Proliferative Pathways of Vascular Smooth Muscle Cells in Response to Intermittent Hypoxia



Abstract
:
{kind=link}
{kind=link}
{kind=link}
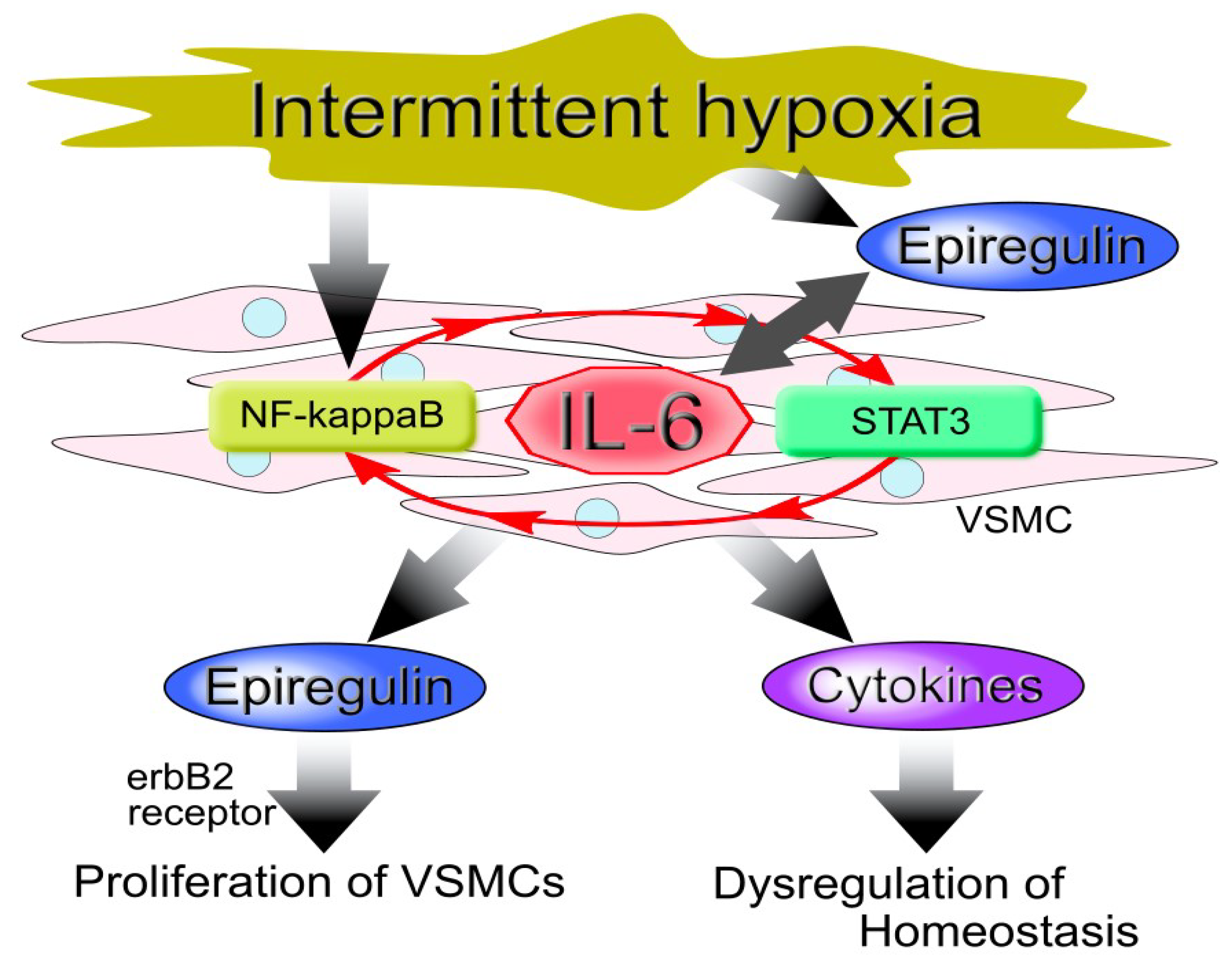{kind=link}
1. Introduction
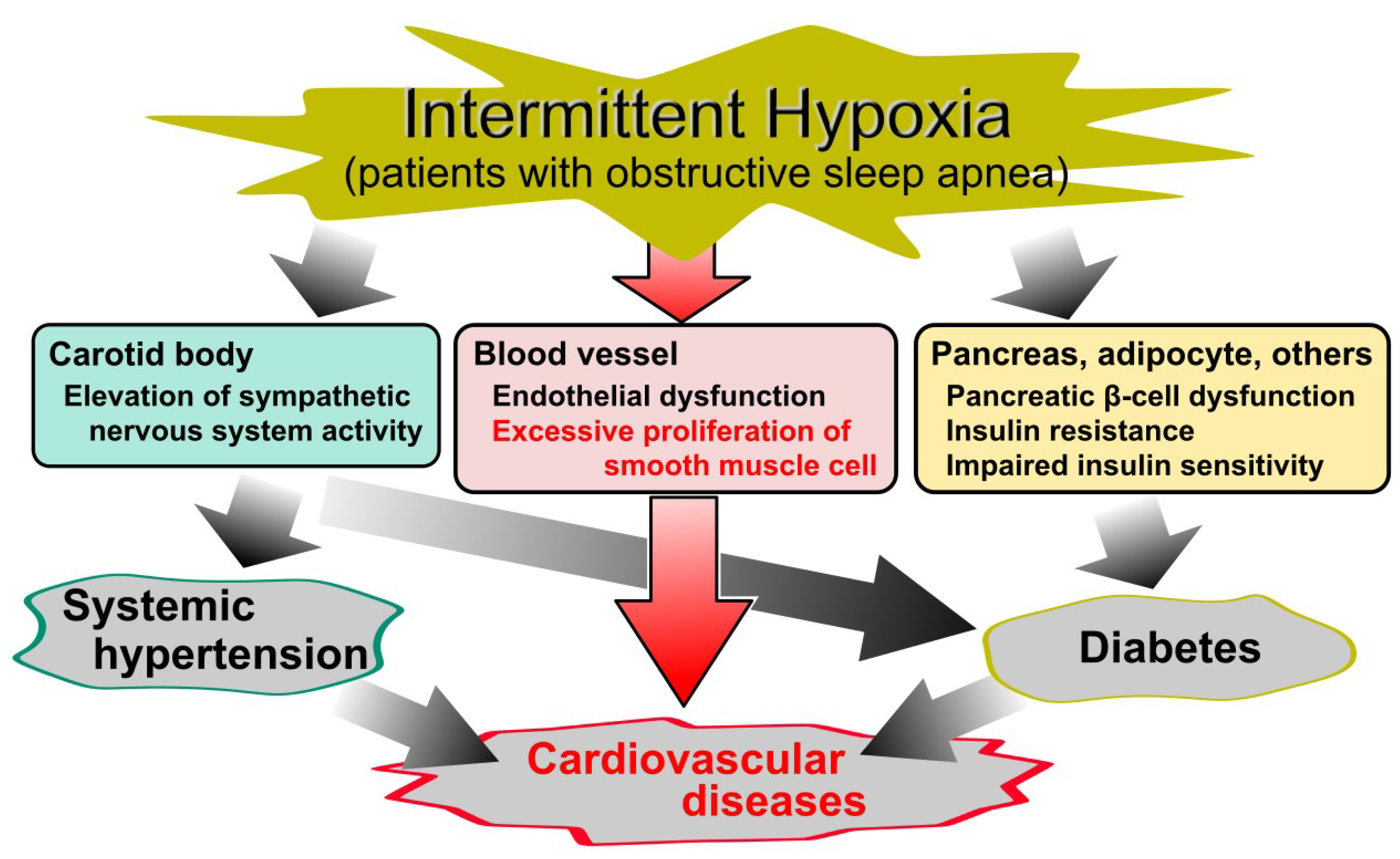
2. Vascular Smooth Muscle Cells (VSMCs) in Atherosclerosis
3. Reactive Oxygen Species (ROS) and Transcriptional Factors in Intermittent Hypoxia (IH)
3.1. Reactive Oxygen Species (ROS)
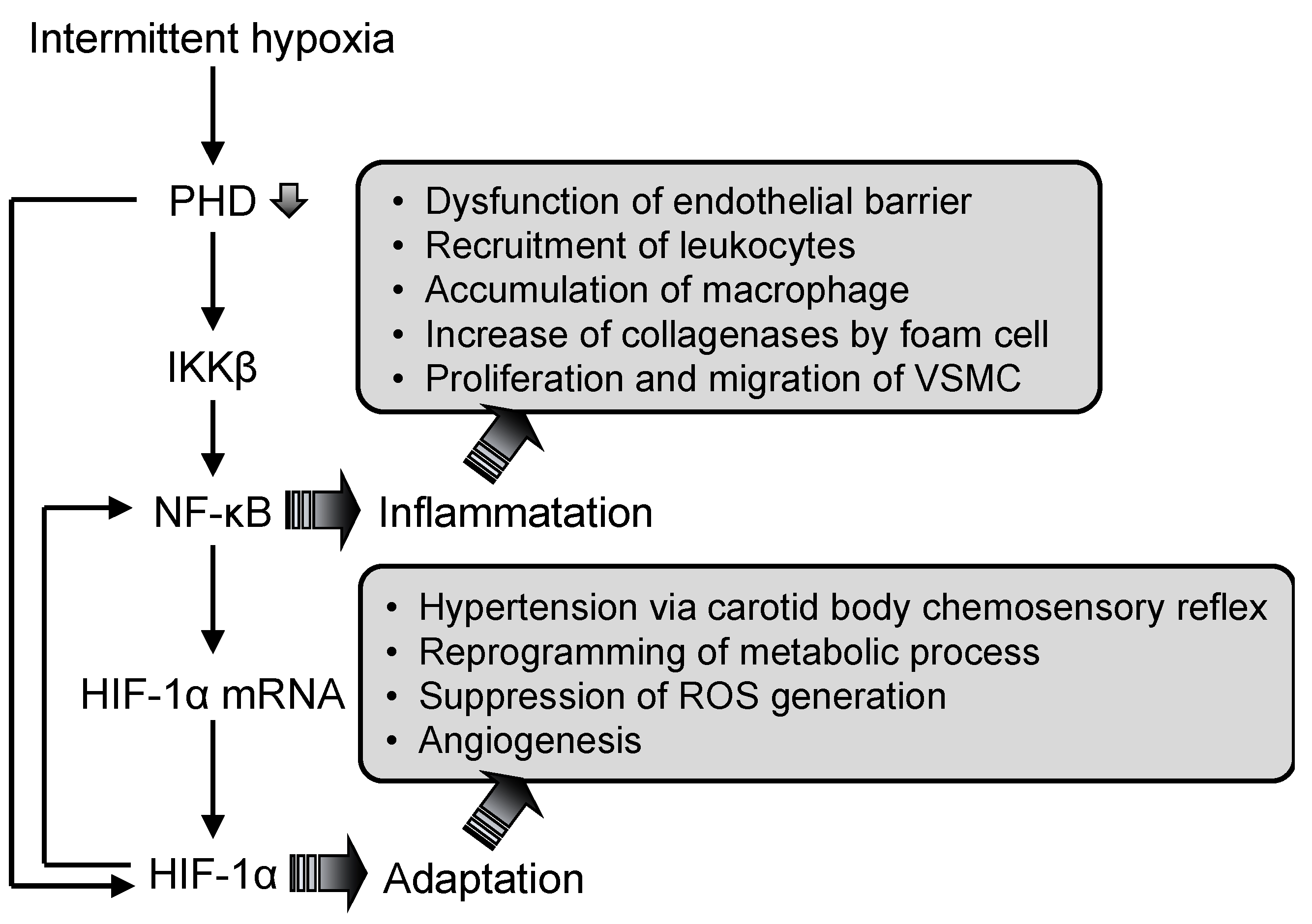
3.2. Nuclear Factor (NF)-κB
3.3. Hypoxia-Inducible Factor (HIF)-1
3.4. Interaction Between Nuclear Factor (NF)-κB and Hypoxia-Inducible Factor (HIF)-1
4. Interleukin (IL)-6
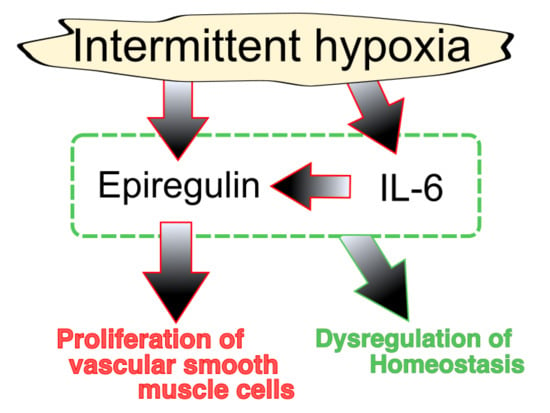
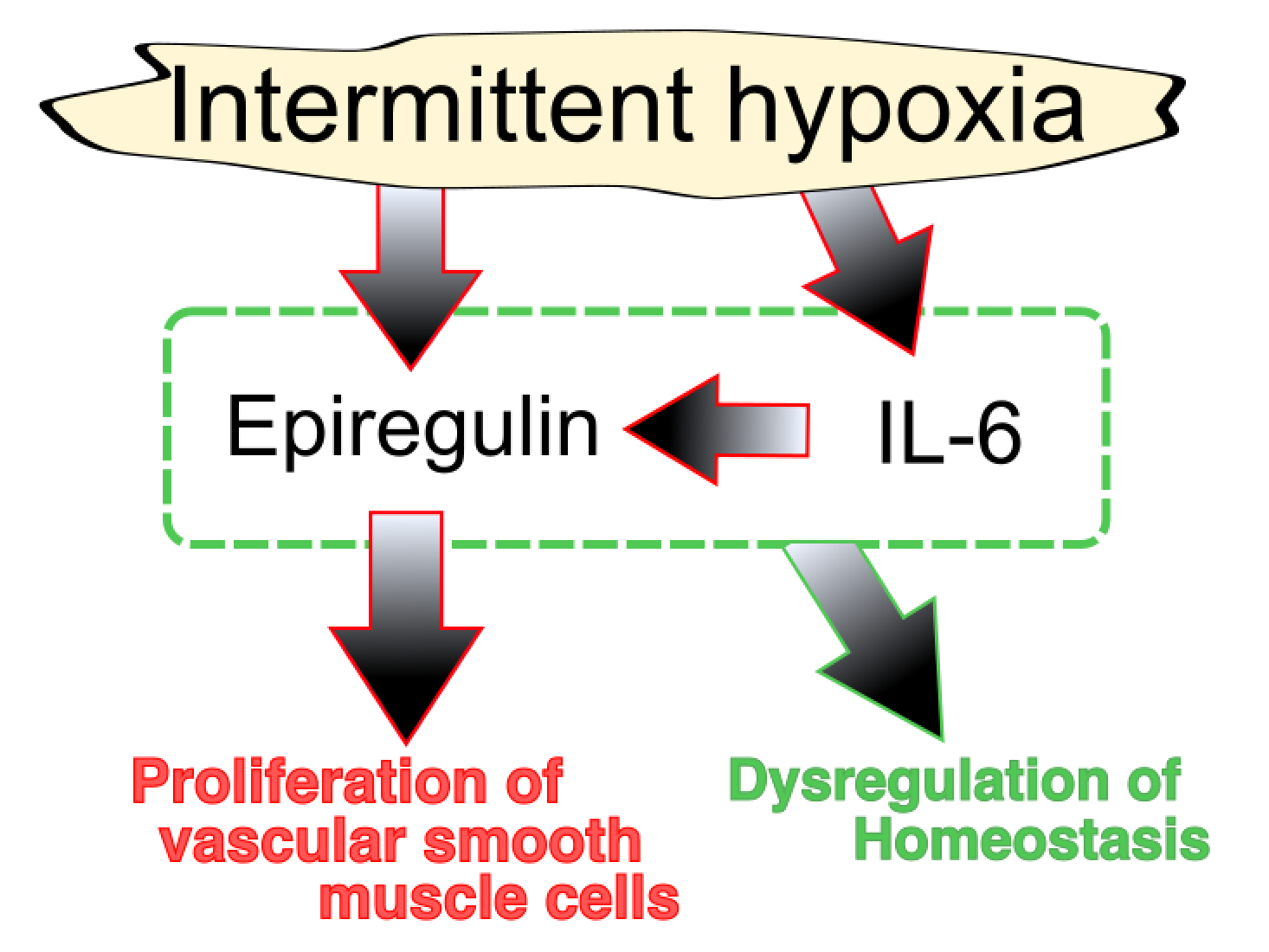
5. Epiregulin
6. Chronic Inflammatory Diseases (CID)
7. Summary and Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | Protein kinase B |
| CID | Chronic inflammatory diseases |
| CPAP | Continuous positive airway pressure |
| DUSP1 | Dual specificity protein phosphatase 1 |
| EGF | Epidermal growth factor |
| erbB | EGF receptor |
| ERK | Extracellular signal-regulated kinase |
| GLUT | Glucose transporter |
| hCASMC | Human coronary artery smooth muscle cell |
| HIF | Hypoxia-inducible factor |
| IH | Intermittent hypoxia |
| I-κB | Inhibitor of NF-κB |
| IKKβ | I-κB kinase-β |
| IL | Interleukin |
| MAPK | Mitogen-activated protein kinase |
| NF-κB | Nucleus factor-κB |
| NRF2 | Nuclear factor (erythroid-derived 2)-like 2 |
| OSA | Obstructive sleep apnea |
| PHD | Prolyl hydroxylases |
| RASMC | Rat aortic smooth muscle cell |
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
| STAT3 | Signal transducer and activator of transcription 3 |
| VSMC | Vascular smooth muscle cell |
References
- Kapur, V.K.; Auckley, D.H.; Chowdhuri, S.; Kuhlmann, D.C.; Mehra, R.; Ramar, K.; Harrod, C.G. Clinical Practice Guideline for Diagnostic Testing for Adult Obstructive Sleep Apnea: An American Academy of Sleep Medicine Clinical Practice Guideline. J. Clin. Sleep Med. 2017, 13, 479–504. [Google Scholar] [CrossRef] [PubMed]
- Laratta, C.R.; Ayas, N.T.; Povitz, M.; Pendharkar, S.R. Diagnosis and treatment of obstructive sleep apnea in adults. CMAJ. 2017, 189, E1481–E1488. [Google Scholar] [CrossRef] [Green Version]
- Peppard, P.E.; Young, T.; Barnet, J.H.; Palta, M.; Hagen, E.W.; Hla, K.M. Increased prevalence of sleep-disordered breathing in adults. Am. J. Epidemiol. 2013, 177, 1006–1014. [Google Scholar] [CrossRef]
- Young, T.; Palta, M.; Dempsey, J.; Skatrud, J.; Weber, S.; Badr, S. The occurrence of sleep-disordered breathing among middle-aged adults. N. Engl. J. Med. 1993, 328, 1230–1235. [Google Scholar] [CrossRef] [PubMed]
- Kent, B.D.; Grote, L.; Ryan, S.; Pépin, J.-L.; Bonsignore, M.R.; Tkacova, R.; Saaresranta, T.; Verbraecken, J.; Lévy, P.; Hedner, J.; et al. Diabetes mellitus prevalence and control in sleep-disordered breathing. Chest 2014, 146, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bi, Y.; Zhang, Q.; Pan, F. Obstructive sleep apnoea and the risk of Type 2 diabetes: A meta-analysis of prospective cohort studies: OSA and Type 2 diabetes. Respirology 2013, 18, 140–146. [Google Scholar] [CrossRef]
- Sahlin, C.; Sandberg, O.; Gustafson, Y.; Bucht, G.; Carlberg, B.; Stenlund, H.; Franklin, K.A. Obstructive sleep apnea is a risk factor for death in patients with stroke: A 10-year follow-up. Arch. Intern. Med. 2008, 168, 297. [Google Scholar] [CrossRef]
- Sorajja, D.; Gami, A.S.; Somers, V.K.; Behrenbeck, T.R.; Garcia-Touchard, A.; Lopez-Jimenez, F. Independent association between obstructive sleep apnea and subclinical coronary artery disease. Chest 2008, 133, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Durán, J.; Esnaola, S.; Rubio, R.; Iztueta, Á. Obstructive sleep apnea–hypopnea and related clinical features in a population-based sample of subjects aged 30 to 70 yr. Am. J. Respir. Crit. Care Med. 2001, 163, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Nieto, F.J. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. JAMA 2000, 283, 1829. [Google Scholar] [CrossRef] [PubMed]
- Peppard, P.E. Longitudinal study of moderate weight change and sleep-disordered breathing. JAMA 2000, 284, 3015. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S. Adipose tissue inflammation by intermittent hypoxia: Mechanistic link between obstructive sleep apnoea and metabolic dysfunction. J. Physiol. 2017, 595, 2423–2430. [Google Scholar] [CrossRef]
- Conde, S.V.; Sacramento, J.F.; Guarino, M.P.; Gonzalez, C.; Obeso, A.; Diogo, L.N.; Monteiro, E.C.; Ribeiro, M.J. Carotid body, insulin, and metabolic diseases: Unraveling the links. Front. Physiol. 2014, 5, 418. [Google Scholar] [CrossRef] [PubMed]
- Garvey, J.F.; Taylor, C.T.; McNicholas, W.T. Cardiovascular disease in obstructive sleep apnoea syndrome: The role of intermittent hypoxia and inflammation. Eur. Respir. J. 2009, 33, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Kanagy, N.L. Vascular effects of intermittent hypoxia. ILAR J. 2009, 50, 282–288. [Google Scholar] [CrossRef]
- Prabhakar, N.R.; Semenza, G.L. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol. Rev. 2012, 92, 967–1003. [Google Scholar] [CrossRef]
- Azarbarzin, A.; Sands, S.A.; Stone, K.L.; Taranto-Montemurro, L.; Messineo, L.; Terrill, P.I.; Ancoli-Israel, S.; Ensrud, K.; Purcell, S.; White, D.P.; et al. The hypoxic burden of sleep apnoea predicts cardiovascular disease-related mortality: The osteoporotic fractures in men study and the sleep heart health study. Eur. Heart J. 2019, 40, 1149–1157. [Google Scholar] [CrossRef]
- Young, T.; Finn, L.; Peppard, P.E.; Szklo-Coxe, M.; Austin, D.; Nieto, F.J.; Stubbs, R.; Hla, K.M. Sleep disordered breathing and mortality: Eighteen-year follow-up of the Wisconsin sleep cohort. Sleep 2008, 31, 1071–1078. [Google Scholar]
- Martínez-Cerón, E.; Barquiel, B.; Bezos, A.-M.; Casitas, R.; Galera, R.; García-Benito, C.; Hernanz, A.; Alonso-Fernández, A.; Garcia-Rio, F. Effect of continuous positive airway pressure on glycemic control in patients with obstructive sleep apnea and Type 2 diabetes. A randomized clinical trial. Am. J. Respir. Crit. Care Med. 2016, 194, 476–485. [Google Scholar] [CrossRef]
- Barbé, F.; Durán-Cantolla, J.; Capote, F.; de la Peña, M.; Chiner, E.; Masa, J.F.; Gonzalez, M.; Marín, J.M.; Garcia-Rio, F.; de Atauri, J.D.; et al. Long-term effect of continuous positive airway pressure in hypertensive patients with sleep apnea. Am. J. Respir. Crit. Care Med. 2010, 181, 718–726. [Google Scholar] [CrossRef]
- Gottlieb, D.J.; Punjabi, N.M.; Mehra, R.; Patel, S.R.; Quan, S.F.; Babineau, D.C.; Tracy, R.P.; Rueschman, M.; Blumenthal, R.S.; Lewis, E.F.; et al. CPAP versus oxygen in obstructive sleep apnea. N. Engl. J. Med. 2014, 370, 2276–2285. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.M.; Carrizo, S.J.; Vicente, E.; Agusti, A.G. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: An observational study. Lancet 2005, 365, 1046–1053. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, Z.; McEvoy, R.D.; Anderson, C.S.; Rodgers, A.; Perkovic, V.; Neal, B. Association of positive airway pressure with cardiovascular events and death in adults with sleep apnea: A systematic review and meta-analysis. JAMA 2017, 318, 156. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.E.; Punjabi, N.M.; Naughton, M.T.; Willes, L.; Bergenstal, R.M.; Cistulli, P.A.; Fulcher, G.R.; Richards, G.N.; Zimmet, P.Z. The effect of treatment of obstructive sleep apnea on glycemic control in Type 2 diabetes. Am. J. Respir. Crit. Care Med. 2016, 194, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Myhill, P.C.; Davis, W.A.; Peters, K.E.; Chubb, S.A.P.; Hillman, D.; Davis, T.M.E. Effect of continuous positive airway pressure therapy on cardiovascular risk factors in patients with Type 2 diabetes and obstructive sleep apnea. J. Clin. Endocrinol. Metab. 2012, 97, 4212–4218. [Google Scholar] [CrossRef]
- Barbé, F.; Durán-Cantolla, J.; Sánchez-de-la-Torre, M.; Martínez-Alonso, M.; Carmona, C.; Barceló, A.; Chiner, E.; Masa, J.F.; Gonzalez, M.; Marín, J.M.; et al. Effect of continuous positive airway pressure on the incidence of hypertension and cardiovascular events in nonsleepy patients with obstructive sleep apnea: A randomized controlled trial. JAMA 2012, 307, 2161–2168. [Google Scholar] [CrossRef]
- Campos-Rodriguez, F.; Martinez-Alonso, M.; Sanchez-de-la-Torre, M.; Barbe, F. Long-term adherence to continuous positive airway pressure therapy in non-sleepy sleep apnea patients. Sleep Med. 2016, 17, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.F.; Irfan, M.; Waheed, Z.; Alam, N.; Mansoor, S.; Islam, M. Compliance with continuous positive airway pressure (CPAP) therapy for obstructive sleep apnea among privately paying patients- a cross sectional study. BMC Pulm. Med. 2014, 14, 188. [Google Scholar] [CrossRef]
- Sawyer, A.M.; Gooneratne, N.S.; Marcus, C.L.; Ofer, D.; Richards, K.C.; Weaver, T.E. A systematic review of CPAP adherence across age groups: Clinical and empiric insights for developing CPAP adherence interventions. Sleep Med. Rev. 2011, 15, 343–356. [Google Scholar] [CrossRef] [Green Version]
- Russell, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Levy, P.; Ryan, S.; Oldenburg, O.; Parati, G. Sleep apnoea and the heart. Eur. Respir. Rev. 2013, 22, 333–352. [Google Scholar] [CrossRef] [Green Version]
- Ryan, S.; Taylor, C.T.; McNicholas, W.T. Systemic inflammation: A key factor in the pathogenesis of cardiovascular complications in obstructive sleep apnoea syndrome? Postgrad. Med. J. 2009, 85, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Kyotani, Y.; Ota, H.; Itaya-Hironaka, A.; Yamauchi, A.; Sakuramoto-Tsuchida, S.; Zhao, J.; Ozawa, K.; Nagayama, K.; Ito, S.; Takasawa, S.; et al. Intermittent hypoxia induces the proliferation of rat vascular smooth muscle cell with the increases in epidermal growth factor family and ErbB2 receptor. Exp. Cell Res. 2013, 319, 3042–3050. [Google Scholar] [CrossRef] [PubMed]
- Kyotani, Y.; Itaya-Hironaka, A.; Yamauchi, A.; Sakuramoto-Tsuchida, S.; Makino, M.; Takasawa, S.; Yoshizumi, M. Intermittent hypoxia-induced epiregulin expression by IL-6 production in human coronary artery smooth muscle cells. FEBS Open Bio. 2018, 8, 868–876. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Vascular smooth muscle cell in atherosclerosis. Acta Physiol. 2015, 214, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular smooth muscle cells in atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Semenza, G.L.; Prabhakar, N.R. The role of hypoxia-inducible factors in carotid body (patho) physiology: HIF and the carotid body. J. Physiol. 2018, 596, 2977–2983. [Google Scholar] [CrossRef]
- Peng, Y.-J.; Nanduri, J.; Yuan, G.; Wang, N.; Deneris, E.; Pendyala, S.; Natarajan, V.; Kumar, G.K.; Prabhakar, N.R. NADPH oxidase is required for the sensory plasticity of the carotid body by chronic intermittent hypoxia. J. Neurosci. 2009, 29, 4903–4910. [Google Scholar] [CrossRef]
- Peng, Y.-J.; Overholt, J.L.; Kline, D.; Kumar, G.K.; Prabhakar, N.R. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: Implications for recurrent apneas. Proc. Natl. Acad. Sci. 2003, 100, 10073–10078. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.-J.; Yuan, G.; Ramakrishnan, D.; Sharma, S.D.; Bosch-Marce, M.; Kumar, G.K.; Semenza, G.L.; Prabhakar, N.R. Heterozygous HIF-1α deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia: HIF-1 and intermittent hypoxia. J. Physiol. 2006, 577, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, H.; Ye, X.; Wilson, D.; Htoo, A.K.; Hendersen, T.; Liu, S.F. Chronic intermittent hypoxia activates nuclear factor-κB in cardiovascular tissues in vivo. Biochem. Biophys. Res. Commun. 2006, 343, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.; McNicholas, W.T.; Taylor, C.T. A critical role for p38 map kinase in NF-κB signaling during intermittent hypoxia/reoxygenation. Biochem. Biophys. Res. Commun. 2007, 355, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Recoquillon, S.; Gómez-Guzmán, M.; Rodier, M.; Koffi, C.; Nitiéma, M.; Gagnadoux, F.; Martínez, M.C.; Andriantsitohaina, R. Non-muscular myosin light chain kinase triggers intermittent hypoxia-induced interleukin-6 release, endothelial dysfunction and permeability. Sci. Rep. 2017, 7, 13664. [Google Scholar] [CrossRef]
- Imano, H.; Kato, R.; Tanikawa, S.; Yoshimura, F.; Nomura, A.; Ijiri, Y.; Yamaguchi, T.; Izumi, Y.; Yoshiyama, M.; Hayashi, T. Factor Xa inhibition by rivaroxaban attenuates cardiac remodeling due to intermittent hypoxia. J. Pharmacol. Sci. 2018, 137, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, E.; Bakker, J.P.; Clarke, D.N.; Csizmadia, E.; Kocher, O.; Veves, A.; Tecilazich, F.; O’Donnell, C.P.; Ferran, C.; Malhotra, A. Molecular biomarkers of vascular dysfunction in obstructive sleep apnea. PLoS ONE 2013, 8, e70559. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Prabhakar, N.R.; Peng, Y.-J.; Yuan, G.; Nanduri, J. Reactive oxygen radicals and gaseous transmitters in carotid body activation by intermittent hypoxia. Cell Tissue Res. 2018, 372, 427–431. [Google Scholar] [CrossRef]
- Semenza, G.L.; Prabhakar, N.R. Neural regulation of hypoxia-inducible factors and redox state drives the pathogenesis of hypertension in a rodent model of sleep apnea. J. Appl. Physiol. 2015, 119, 1152–1156. [Google Scholar] [CrossRef] [Green Version]
- Dyugovskaya, L.; Lavie, P.; Lavie, L. Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. Am. J. Respir. Crit. Care Med. 2002, 165, 934–939. [Google Scholar] [CrossRef]
- Schulz, R.; Mahmoudi, S.; Hattar, K.; Sibelius, U.; Olschewski, H.; Mayer, K.; Seeger, W.; Grimminger, F. Enhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea: Impact of continuous positive airway pressure therapy. Am. J. Respir. Crit. Care Med. 2000, 162, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative stress in atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.S.; Singh, P.; Wolk, R.; Romero–Corral, A.; Raghavakaimal, S.; Somers, V.K. Microarray studies of genomic oxidative stress and cell cycle responses in obstructive sleep apnea. Antioxid. Redox Signal. 2007, 9, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Makarenko, V.V.; Usatyuk, P.V.; Yuan, G.; Lee, M.M.; Nanduri, J.; Natarajan, V.; Kumar, G.K.; Prabhakar, N.R. Intermittent hypoxia-induced endothelial barrier dysfunction requires ROS-dependent MAP kinase activation. Am. J. Physiol.-Cell Physiol. 2014, 306, C745–C752. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.S.; Singh, P.; Wolk, R.; Narkiewicz, K.; Somers, V.K. Obstructive sleep apnea and intermittent hypoxia increase expression of dual specificity phosphatase 1. Atherosclerosis 2013, 231, 378–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, M. Evidence that reactive oxygen species do not mediate NF-κB activation. EMBO J. 2003, 22, 3356–3366. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Li, Q.; Verma, I.M. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef]
- Son, Y.-H.; Jeong, Y.-T.; Lee, K.-A.; Choi, K.-H.; Kim, S.-M.; Rhim, B.-Y.; Kim, K. Roles of MAPK and NF-κB in interleukin-6 induction by lipopolysaccharide in vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 2008, 51, 71–77. [Google Scholar] [CrossRef]
- Kunsch, C.; Rosen, C.A. NF-kappa B subunit-specific regulation of the interleukin-8 promoter. Mol. Cell. Biol. 1993, 13, 6137–6146. [Google Scholar] [CrossRef]
- Ryan, S.; Taylor, C.T.; McNicholas, W.T. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation 2005, 112, 2660–2667. [Google Scholar] [CrossRef] [PubMed]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. p38MAPK: Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Kyotani, Y.; Ota, H.; Itaya-Hironaka, A.; Yamauchi, A.; Sakuramoto-Tsuchida, S.; Zhao, J.; Nagayama, K.; Ozawa, K.; Takasawa, S.; Kimura, H.; et al. Sleep apnea syndrome as an emerging risk factor for type 2 diabetes and atherosclerosis: Evidence and underlying mechanism. In Proceedings of the 9th Metabolic Syndrome, Type 2 Diabetes and Atherosclerosis Congress (MSDA 2014), Kyoto, Japan, 12–14 September 2014. [Google Scholar]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Madrid, L.V.; Mayo, M.W.; Reuther, J.Y.; Baldwin, A.S. Akt stimulates the transactivation potential of the RelA/p65 subunit of NF-κB through utilization of the IκB kinase and activation of the mitogen-activated protein kinase p38. J. Biol. Chem. 2001, 276, 18934–18940. [Google Scholar] [CrossRef]
- Minet, E.; Arnould, T.; Michel, G.; Roland, I.; Mottet, D.; Raes, M.; Remacle, J.; Michiels, C. ERK activation upon hypoxia: Involvement in HIF-1 activation. FEBS Lett. 2000, 468, 53–58. [Google Scholar] [CrossRef]
- Polotsky, V.Y.; Savransky, V.; Bevans-Fonti, S.; Reinke, C.; Li, J.; Grigoryev, D.N.; Shimoda, L.A. Intermittent and sustained hypoxia induce a similar gene expression profile in human aortic endothelial cells. Physiol. Genomics 2010, 41, 306–314. [Google Scholar] [CrossRef] [PubMed]
- BelAiba, R.S.; Bonello, S.; Zähringer, C.; Schmidt, S.; Hess, J.; Kietzmann, T.; Görlach, A. Hypoxia up-regulates hypoxia-inducible factor-1α transcription by involving phosphatidylinositol 3-kinase and nuclear factor κB in pulmonary artery smooth muscle cells. Mol. Biol. Cell 2007, 18, 4691–4697. [Google Scholar] [CrossRef] [PubMed]
- Van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1α by NF-κB. Biochem. J. 2008, 412, 477–484. [Google Scholar] [CrossRef]
- Cummins, E.P.; Berra, E.; Comerford, K.M.; Ginouves, A.; Fitzgerald, K.T.; Seeballuck, F.; Godson, C.; Nielsen, J.E.; Moynagh, P.; Pouyssegur, J.; et al. Prolyl hydroxylase-1 negatively regulates IκB kinase-β, giving insight into hypoxia-induced NFκB activity. Proc. Natl. Acad. Sci. USA 2006, 103, 18154–18159. [Google Scholar] [CrossRef] [PubMed]
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxia-induced neutrophil survival is mediated by HIF-1α–dependent NF-κB activity. J. Exp. Med. 2005, 201, 105–115. [Google Scholar] [CrossRef]
- Scortegagna, M.; Cataisson, C.; Martin, R.J.; Hicklin, D.J.; Schreiber, R.D.; Yuspa, S.H.; Arbeit, J.M. HIF-1 regulates epithelial inflammation by cell autonomous NFκB activation and paracrine stromal remodeling. Blood 2008, 111, 3343–3354. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T. Interdependent roles for hypoxia inducible factor and nuclear factor-κB in hypoxic inflammation. J. Physiol. 2008, 586, 4055–4059. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, R.; Molnar, J.; Madbouly, E.M.; Nida, M.; Aggarwal, S.; Sajid, H.; Naseem, J.; Loomba, R. Serum inflammatory markers in obstructive sleep apnea: A meta-analysis. J. Clin. Sleep Med. 2013, 9, 1003–1012. [Google Scholar] [CrossRef]
- Wang, J.; Yu, W.; Gao, M.; Zhang, F.; Gu, C.; Yu, Y.; Wei, Y. Impact of obstructive sleep apnea syndrome on endothelial function, arterial stiffening, and serum inflammatory markers: An updated meta-analysis and metaregression of 18 studies. J. Am. Heart Assoc. 2015, 4, e002454. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, M.; Scicchitano, P.; Zito, A.; Cortese, F.; Boninfante, B.; Falcone, V.; Quaranta, V.; Ventura, V.; Zucano, A.; Di Serio, F.; et al. Correlation between inflammatory markers of atherosclerosis and carotid intima-media thickness in obstructive sleep apnea. Molecules 2014, 19, 1651–1662. [Google Scholar] [CrossRef] [PubMed]
- Minoguchi, K.; Yokoe, T.; Tazaki, T.; Minoguchi, H.; Tanaka, A.; Oda, N.; Okada, S.; Ohta, S.; Naito, H.; Adachi, M. Increased carotid intima-media thickness and serum inflammatory markers in obstructive sleep apnea. Am. J. Respir. Crit. Care Med. 2005, 172, 625–630. [Google Scholar] [CrossRef]
- Kaptoge, S.; Seshasai, S.R.K.; Gao, P.; Freitag, D.F.; Butterworth, A.S.; Borglykke, A.; Di Angelantonio, E.; Gudnason, V.; Rumley, A.; Lowe, G.D.O.; et al. Inflammatory cytokines and risk of coronary heart disease: New prospective study and updated meta-analysis. Eur. Heart J. 2014, 35, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Stefaniak, J.; Hafner, C.; Schramel, J.P.; Kaun, C.; Wojta, J.; Ullrich, R.; Tretter, V.E.; Markstaller, K.; Klein, K.U. Intermittent hypoxia causes inflammation and injury to human adult cardiac myocytes. Anesth. Analg. 2016, 122, 373–380. [Google Scholar] [CrossRef]
- Ogura, H.; Murakami, M.; Okuyama, Y.; Tsuruoka, M.; Kitabayashi, C.; Kanamoto, M.; Nishihara, M.; Iwakura, Y.; Hirano, T. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 2008, 29, 628–636. [Google Scholar] [CrossRef]
- Murakami, M.; Okuyama, Y.; Ogura, H.; Asano, S.; Arima, Y.; Tsuruoka, M.; Harada, M.; Kanamoto, M.; Sawa, Y.; Iwakura, Y.; et al. Local microbleeding facilitates IL-6- and IL-17-dependent arthritis in the absence of tissue antigen recognition by activated T cells. J. Exp. Med. 2011, 208, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Nakagiri, T.; Kamimura, D.; Harada, M.; Oto, T.; Susaki, Y.; Shintani, Y.; Inoue, M.; Miyoshi, S.; Morii, E.; et al. IL-6 amplifier activation in epithelial regions of bronchi after allogeneic lung transplantation. Int. Immunol. 2013, 25, 319–332. [Google Scholar] [CrossRef] [Green Version]
- Takeda, N.; Manabe, I.; Shindo, T.; Iwata, H.; Iimuro, S.; Kagechika, H.; Shudo, K.; Nagai, R. Synthetic retinoid Am80 reduces scavenger receptor expression and atherosclerosis in mice by inhibiting IL-6. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Riese, D.J.; Cullum, R.L. Epiregulin: Roles in normal physiology and cancer. Semin. Cell Dev. Biol. 2014, 28, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Carpenter, G.; Coffey, R.J. EGF receptor ligands: Recent advances. F1000Research 2016, 5, 2270. [Google Scholar] [CrossRef] [PubMed]
- Dreux, A.C.; Lamb, D.J.; Modjtahedi, H.; Ferns, G.A.A. The epidermal growth factor receptors and their family of ligands: Their putative role in atherogenesis. Atherosclerosis 2006, 186, 38–53. [Google Scholar] [CrossRef]
- Takahashi, M.; Hayashi, K.; Yoshida, K.; Ohkawa, Y.; Komurasaki, T.; Kitabatake, A.; Ogawa, A.; Nishida, W.; Yano, M.; Monden, M.; et al. Epiregulin as a major autocrine/paracrine factor released from ERK- and p38MAPK-activated vascular smooth muscle cells. Circulation 2003, 108, 2524–2529. [Google Scholar] [CrossRef] [PubMed]
- Kyotani, Y.; Zhao, J.; Itaya-Hironaka, A.; Yamauchi, A.; Sakuramoto-Tsuchida, S.; Makino, M.; Takasawa, S.; Yoshizumi, M. Intermittent hypoxia-induced cell proliferation via ipregulations of interleukin-6 and epiregulin. In Proceedings of the 18th World Congress of Basic and Clinical Pharmacology (WCP2018), Kyoto, Japan, 1–6 July 2018. [Google Scholar]
- Caunt, C.J.; Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signalling. FEBS J. 2013, 280, 489–504. [Google Scholar] [CrossRef]
- Murakami, M.; Harada, M.; Kamimura, D.; Ogura, H.; Okuyama, Y.; Kumai, N.; Okuyama, A.; Singh, R.; Jiang, J.-J.; Atsumi, T.; et al. Disease-association analysis of an inflammation-related feedback loop. Cell Rep. 2013, 3, 946–959. [Google Scholar] [CrossRef] [PubMed]
- Lacedonia, D.; Scioscia, G.; Palladino, G.P.; Gallo, C.; Carpagnano, G.E.; Sabato, R.; Barbaro, M.P.F. MicroRNA expression profile during different conditions of hypoxia. Oncotarget 2018, 9, 35114–35122. [Google Scholar] [CrossRef]
- Liu, K.-X.; Chen, Q.; Chen, G.-P.; Huang, J.-C.; Huang, J.-F.; He, X.-R.; Lin, T.; Lin, Q.-C. Inhibition of MicroRNA-218 reduces HIF-1α by targeting on Robo1 in mice aortic endothelial cells under intermittent hypoxia. Oncotarget 2017, 8, 104359–104366. [Google Scholar] [PubMed]
- Uchiyama, T.; Ota, H.; Itaya-Hironaka, A.; Shobatake, R.; Yamauchi, A.; Sakuramoto-Tsuchida, S.; Makino, M.; Kimura, H.; Takeda, M.; Ohbayashi, C.; et al. Up-regulation of Selenoprotein P and HIP/PAP mRNAs in hepatocytes by intermittent hypoxia via down-regulation of miR-203. Biochem. Biophys. Rep. 2017, 11, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, T.; Itaya-Hironaka, A.; Yamauchi, A.; Makino, M.; Sakuramoto-Tsuchida, S.; Shobatake, R.; Ota, H.; Takeda, M.; Ohbayashi, C.; Takasawa, S. Intermittent hypoxia up-regulates CCL2, RETN, and TNFα mRNAs in adipocytes via down-regulation of miR-452. Int. J. Mol. Sci. 2019, 20, 1960. [Google Scholar] [CrossRef]
- Browning, L.; Patel, M.R.; Horvath, E.B.; Tawara, K.; Jorcyk, C.L. IL-6 and ovarian cancer: Inflammatory cytokines in promotion of metastasis. Cancer Manag. Res. 2018, 10, 6685–6693. [Google Scholar] [CrossRef]
- Nguyen, D.P.; Li, J.; Tewari, A.K. Inflammation and prostate cancer: The role of interleukin 6 (IL-6). BJU Int. 2014, 113, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Maggio, M.; Guralnik, J.M.; Longo, D.L.; Ferrucci, L. Interleukin-6 in aging and chronic disease: A magnificent pathway. J. Gerontol. A. Biol. Sci. Med. Sci. 2006, 61, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Rincon, M.; Irvin, C.G. Role of IL-6 in asthma and other inflammatory pulmonary diseases. Int. J. Biol. Sci. 2012, 8, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Martínez-García, M.Á.; Campos-Rodriguez, F.; Barbé, F. Cancer and OSA: Current evidence from human studies. Chest 2016, 150, 451–463. [Google Scholar] [CrossRef]
- Gozal, D.; Farré, R.; Nieto, F.J. Obstructive sleep apnea and cancer: Epidemiologic links and theoretical biological constructs. Sleep Med. Rev. 2016, 27, 43–55. [Google Scholar] [CrossRef]
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1947–1952. [Google Scholar] [CrossRef]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumor Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Fujieda, K.; Senju, S.; Ikeda, T.; Oshiumi, H.; Nishimura, Y. Immune-suppressive effects of interleukin-6 on T-cell-mediated anti-tumor immunity. Cancer Sci. 2018, 109, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, T.; Singh, R.; Sabharwal, L.; Bando, H.; Meng, J.; Arima, Y.; Yamada, M.; Harada, M.; Jiang, J.-J.; Kamimura, D.; et al. Inflammation amplifier, a new paradigm in cancer biology. Cancer Res. 2014, 74, 8–14. [Google Scholar] [CrossRef]
- Li, C.; Kuemmerle, J.F. Mechanisms that mediate the development of fibrosis in patients with Crohn’s disease. Inflamm. Bowel Dis. 2014, 20, 1250–1258. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kyotani, Y.; Takasawa, S.; Yoshizumi, M. Proliferative Pathways of Vascular Smooth Muscle Cells in Response to Intermittent Hypoxia. Int. J. Mol. Sci. 2019, 20, 2706. https://doi.org/10.3390/ijms20112706
Kyotani Y, Takasawa S, Yoshizumi M. Proliferative Pathways of Vascular Smooth Muscle Cells in Response to Intermittent Hypoxia. International Journal of Molecular Sciences. 2019; 20(11):2706. https://doi.org/10.3390/ijms20112706
Chicago/Turabian StyleKyotani, Yoji, Shin Takasawa, and Masanori Yoshizumi. 2019. "Proliferative Pathways of Vascular Smooth Muscle Cells in Response to Intermittent Hypoxia" International Journal of Molecular Sciences 20, no. 11: 2706. https://doi.org/10.3390/ijms20112706
APA StyleKyotani, Y., Takasawa, S., & Yoshizumi, M. (2019). Proliferative Pathways of Vascular Smooth Muscle Cells in Response to Intermittent Hypoxia. International Journal of Molecular Sciences, 20(11), 2706. https://doi.org/10.3390/ijms20112706