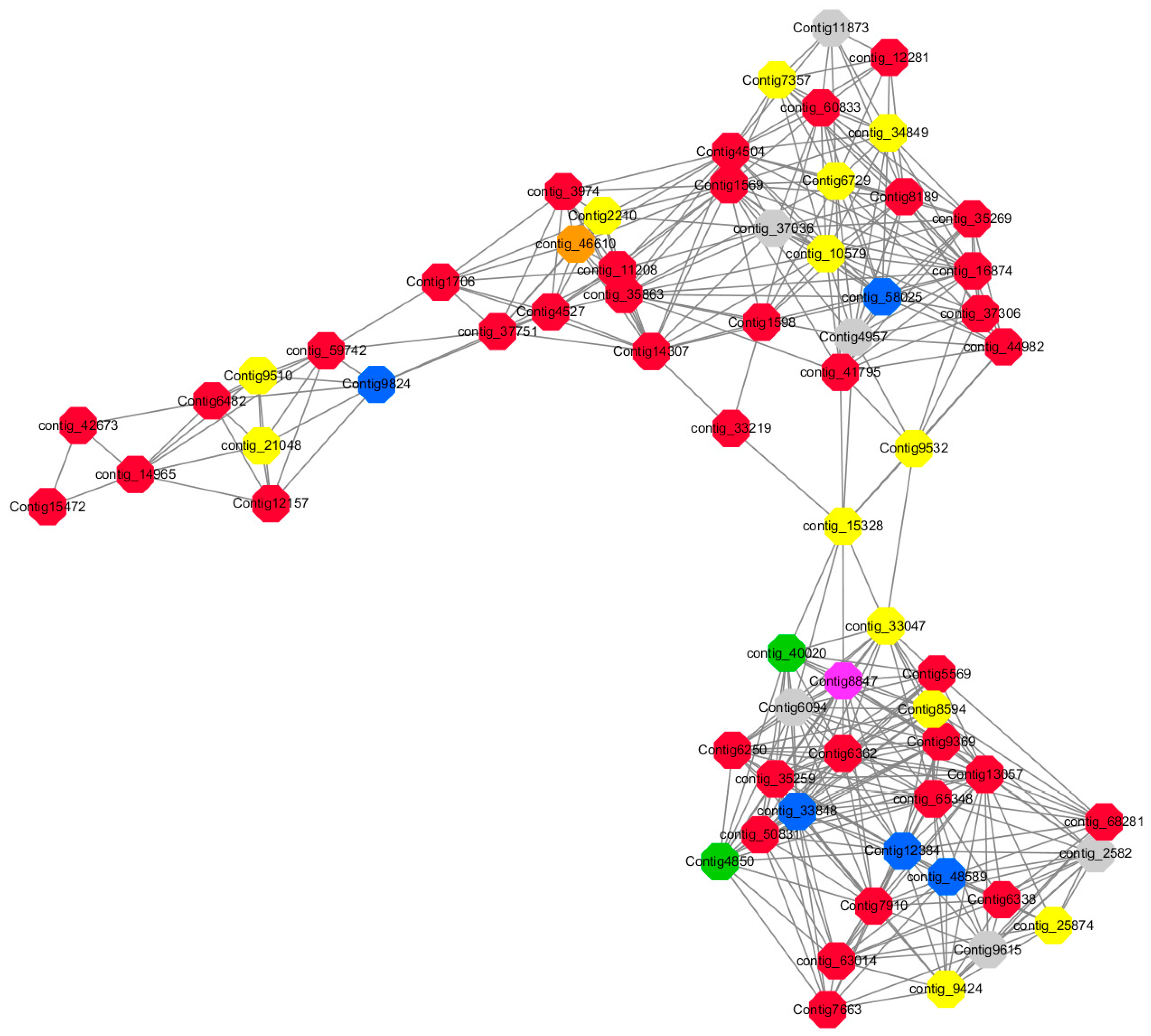
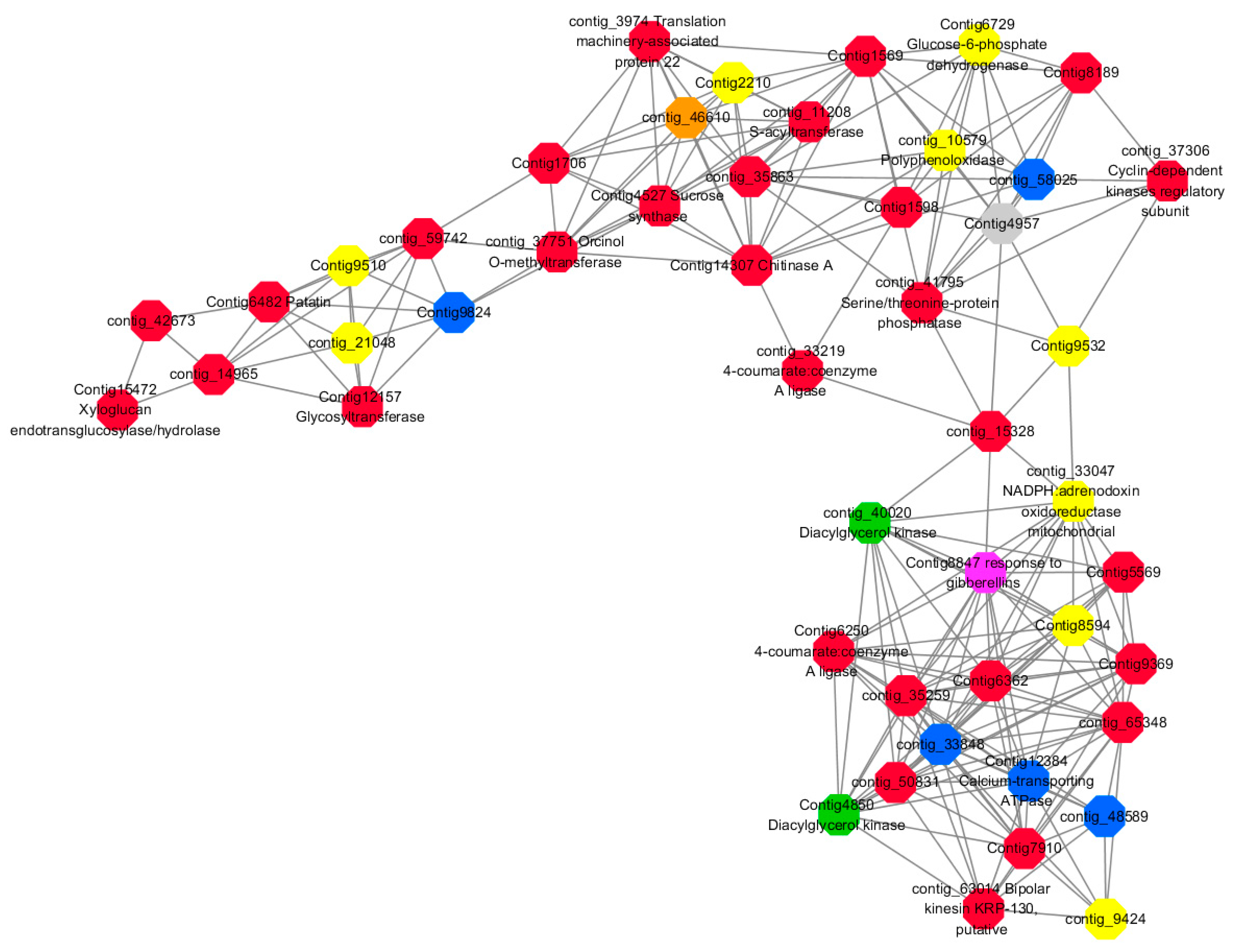
Module 2 consisted of 185 gene nodes and is highly enriched with metabolic process (GO:0008152, FDR = 0.0014), oxidation-reduction process (GO:0055114, FDR = 0.045), cation-transport (GO:0006812, FDR = 0.04), G-protein coupled receptor signaling pathway (GO:0007186, FDR = 0.001), gibberellic acid mediated signaling pathway (GO:0009740, FDR = 0.007), histone phosphorylation (GO:0016572, FDR = 0.03). 83% of genes in the module are upregulated by GA
3, while rest of the genes are downregulated (
Figure 15). A total of 66 genes associated with various kinds of biological process (FDR ≤ 0.05), 23 of which were annotated genes in this module (
Figure S10). The expression of genes in Module 2 was presented through the heatmap (
Figure S10).
Among them, red represents the related genes enriched in the metabolic process (contig_12281, contig_14965, contig_15328, contig_16874, contig_35259, contig_35269, contig_35863, contig_42673, contig_44982, contig_50831, contig_59742, contig_60833, contig_65348, contig_68281, Congtig13057, Congtig1569, Congtig4504, Congtig5569, Congtig6338, Congtig6362, Congtig7663, Congtig7910, Congtig8189, Congtig9369, Congtig1598, Congtig1706 transcriptome analysis in these genes were not being functional annotation, contig_11208 S -acyltransferase, contig_33219 4- coumarate-CoA ligase -A-, contig_37306 regulation of cyclin-dependent kinases, contig_37751 moss melanin methyltransferase, contig_3974 translator correlators, contig_41795 serine/threonine protein phosphatase, contig_63014 bipolar kinesin KRP130, Congtig12157 glycosyltransferase, Congtig14307 chitinase A, Congtig15472 xyloglucan, Congtig4527 sucrose synthase, Congtig6250 4- coumarate-CoA ligase -A-, Congtig6482 patatin).
Yellow represents the related genes enriched in the redox process (contig_21048, contig_25874, contig_34849, contig_9424, Congtig2210, Congtig7357, Congtig8594, Congtig9510, Congtig9532 These genes are not functionally annotated in the transcriptome, contig_1057 polyphenol, contig_33047 NADPH adrenaline redox Enzyme, Congtig6729 Glucose-6-phosphate dehydrogenase).
Blue represents the relevant genes enriched into the cation transport process (contig_48589, Congtig9824 has no functional annotation in the transcriptome, contig_33848 Ammonium transporter, contig_58025 plasma membrane ATPase, Congtig12384 calcium transport ATPase).
Green represents the related gene enriched in the G protein pathway (contig_40020, Congtig4850 diacylglycerol kinase).
Pink represents enrichment Related genes in gibberellin signal transduction (not functionally annotated in the Congtig 8847 transcriptome).
Orange representing genes enriched in histone phosphorylation (contig_46610 transcriptome Commented by the function).
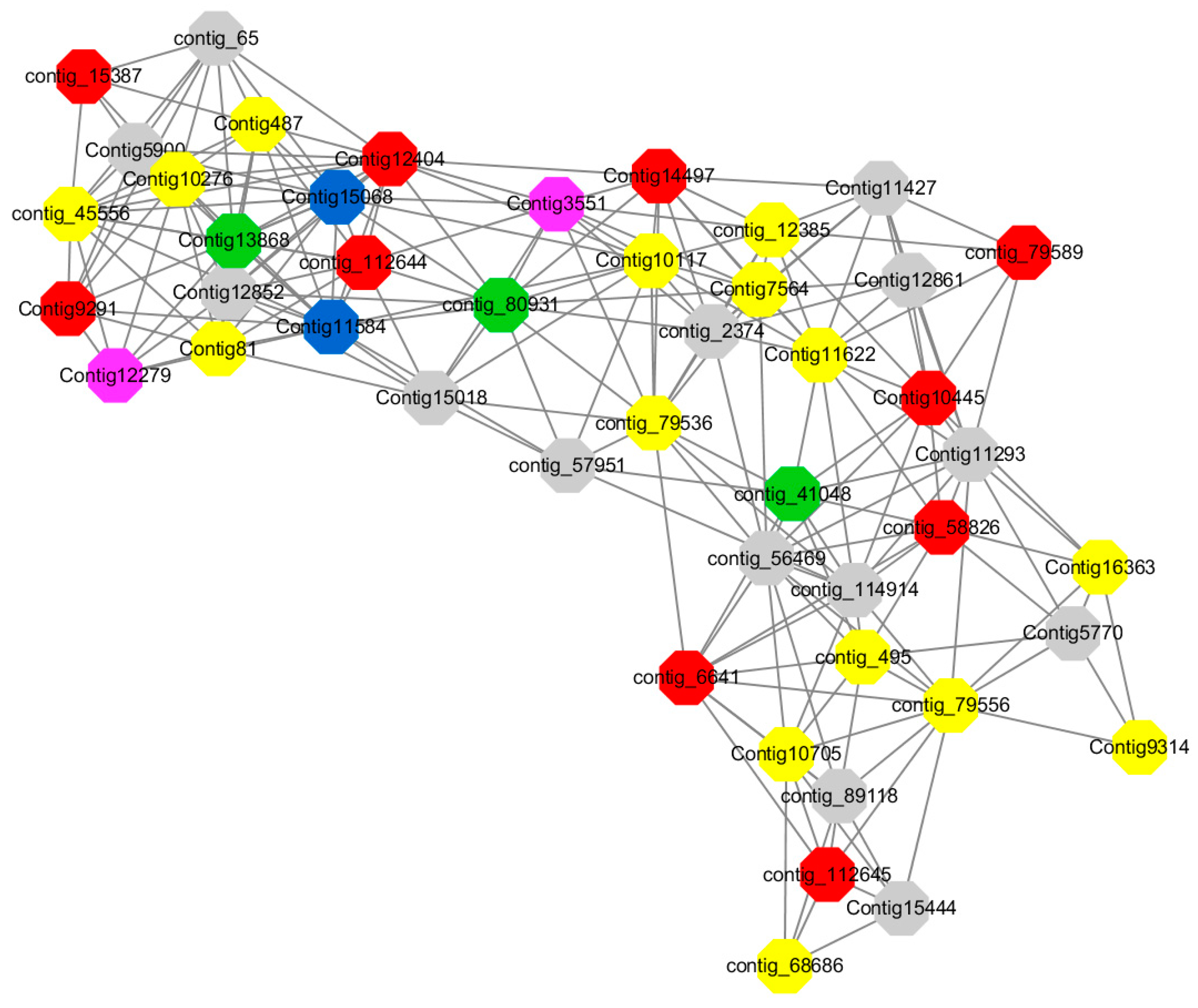
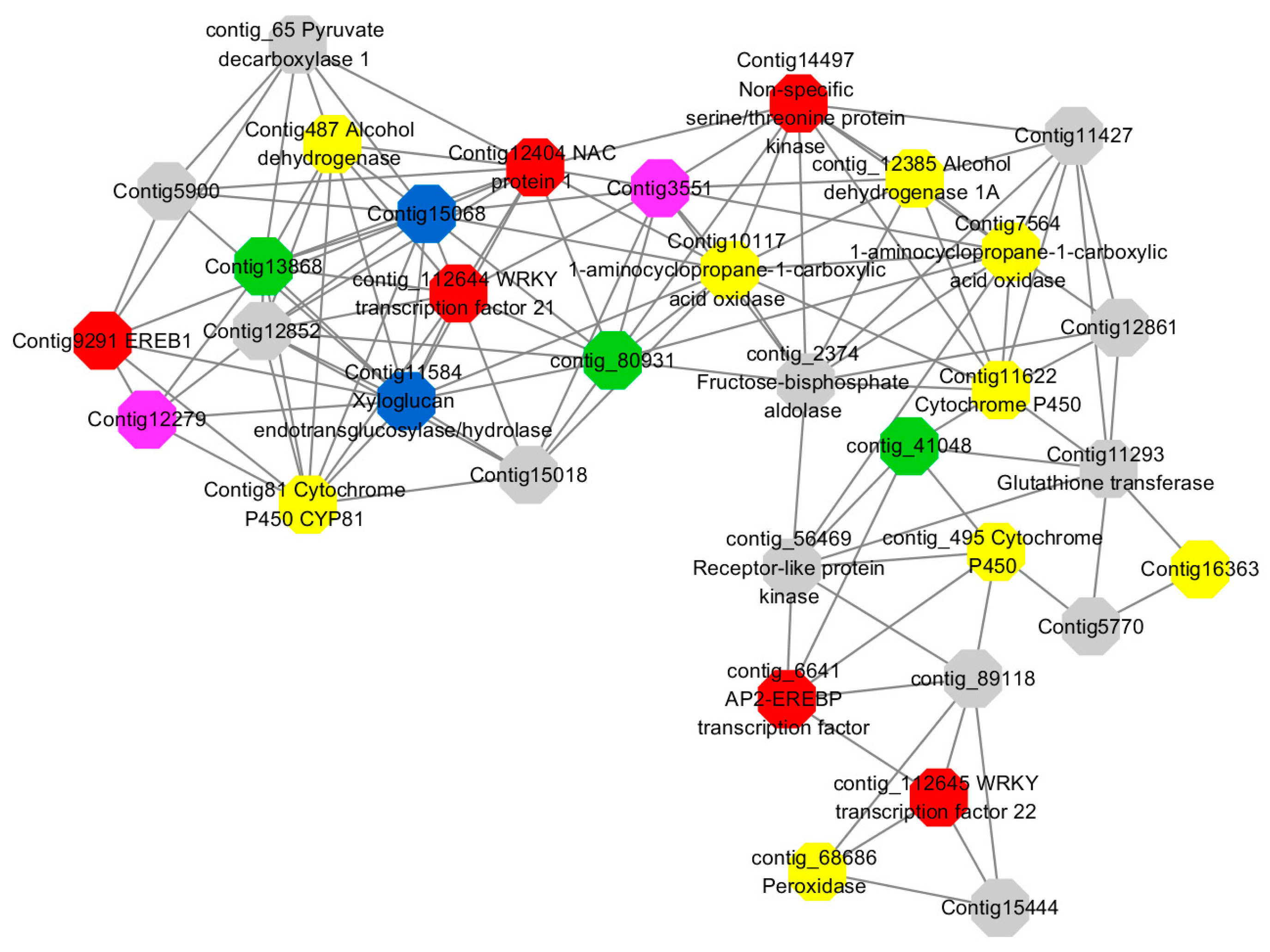
Module 3 consisted of 113 gene nodes and is highly enriched with regulation of cellular process (GO:0050794, FDR = 0.02), oxidation-reduction process (GO:0055114, FDR = 2.12E−05), dephosphorylation (GO:0016311, FDR = 0.025), response to oxygen-containing compound (GO:1901700, FDR = 0.034) and response to cold (GO:0009409, FDR = 0.039). 80% of genes in the module are upregulated by GA
3, while rest of the genes is down regulated (
Figure 12). A total of 46 genes associated with various kinds of biological process (FDR ≤ 0.05), 21 of which were annotated genes in this module. Label genes with different colors and co-expressing networks to show inter-genetic interactions (
Figure 16). The expression of genes in Module 3 was presented through the heatmap (
Figure S11).
Among them, red represents genes enriched in biological process regulation (contig_15387, contig_588269, contig_79589, Congtig10445 are genes without functional annotation in transcriptome analysis, contig_112644 transcription factor WRKY21, contig_112645 transcription factor WRKY22, contig_6641 transcription factor AP2-EREBP, Congtig12404 NAC1, Congtig14497 non-specific serine/threonine protein kinase, Congtig9291 EREB1).
Yellow represents genes enriched in the redox process (contig_45556, contig_79536, contig_79556, Congtig10276, Congtig10705, Congtig16363, Congtig9314 are all genes with no functional annotation in transcriptome analysis, contig_12385 alcohol dehydrogenase 1A, contig_495 Cytochrome P450, contig _68686 Peroxidase, Congtig10117 1-Aminocyclopropane-1-carboxylic acid oxidase, Congtig11622 cytochrome P450, Congtig487 alcohol dehydrogenase, Congtig75641-aminocyclopropane-1-carboxylic acid oxidase, Congtig81 cytochrome P450 CYP81).
Green represents the gene enriched in dephosphorylation (contig_80931, Congtig13868 transcriptome has no gene function annotation, contig_41048 cytoplasmic 5’-nucleotidase III).
Pink represents genes enriched in response to oxygenates (Congtig12279, Congtig3551 transcriptome analysis has no relevant functional annotations).
Blue represents genes enriched in response to low temperatures (no functional annotations in theCongtig15068 transcriptome, Congtig11584 xyloglucan endotransglucosyl hydrolase).
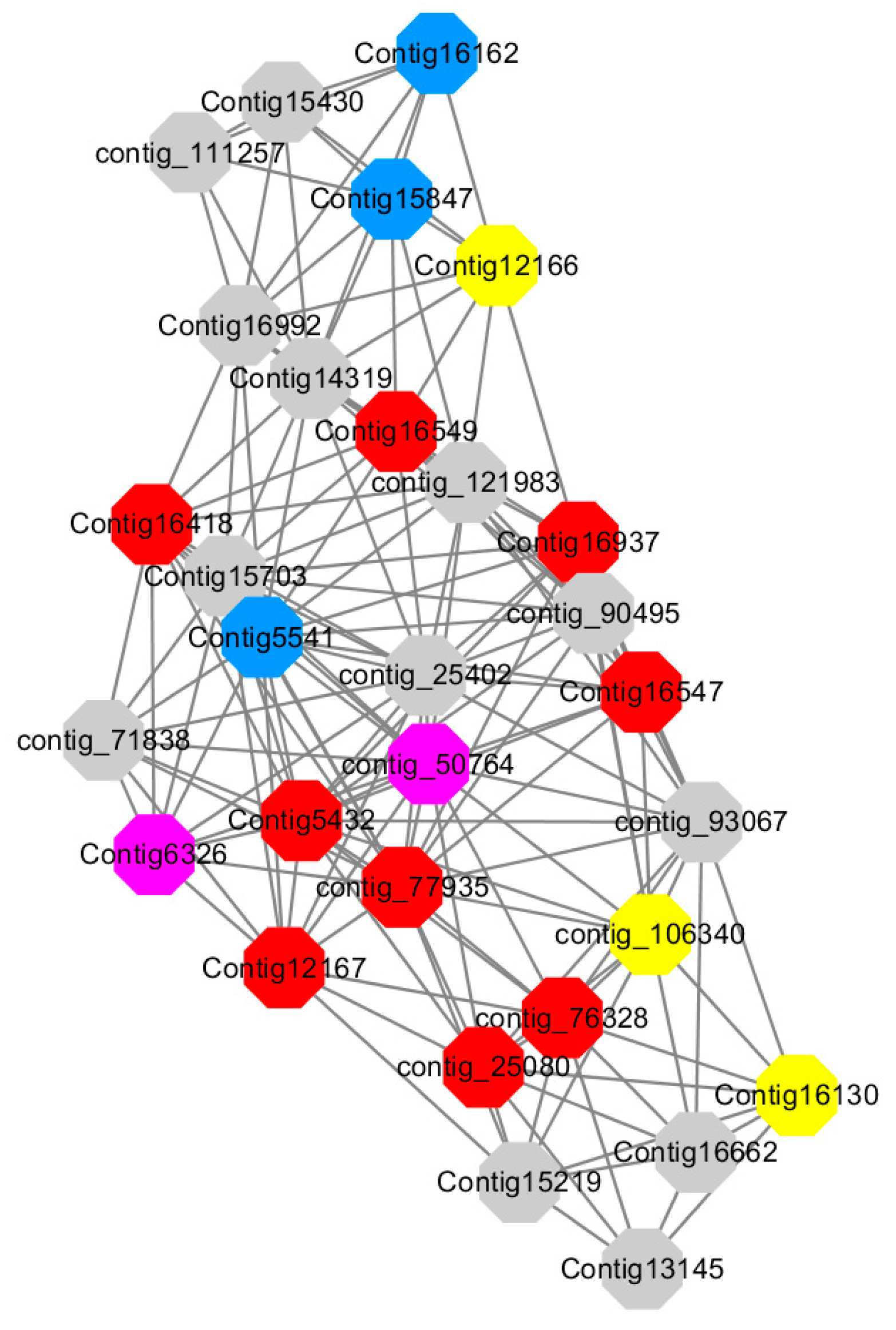
Module 5 consisted of 66 gene nodes and is highly enriched with cellular protein modification process (GO:0006464, FDR = 0.04), response to hormone (GO:0009725, FDR = 0.03), response to stress (GO:0006950, FDR = 0.04), polysaccharide metabolic process (GO:0005976, FDR = 0.008). 82% of genes in the module are upregulated by GA
3, while rest of the genes is downregulated (
Figure 12). A total of 30 genes associated with various kinds of biological process (FDR ≤ 0.05), 14 of which were annotated genes in this module. Label genes with different colors and co-expressing networks to show inter-genetic interactions (
Figure 17). The expression of genes in Module 5 was presented through the heatmap (
Figure S12).
Among them, red represents genes enriched in cellular protein modification (Congtig16549, Congtig16937, Congtig16547, Congtig12167, Congtig5432, contig_76328, contig_77935, contig_25080 is a gene with no functional annotation in transcriptome analysis, Congtig16418 serine/threonine protein kinase).
Yellow represents a gene enriched in response hormone (contig_106340 transcriptome analysis functionally annotated genes, Congtig12166 pectin gene, Congtig16130 transcription factor AP2).
Pink represents genes enriched in response stress (Congtig6326 pectin, contig_50764 hexosyltransferase).
Blue represents Genes enriched in polysaccharide metabolism (Congtig15847, Congtig5541 transcriptome analysis, no functionally annotated genes, Congtig16162 peroxidase).
Module 12 consisted of 34 gene nodes and is highly enriched with regulation of biological processes (GO: 0050789), vegetative growth to reproductive growth (GO: 0010228), L-phenylalanine metabolism (GO: 0006558). 50% of genes in the module were upregulated (
Figure 12). Labeled genes with different colors and co-expressing networks to show inter-genetic interactions (
Figure 18). The expression of genes in Module 12 was presented through the heatmap (
Figure S13).
Red represents genes enriched for biological process regulation (first no functionally annotated genes in the Congtig10652 transcriptome analysis, contig_55686, contig_90494 transcriptome analysis without functional annotation Gene, contig_58912 transcription factor WRKY27, contig_98142 thioredoxin gene).
Yellow represents the gene enriched into vegetative growth to reproductive growth (contig_66570, contig_55519, Congtig10396 are all transcriptome analysis without functional annotation gene).
Blue represents enrichment to L-phenylalanine metabolism (contig_55519 phenylalanine ammonia lyase).
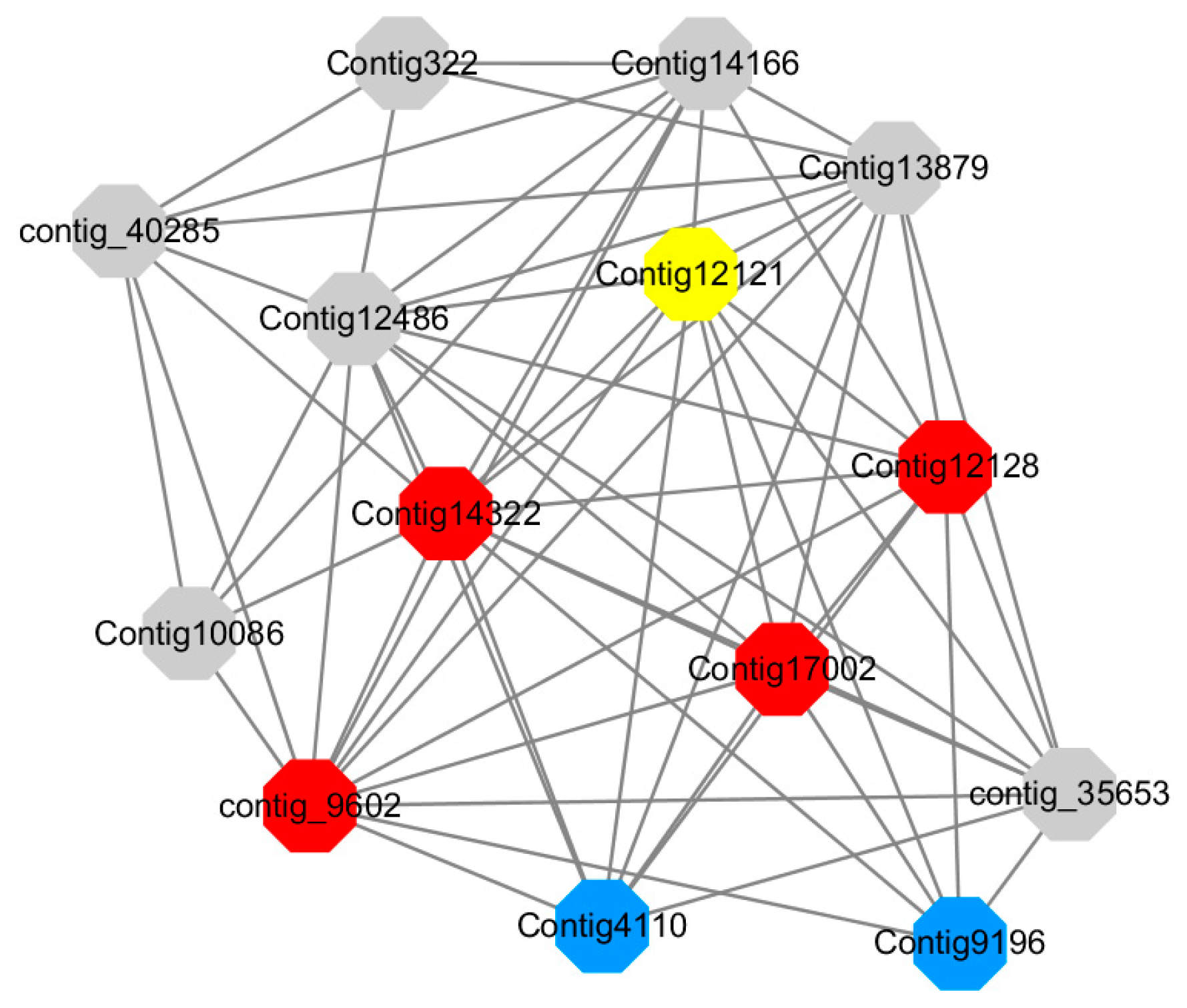
Module 14 consisted of 31 gene nodes and is highly enriched with protein phosphorylation (GO: 0006468), cell recognition (GO: 0008037), response to gibberellin (GO: 0009739). All genes in the module were downregulated (
Figure 12). Label genes with different colors and co-expressing networks to show inter-genetic interactions (
Figure 19). The expression of genes in Module 14 was presented through the heatmap (
Figure S14).
Red represents genes enriched for protein phosphorylation (Congtig14322, Congtig12128, Congtig17002 are genes with no functional annotation in transcriptome analysis, contig_9602 is a serine/threonine protein kinase). Yellow represents rich genes that are integrated into cell recognition (Congtig12121, Congtig4110 are genes that have no functional annotation in transcriptome analysis). Blue represents a gene that is enriched in response to gibberellin (a gene that has no functional annotation in transcriptome analysis atCongtig9196).
2.9. qRT-PCR Verifing Gene Expression Profiles
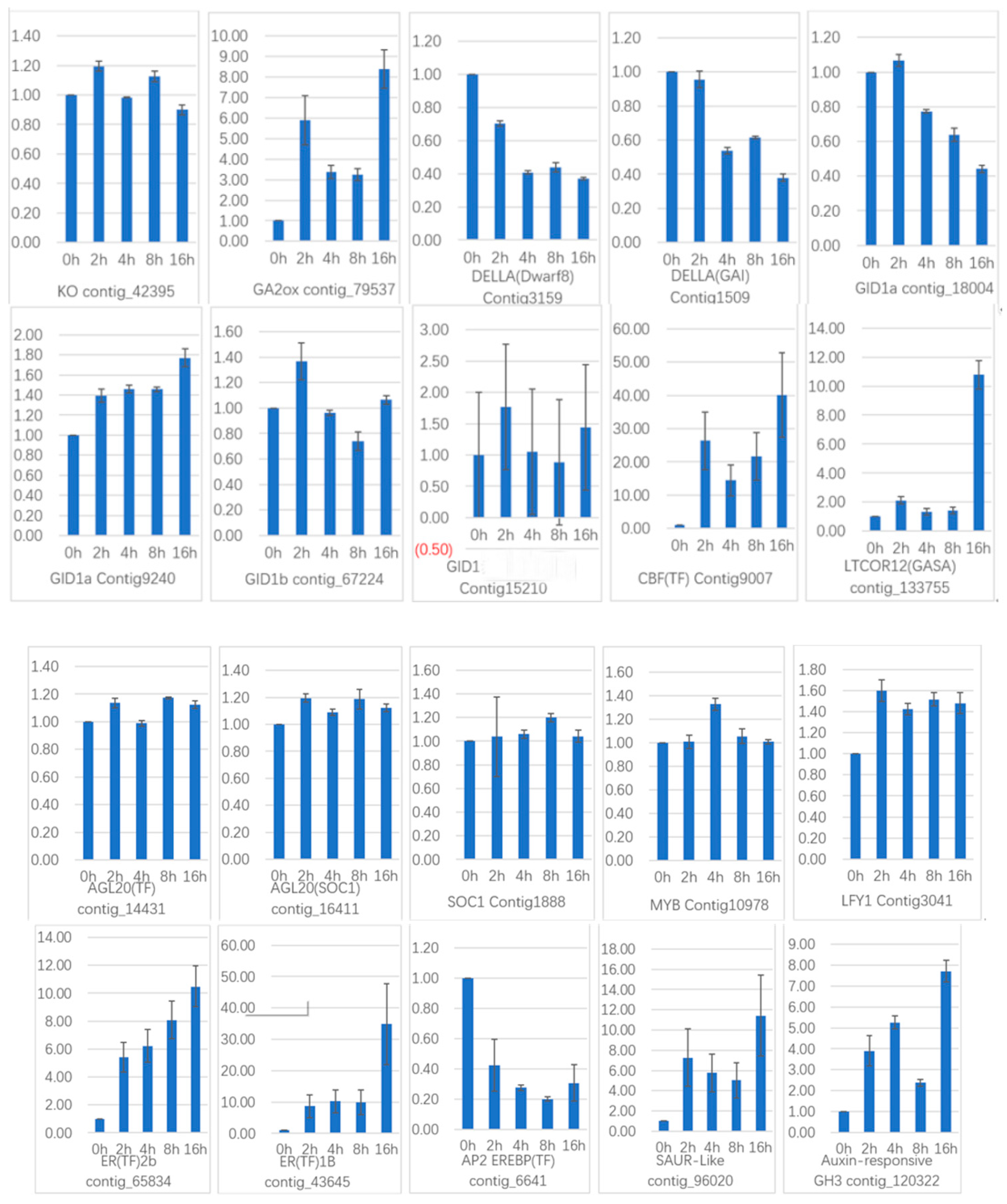
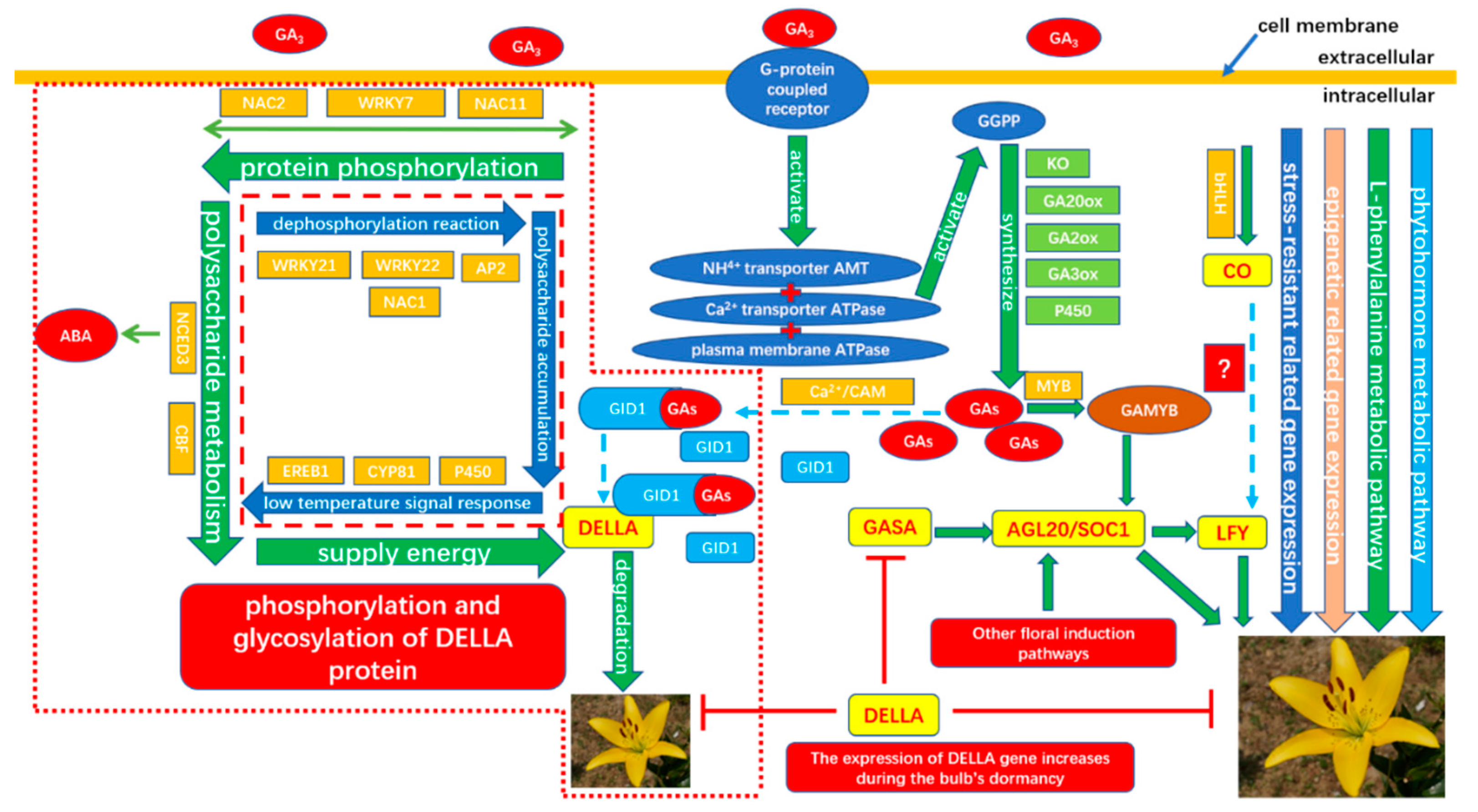
To verify the gene expression in our Illumina sequencing analyses, 20 DEGs containing two candidate genes (Gibberellic acid insensitive, GAI contig 1509; GA-insensitive dwarf mutant1a, GID1a contig_18004) related to the gibberellin signal transduction were selected for qRT-PCR using samples of 0, 2, and 4 h stages originally used for RNA-Seq, all of which are known to be related to Gibberellins signal, including the genes encoding GA2ox (gibberellin 2-oxidase, Contig_79537), DELLA (Dwarf8, Contig3159), CBF (C-repeat binding factor, Contig9007), LTCOR12 (gibberellic acid-stimulated in Arabidopsis, GASA, Contig_133755), ER (estrogen receptor, Contig_43645), AP2-EREBP (Apetala2/ethylene responsive element binding protein, Contig_6641), SAUR-like (small auxin-up RNA-like, Contig_96020), auxin-responsive GH3 (auxin-responsive Gretchen Hagen 3, Contig_120322), DELLA (GAI, Contig1509), GID1a (Contig18004) and so on. The Ct values of the LoTIP (tonoplast intrinsic protein) in all samples ranged from 24.0 to 28.0. All transcripts showed the same expression pattern as the in silico differential analysis results from high-throughput sequencing.
These genes were selected for their key roles in regulating GA signal transcription, endogenous plant hormones responses, and cold responses. The results presented in
Figure 20 showed that the expression levels of
DELLA (
Dwarf8 Contig3159,
GAI Contig1509) decreased sharply in 2 h and 4 h samples than in the 0 h sample, which indicated that the two genes may play key roles in GA signal response and may delay the floral transition. The expression of transcription factor genes
CBF (Contig9007) and
ER (contig_43645) increased sharply in the 2 h sample than in the untreated sample (0 h), which indicated that these TF genes may play important roles in GA signal response and transduction process. The expression of
GA2ox (contig_79537) was higher in 2 h and 4 h stages than the 0 h stage, which indicated that
GA2ox has a passivation effect to gibberellin.
AP2-EREBP (contig_6641),
SAUR-like (contig_96020), and
auxin-responsive GH3 (Gretchen Hagen 3,contig_120322) are related to endogenous hormone. The expression of
AP2-EREBP (contig_6641) continued to drop sharply in 2, 4, and 8 h, and increased a little in 16 h, which was still less than half of the expression in 0 h. The expression of
SAUR-like (contig_96020) upregulated dramatically in 2 h, and continued to downregulate in 4 h and 8 h, following a sharp recovery in 16 h, which was almost 11 times the original expression level in 0 h. The
auxin-responsive GH3 (contig_120322) continued to upregulate dramatically except a sharp fall in 8 h. The cold-regulated gibberellin-regulated gene
LTCOR12 (
GASA contig_133755) expressed more in 2 h and 4 h stages than 0 h and expressed highest after being treated by. GA for 16 h, which showed an obvious delay in expression contrast to the genes mentioned above. The expression of the gene
GAI (Contig1509) also encoding
DELLA protein and a gibberellin signal receptor gene
GID1a (contig_18004) did not show differential in the first three stages (0, 2, and 4 h), but decreased significantly in the 16 h stage. We predicted that the bulb has played an important role in for the LA hybrid lily adaptation to GA and releasing the dormancy.
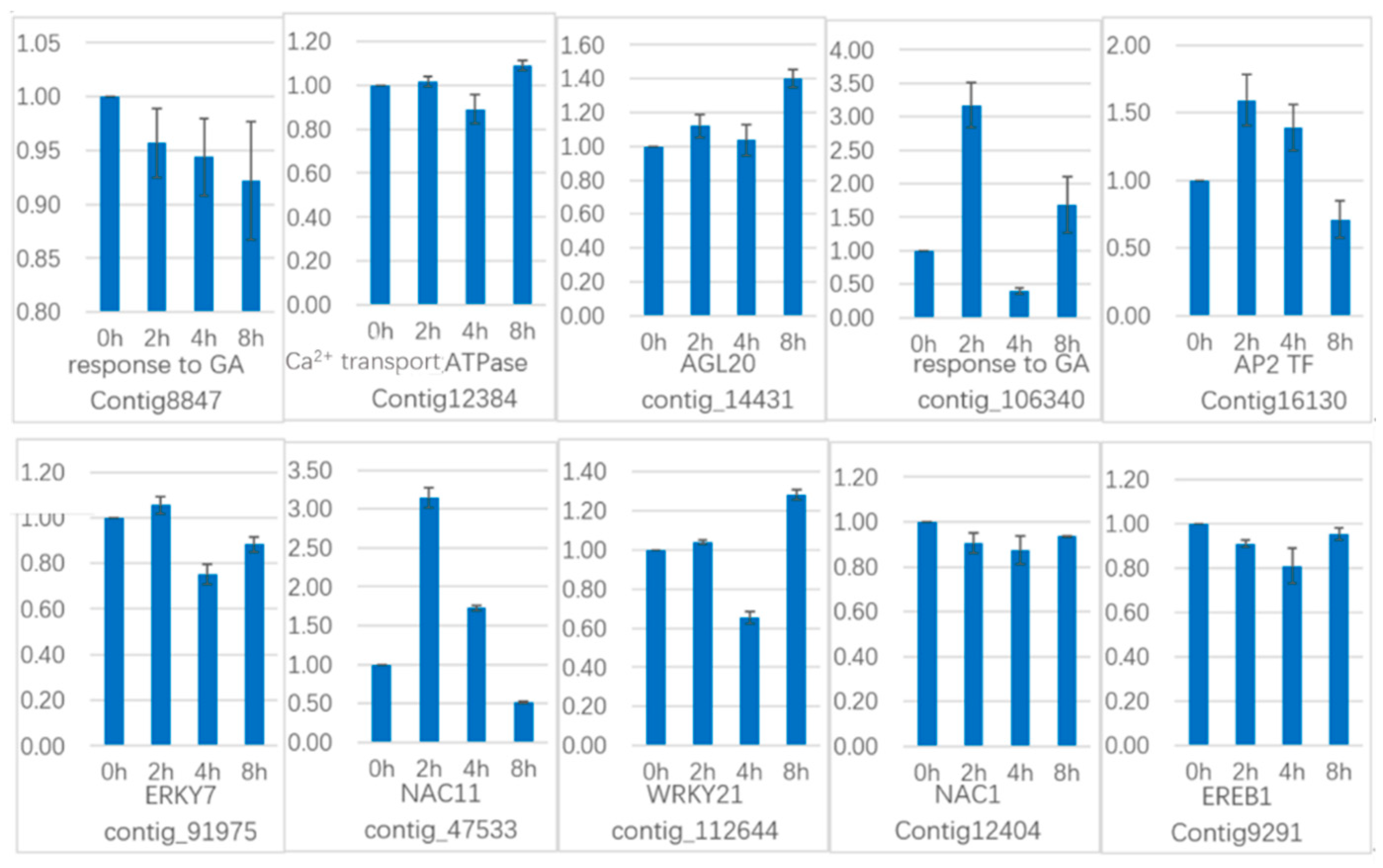
Another 10 genes were selected for qRT-PCR (
Figure 21) in GCN modules. Three genes in Module 2:Contig8847 is a key gene that responds to gibberellin by gene co-expression clustering and GO function enrichment, but it is not annotated in transcriptome analysis. The expression level increased slightly in 2 h, and the content in 4 h increased sharply. In the qRT-PCR, the content decreased continuously during the treatment time.
Calcium transport ATPase-related gene (Contig12384) showed a significant increase in transcriptome results. It also showed an upward trend in qRT-PCR. The transcription factor
AGL20 (
Agamousclike 20, contig_14431) was significantly upregulated in the transcriptome results, and the expression of 4 h increased sharply. In the qRT-PCR, the expression increased sharply at 2 h, decreased slightly at 4 h, and increased sharply at 8 h. Rising expression trends.
Two genes in Module 5: contig_106340 is also a key gene in response to gibberellin analyzed by gene co-expression clustering and GO function enrichment, but it is not in transcriptome analysis. In the transcriptome data, the expression level increased sharply at 2 h, but decreased at 4 h, but it still showed upregulated expression. In the qRT-PCR, it increased sharply at 2 h, decreased at 4 h, and increased sharply at 8 h, and the whole expression was upregulated. The transcription factor AP2 (Contig16130) was upregulated in the transcriptome results and was upregulated in 2h, and decreased in 4 h, and upregulated in the whole. In the qRT-PCR results, the expression was upregulated at 2 h and 4 h, and downregulated at 8 h.
Two genes in Module 1: the transcription factor WRKY7 (contig_91975) has a sustained downregulated expression in the transcriptome, and there is a slight increase in qRT-PCR results in 2 h, followed by downregulation. The transcription factor NAC11 (NAM-ATAF-CUC 11, contig_47533) continued to be upregulated in the transcriptome data, it was upregulated in the qRT-PCR results until 4 h, and downregulated in 8 h.
There are three genes in Module 3: the transcription factor WRKY21 (contig_112644) is continuously upregulated in the transcriptome data and is downregulated in the qRT-PCR analysis at 4 h and upregulated in 8 h. The transcription factor NAC1 (Contig12404) continued to be significantly upregulated in transcriptome data and was downregulated in qRT-PCR analysis. The transcription factor EREB1 (ineversible electrical break down 1, Contig9291) continued to be significantly upregulated in the transcriptome results, with a slight downregulation in qRT-PCR results.

































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}