E-cadherin Beyond Structure: A Signaling Hub in Colon Homeostasis and Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. The Adherens Junctions
1.2. The Colonic Crypt
2. Colorectal Cancer and E-cadherin
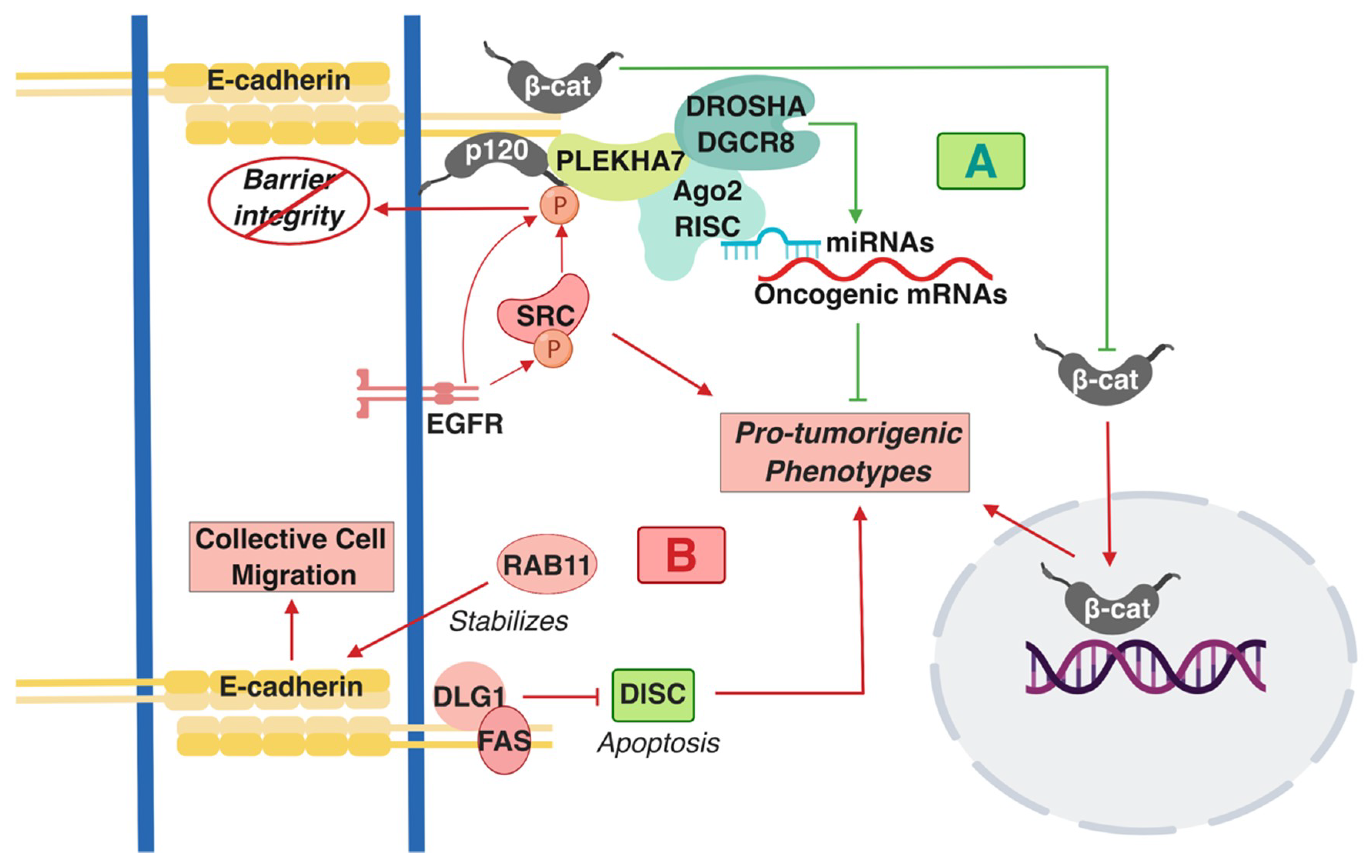
2.1. E-cadherin is a Double-Faced Signaling Molecule in the Colon
2.2. E-cadherin as a Colon Cancer Biomarker?
2.3. The Role of Other Cadherins in Colon Tumorigenesis
3. E-cadherin in Inflammatory Bowel Disease
4. E-cadherin Interacts with the Colon Microbiome
5. E-cadherin as a Sensor of Physical Strain in the Colon
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AJ | Adherens Junction |
| CD | Crohn’s Disease |
| CRC | Colorectal Cancer |
| DISC | death-inducing signaling complex |
| DSS | Dextran Sulfate Sodium |
| ECM | extracellular matrix |
| EMT | Epithelial to Mesenchymal Transition |
| IBD | Inflammatory Bowel Disease |
| NE | neutrophil elastase |
| p120 | p120 catenin |
| PMNs | Polymorphonuclear neutrophils |
| qRT-PCR | quantitative reverse transcription polymerase chain reaction |
| RNAi | RNA interference |
| RISC | RNA-induced silencing complex |
| TEER | trans-epithelial electrical resistance |
| TJ | tight junction |
| UC | Ulcerative Colitis |
| ZA | zonula adherens |
References
- Takeichi, M. Dynamic Contacts: Rearranging Adherens Junctions to Drive Epithelial Remodelling. Nat. Rev. Mol. Cell Biol. 2014, 15, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.J.C.; Tepass, U. Adherens Junctions: From Molecules to Morphogenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Ogita, H.; Rikitake, Y.; Miyoshi, J.; Takai, Y. Cell Adhesion Molecules Nectins and Associating Proteins: Implications for Physiology and Pathology. Proc. Jpn. Acad. Ser. B 2010, 86, 621–629. [Google Scholar] [CrossRef]
- Noemi, R.; Dejana, E. Adherens Junctions. Curr. Biol. 2008, 18, R1080–R1082. [Google Scholar]
- van Roy, F. Beyond E-Cadherin: Roles of Other Cadherin Superfamily Members in Cancer. Nat. Rev. Cancer 2014, 14, 121–134. [Google Scholar] [CrossRef]
- Hartsock, A.; Nelson, W.J. Adherens and Tight Junctions: Structure, Function and Connections to the Actin Cytoskeleton. Biochim. Biophys. Acta BBA Biomembr. 2008, 1778, 660–669. [Google Scholar] [CrossRef]
- Shapiro, L.; Weis, W.I. Structure and Biochemistry of Cadherins and Catenins. Cold Spring Harb. Perspect. Biol. 2009, 1, a003053. [Google Scholar] [CrossRef] [PubMed]
- Nelson, W.J. Regulation of Cell–Cell Adhesion by the Cadherin–Catenin Complex. Biochem. Soc. Trans. 2008, 36, 149–155. [Google Scholar] [CrossRef]
- Anastasiadis, P.Z. P120-Ctn: A Nexus for Contextual Signaling via Rho GTPases. Biochim. Biophys. Acta BBA Mol. Cell Res. 2007, 1773, 34–46. [Google Scholar] [CrossRef]
- Xiao, K.; Oas, R.G.; Chiasson, C.M.; Kowalczyk, A.P. Role of P120-Catenin in Cadherin Trafficking. Biochim. Biophys. Acta BBA Mol. Cell Res. 2007, 1773, 8–16. [Google Scholar] [CrossRef]
- Hartsock, A.; Nelson, W.J. Competitive Regulation of E-Cadherin JuxtaMembrane Domain Degradation by P120-Catenin Binding and Hakai-Mediated Ubiquitination. PLoS ONE 2012, 7, e37476. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, N.; Lee, S.-H.; Liu, S.; Li, G.-Y.; Smith, M.J.; Reichardt, L.F.; Ikura, M. Dynamic and Static Interactions between P120 Catenin and E-Cadherin Regulate the Stability of Cell-Cell Adhesion. Cell 2010, 141, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Ireton, R.C.; Davis, M.A.; van Hengel, J.; Mariner, D.J.; Barnes, K.; Thoreson, M.A.; Anastasiadis, P.Z.; Matrisian, L.; Bundy, L.M.; Sealy, L.; et al. A Novel Role for P120 Catenin in E-Cadherin Function. J. Cell Biol. 2002, 159, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Mushika, Y.; Ichii, T.; Takeichi, M. Anchorage of Microtubule Minus Ends to Adherens Junctions Regulates Epithelial Cell-Cell Contacts. Cell 2008, 135, 948–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourtidis, A.; Anastasiadis, P.Z. PLEKHA7 Defines an Apical Junctional Complex with Cytoskeletal Associations and MiRNA-Mediated Growth Implications. Cell Cycle 2016, 15, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Kourtidis, A.; Ngok, S.P.; Pulimeno, P.; Feathers, R.W.; Carpio, L.R.; Baker, T.R.; Carr, J.M.; Yan, I.K.; Borges, S.; Perez, E.A.; et al. Distinct E-Cadherin-Based Complexes Regulate Cell Behaviour through MiRNA Processing or Src and P120 Catenin Activity. Nat. Cell Biol. 2015, 17, 1145–1157. [Google Scholar] [CrossRef] [PubMed]
- Paschoud, S.; Jond, L.; Guerrera, D.; Citi, S. PLEKHA7 Modulates Epithelial Tight Junction Barrier Function. Tissue Barriers 2014, 2, e28755. [Google Scholar] [CrossRef]
- Matter, K.; Balda, M.S. Signalling to and from Tight Junctions. Nat. Rev. Mol. Cell Biol. 2003, 4, 225–236. [Google Scholar] [CrossRef]
- Cunningham, K.E.; Turner, J.R. Myosin Light Chain Kinase: Pulling the Strings of Epithelial Tight Junction Function. Ann. N. Y. Acad. Sci. 2012, 1258, 34–42. [Google Scholar] [CrossRef]
- Nita-Lazar, M.; Rebustini, I.; Walker, J.; Kukuruzinska, M.A. Hypoglycosylated E-Cadherin Promotes the Assembly of Tight Junctions through the Recruitment of PP2A to Adherens Junctions. Exp. Cell Res. 2010, 316, 1871–1884. [Google Scholar] [CrossRef]
- Tunggal, J.A.; Helfrich, I.; Schmitz, A.; Schwarz, H.; Günzel, D.; Fromm, M.; Kemler, R.; Krieg, T.; Niessen, C.M. E-Cadherin Is Essential for in Vivo Epidermal Barrier Function by Regulating Tight Junctions. EMBO J. 2005, 24, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Humphries, A.; Wright, N.A. Colonic Crypt Organization and Tumorigenesis. Nat. Rev. Cancer 2008, 8, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Beumer, J.; Clevers, H. Regulation and Plasticity of Intestinal Stem Cells during Homeostasis and Regeneration. Development 2016, 143, 3639–3649. [Google Scholar] [CrossRef] [PubMed]
- Gregorieff, A. Wnt Signaling in the Intestinal Epithelium: From Endoderm to Cancer. Genes Dev. 2005, 19, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Farin, H.F.; Jordens, I.; Mosa, M.H.; Basak, O.; Korving, J.; Tauriello, D.V.F.; de Punder, K.; Angers, S.; Peters, P.J.; Maurice, M.M.; et al. Visualization of a Short-Range Wnt Gradient in the Intestinal Stem-Cell Niche. Nature 2016, 530, 340. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-H.; Huels, D.J.; Ridgway, R.A.; Sansom, O.J.; Kholodenko, B.N.; Kolch, W.; Cho, K.-H. The APC Network Regulates the Removal of Mutated Cells from Colonic Crypts. Cell Rep. 2014, 7, 94–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondow, B.J.; Faber, M.L.; Wojta, K.J.; Walker, E.M.; Battle, M.A. E-Cadherin Is Required for Intestinal Morphogenesis in the Mouse. Dev. Biol. 2012, 371, 1–12. [Google Scholar] [CrossRef]
- Tan, C.W.; Hirokawa, Y.; Gardiner, B.S.; Smith, D.W.; Burgess, A.W. Colon Cryptogenesis: Asymmetric Budding. PLoS ONE 2013, 8, e78519. [Google Scholar] [CrossRef]
- Solanas, G.; Cortina, C.; Sevillano, M.; Batlle, E. Cleavage of E-Cadherin by ADAM10 Mediates Epithelial Cell Sorting Downstream of EphB Signalling. Nat. Cell Biol. 2011, 13, 1100–1107. [Google Scholar] [CrossRef]
- Wang, F.; Scoville, D.; He, X.C.; Mahe, M.M.; Box, A.; Perry, J.M.; Smith, N.R.; Lei, N.Y.; Davies, P.S.; Fuller, M.K.; et al. Isolation and Characterization of Intestinal Stem Cells Based on Surface Marker Combinations and Colony-Formation Assay. Gastroenterology 2013, 145, 383–395.e2. [Google Scholar] [CrossRef]
- Tang, Y.; Yang, G.; Zhang, J.; Li, X.; Zhang, C.; Wang, Y.; Xu, J.; Chen, Y.; Teng, Y.; Yang, X. E-Cadherin Is Required for the Homeostasis of Lgr5 + Gastric Antral Stem Cells. Int. J. Biol. Sci. 2019, 15, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Guebel, D.V.; Schmitz, U.; Wolkenhauer, O.; Vera, J. Analysis of Cell Adhesion during Early Stages of Colon Cancer Based on an Extended Multi-Valued Logic Approach. Mol. Biosyst. 2012, 8, 1230–1242. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA. Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Siegel, R.L.; Rosenberg, P.S.; Jemal, A. Emerging Cancer Trends among Young Adults in the USA: Analysis of a Population-Based Cancer Registry. Lancet Public Health 2019, 4, e137–e147. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Colorectal Cancer Mortality Rates in Adults Aged 20 to 54 Years in the United States, 1970-2014. JAMA 2017, 318, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Levy, M.; Grosheva, I.; Zheng, D.; Soffer, E.; Blacher, E.; Braverman, S.; Tengeler, A.C.; Barak, O.; Elazar, M.; et al. Hyperglycemia Drives Intestinal Barrier Dysfunction and Risk for Enteric Infection. Science 2018, 359, 1376–1383. [Google Scholar] [CrossRef] [PubMed]
- Devaux, M.; Graf, S.; Goryakin, Y.; Cecchini, M.; Huber, H.; Colombo, F. OECD Obesity Update. In The Organisation for Economic Co-operation and Development; OECD: London, UK, 2017. [Google Scholar]
- Kinzler, K.W.; Nilbert, M.C.; Su, L.K.; Vogelstein, B.; Bryan, T.M.; Levy, D.B.; Smith, K.J.; Preisinger, A.C.; Hedge, P.; McKechnie, D. Identification of FAP Locus Genes from Chromosome 5q21. Science 1991, 253, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e3. [Google Scholar] [CrossRef]
- Korinek, V.; Barker, N.; Morin, P.J.; van Wichen, D.; de Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive Transcriptional Activation by a Beta-Catenin-Tcf Complex in APC-/- Colon Carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/β-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef]
- Huels, D.J.; Ridgway, R.A.; Radulescu, S.; Leushacke, M.; Campbell, A.D.; Biswas, S.; Leedham, S.; Serra, S.; Chetty, R.; Moreaux, G.; et al. E-Cadherin Can Limit the Transforming Properties of Activating β-Catenin Mutations. EMBO J. 2015, 34, 2321–2333. [Google Scholar] [CrossRef] [PubMed]
- Irby, R.B.; Malek, R.L.; Bloom, G.; Tsai, J.; Letwin, N.; Frank, B.C.; Verratti, K.; Yeatman, T.J.; Lee, N.H. Iterative Microarray and RNA Interference-Based Interrogation of the SRC-Induced Invasive Phenotype. Cancer Res. 2005, 65, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Cordero, J.B.; Ridgway, R.A.; Valeri, N.; Nixon, C.; Frame, M.C.; Muller, W.J.; Vidal, M.; Sansom, O.J. C-Src Drives Intestinal Regeneration and Transformation. EMBO J. 2014, 33, 1474–1491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S.; et al. Proteogenomic Characterization of Human Colon and Rectal Cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef]
- Mariner, D.J.; Anastasiadis, P.; Keilhack, H.; Böhmer, F.D.; Wang, J.; Reynolds, A.B. Identification of Src Phosphorylation Sites in the Catenin P120ctn. J. Biol. Chem. 2001, 276, 28006–28013. [Google Scholar] [CrossRef] [PubMed]
- Mariner, D.J.; Davis, M.A.; Reynolds, A.B. EGFR Signaling to P120-Catenin through Phosphorylation at Y228. J. Cell Sci. 2004, 117, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, M.; Ohkubo, T. Tyrosine Phosphorylation of P120(Ctn) in v-Src Transfected L Cells Depends on Its Association with E-Cadherin and Reduces Adhesion Activity. J. Cell Sci. 2001, 114, 503–512. [Google Scholar] [PubMed]
- Irby, R.B.; Yeatman, T.J. Increased Src Activity Disrupts Cadherin/Catenin-Mediated Homotypic Adhesion in Human Colon Cancer and Transformed Rodent Cells. Cancer Res. 2002, 62, 2669–2674. [Google Scholar] [PubMed]
- Dohn, M.R.; Brown, M.V.; Reynolds, A.B. An Essential Role for P120-Catenin in Src- and Rac1-Mediated Anchorage-Independent Cell Growth. J. Cell Biol. 2009, 184, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Short, S.P.; Kondo, J.; Smalley-Freed, W.G.; Takeda, H.; Dohn, M.R.; Powell, A.E.; Carnahan, R.H.; Washington, M.K.; Tripathi, M.; Payne, D.M.; et al. P120-Catenin Is an Obligate Haploinsufficient Tumor Suppressor in Intestinal Neoplasia. J. Clin. Invest. 2017, 127, 4462–4476. [Google Scholar] [CrossRef] [PubMed]
- Gagnoux-Palacios, L.; Awina, H.; Audebert, S.; Rossin, A.; Mondin, M.; Borgese, F.; Planas-Botey, C.; Mettouchi, A.; Borg, J.-P.; Hueber, A.-O. Cell Polarity and Adherens Junction Formation Inhibit Epithelial Fas Cell Death Receptor Signaling. J. Cell Biol. 2018, 217, 3839–3852. [Google Scholar] [CrossRef] [PubMed]
- Druzhkova, I.; Ignatova, N.; Prodanets, N.; Kiselev, N.; Zhukov, I.; Shirmanova, M.; Zagainov, V.; Zagaynova, E. E-Cadherin in Colorectal Cancer: Relation to Chemosensitivity. Clin. Colorectal Cancer 2019, 18, e74–e86. [Google Scholar] [CrossRef] [PubMed]
- Dinicola, S.; Pasqualato, A.; Proietti, S.; Masiello, M.G.; Palombo, A.; Coluccia, P.; Canipari, R.; Catizone, A.; Ricci, G.; Harrath, A.H.; et al. Paradoxical E-Cadherin Increase in 5FU-Resistant Colon Cancer Is Unaffected during Mesenchymal-Epithelial Reversion Induced by γ-Secretase Inhibition. Life Sci. 2016, 145, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Gavert, N.; Sheffer, M.; Raveh, S.; Spaderna, S.; Shtutman, M.; Brabletz, T.; Barany, F.; Paty, P.; Notterman, D.; Domany, E.; et al. Expression of L1-CAM and ADAM10 in Human Colon Cancer Cells Induces Metastasis. Cancer Res. 2007, 67, 7703–7712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, Y.-C.; Wei, W.-C.; Hung, C.-N.; Kuo, J.-F.; Hsu, C.-P.; Chang, K.-J.; Chao, W.-T. Rab11 Collaborates E-Cadherin to Promote Collective Cell Migration and Indicates a Poor Prognosis in Colorectal Carcinoma. Eur. J. Clin. Invest. 2016, 46, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.-C.; Wei, W.-C.; Huang, S.-H.; Shih, C.-M.; Hsu, C.-P.; Chang, K.-J.; Chao, W.-T. Rab11 Regulates E-Cadherin Expression and Induces Cell Transformation in Colorectal Carcinoma. BMC Cancer 2014, 14, 587. [Google Scholar] [CrossRef] [PubMed]
- Kourtidis, A.; Necela, B.; Lin, W.-H.; Lu, R.; Feathers, R.W.; Asmann, Y.W.; Thompson, E.A.; Anastasiadis, P.Z. Cadherin Complexes Recruit MRNAs and RISC to Regulate Epithelial Cell Signaling. J. Cell Biol. 2017, 216, 3073–3085. [Google Scholar] [CrossRef]
- Christou, N.; Perraud, A.; Blondy, S.; Jauberteau, M.-O.; Battu, S.; Mathonnet, M. E-Cadherin: A Potential Biomarker of Colorectal Cancer Prognosis. Oncol. Lett. 2017, 13, 4571–4576. [Google Scholar] [CrossRef]
- Duffy, M.J. Carcinoembryonic Antigen as a Marker for Colorectal Cancer: Is It Clinically Useful? Clin. Chem. 2001, 4, 7. [Google Scholar]
- Yörüker, E.E.; Holdenrieder, S.; Gezer, U. Blood-Based Biomarkers for Diagnosis, Prognosis and Treatment of Colorectal Cancer. Clin. Chim. Acta 2016, 455, 26–32. [Google Scholar] [CrossRef]
- Newton, K.F.; Newman, W.; Hill, J. Review of Biomarkers in Colorectal Cancer: Biomarkers in Colorectal Cancer. Colorectal Dis. 2012, 14, 3–17. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Chen, Z.; Jia, M.; Zhao, X. Downregulated E-Cadherin Expression Indicates Worse Prognosis in Asian Patients with Colorectal Cancer: Evidence from Meta-Analysis. PLoS ONE 2013, 8, e70858. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ma, X.; Li, Y.; He, Y.; Huang, D.; Cai, S.; Peng, J. The Characteristics and Prognostic Effect of E-Cadherin Expression in Colorectal Signet Ring Cell Carcinoma. PLoS ONE 2016, 11, e0160527. [Google Scholar] [CrossRef] [PubMed]
- Bosman, F.T. WHO Classification of Tumours of the Digestive System; International Agency for Research on Cancer, World Health Organization: Lyon, France, 2010. [Google Scholar]
- Weiss, J.V.; Klein-Scory, S.; Kübler, S.; Reinacher-Schick, A.; Stricker, I.; Schmiegel, W.; Schwarte-Waldhoff, I. Soluble E-Cadherin as a Serum Biomarker Candidate: Elevated Levels in Patients with Late-Stage Colorectal Carcinoma and FAP. Int. J. Cancer 2011, 128, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Karamitopoulou, E.; Zlobec, I.; Patsouris, E.; Peros, G.; Lugli, A. Loss of E-Cadherin Independently Predicts the Lymph Node Status in Colorectal Cancer. Pathology 2011, 43, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Tóth, L.; András, C.; Molnár, C.; Tanyi, M.; Csiki, Z.; Molnár, P.; Szántó, J. Investigation of β-Catenin and E-Cadherin Expression in Dukes B2 Stage Colorectal Cancer with Tissue Microarray Method. Is It a Marker of Metastatic Potential in Rectal Cancer? Pathol. Oncol. Res. POR 2012, 18, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Khoursheed, M.A.; Mathew, T.C.; Makar, R.R.; Louis, S.; Asfar, S.K.; Al-Sayer, H.M.; Dashti, H.M.; Al-Bader, A. Expression of E-Cadherin in Human Colorectal Cancer. Surg. J. R. Coll. Surg. Edinb. Irel. 2003, 1, 86–91. [Google Scholar] [CrossRef]
- Ilyas, M.; Tomlinson, I.P.; Rowan, A.; Pignatelli, M.; Bodmer, W.F. Beta-Catenin Mutations in Cell Lines Established from Human Colorectal Cancers. Proc. Natl. Acad. Sci. USA 1997, 94, 10330–10334. [Google Scholar] [CrossRef]
- Ludwig, K.; Tse, E.S.; Wang, J.Y. Colon Cancer Cells Adopt an Invasive Phenotype without Mesenchymal Transition in 3-D but Not 2-D Culture upon Combined Stimulation with EGF and Crypt Growth Factors. BMC Cancer 2013, 13, 221. [Google Scholar] [CrossRef]
- Milicic, A.; Harrison, L.-A.; Goodlad, R.A.; Hardy, R.G.; Nicholson, A.M.; Presz, M.; Sieber, O.; Santander, S.; Pringle, J.H.; Mandir, N.; et al. Ectopic Expression of P-Cadherin Correlates with Promoter Hypomethylation Early in Colorectal Carcinogenesis and Enhanced Intestinal Crypt Fission In Vivo. Cancer Res. 2008, 68, 7760–7768. [Google Scholar] [CrossRef]
- Sun, L.; Hu, H.; Peng, L.; Zhou, Z.; Zhao, X.; Pan, J.; Sun, L.; Yang, Z.; Ran, Y. P-Cadherin Promotes Liver Metastasis and Is Associated with Poor Prognosis in Colon Cancer. Am. J. Pathol. 2011, 179, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Kumara, H.M.C.S.; Bellini, G.A.; Caballero, O.L.; Herath, S.A.C.; Su, T.; Ahmed, A.; Njoh, L.; Cekic, V.; Whelan, R.L. P-Cadherin (CDH3) Is Overexpressed in Colorectal Tumors and Has Potential as a Serum Marker for Colorectal Cancer Monitoring. Oncoscience 2017, 4, 139–147. [Google Scholar] [PubMed]
- Rosivatz, E.; Becker, I.; Bamba, M.; Schott, C.; Diebold, J.; Mayr, D.; Höfler, H.; Becker, K.-F. Neoexpression of N-Cadherin in E-Cadherin Positive Colon Cancers. Int. J. Cancer 2004, 111, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Labernadie, A.; Kato, T.; Brugués, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.; Weston, A.; González-Tarragó, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A Mechanically Active Heterotypic E-Cadherin/N-Cadherin Adhesion Enables Fibroblasts to Drive Cancer Cell Invasion. Nat. Cell Biol. 2017, 19, 224–237. [Google Scholar] [CrossRef] [PubMed]
- De Wever, O.; Westbroek, W.; Verloes, A.; Bloemen, N.; Bracke, M.; Gespach, C.; Bruyneel, E.; Mareel, M. Critical Role of N-Cadherin in Myofibroblast Invasion and Migration in Vitro Stimulated by Colon-Cancer-Cell-Derived TGF-Beta or Wounding. J. Cell Sci. 2004, 117, 4691–4703. [Google Scholar] [CrossRef]
- Zhu, Q.; Wang, Z.; Zhou, L.; Ren, Y.; Gong, Y.; Qin, W.; Bai, L.; Hu, J.; Wang, T. The Role of Cadherin-11 in Microcystin-LR-Induced Migration and Invasion in Colorectal Carcinoma Cells. Oncol. Lett. 2018, 15, 1417–1422. [Google Scholar] [CrossRef] [PubMed]
- Van der Goten, J.; Vanhove, W.; Lemaire, K.; Van Lommel, L.; Machiels, K.; Wollants, W.-J.; De Preter, V.; De Hertogh, G.; Ferrante, M.; Van Assche, G.; et al. Integrated MiRNA and MRNA Expression Profiling in Inflamed Colon of Patients with Ulcerative Colitis. PloS ONE 2014, 9, e116117. [Google Scholar] [CrossRef]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide Incidence and Prevalence of Inflammatory Bowel Disease in the 21st Century: A Systematic Review of Population-Based Studies. Lancet Lond. Engl. 2018, 390, 2769–2778. [Google Scholar] [CrossRef]
- Anbazhagan, A.N.; Priyamvada, S.; Alrefai, W.A.; Dudeja, P.K. Pathophysiology of IBD Associated Diarrhea. Tissue Barriers 2018, 6, e1463897. [Google Scholar] [CrossRef]
- Kim, E.R. Colorectal Cancer in Inflammatory Bowel Disease: The Risk, Pathogenesis, Prevention and Diagnosis. World J. Gastroenterol. 2014, 20, 9872. [Google Scholar] [CrossRef]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.-D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal Permeability—a New Target for Disease Prevention and Therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [PubMed]
- France, M.M.; Turner, J.R. The Mucosal Barrier at a Glance. J. Cell Sci. 2017, 130, 307–314. [Google Scholar] [CrossRef] [PubMed]
- The NIDDK IBD Genetics Consortium; McGovern, D.P.B.; Gardet, A.; Törkvist, L.; Goyette, P.; Essers, J.; Taylor, K.D.; Neale, B.M.; Ong, R.T.H.; Lagacé, C.; et al. Genome-Wide Association Identifies Multiple Ulcerative Colitis Susceptibility Loci. Nat. Genet. 2010, 42, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Lee, J.C.; Lees, C.W.; Prescott, N.J.; Anderson, C.A.; Phillips, A.; Wesley, E.; Parnell, K.; Zhang, H.; Drummond, H.; et al. Genome-Wide Association Study of Ulcerative Colitis Identifies Three New Susceptibility Loci, Including the HNF4A Region. Nat. Genet. 2009, 41, 1330–1334. [Google Scholar]
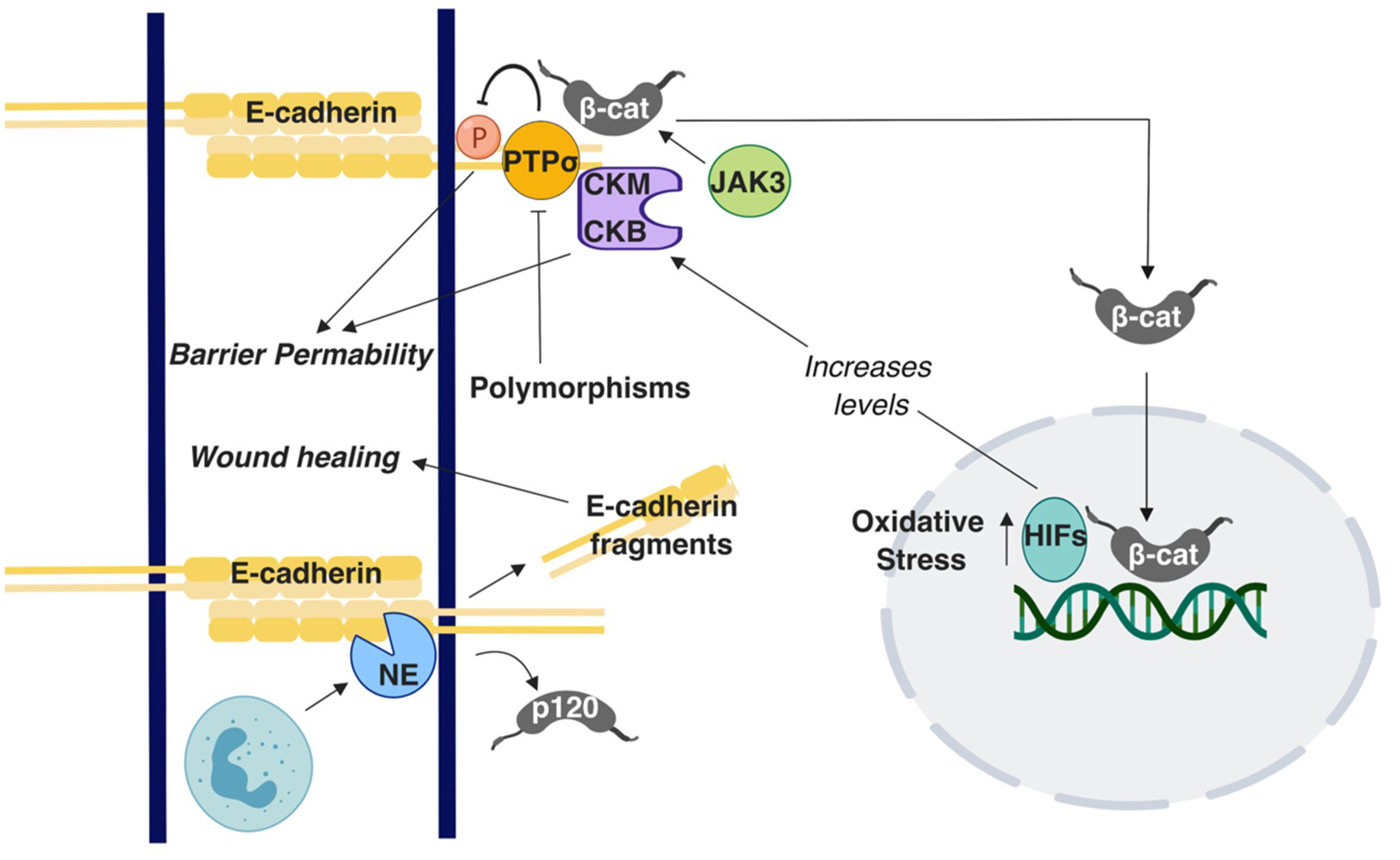
- Muise, A.M.; Walters, T.D.; Glowacka, W.K.; Griffiths, A.M.; Ngan, B.-Y.; Lan, H.; Xu, W.; Silverberg, M.S.; Rotin, D. Polymorphisms in E-Cadherin (CDH1) Result in a Mis-Localised Cytoplasmic Protein That Is Associated with Crohn’s Disease. Gut 2009, 58, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Mohanan, V.; Nakata, T.; Desch, A.N.; Lévesque, C.; Boroughs, A.; Guzman, G.; Cao, Z.; Creasey, E.; Yao, J.; Boucher, G.; et al. C1orf106 Is a Colitis Risk Gene That Regulates Stability of Epithelial Adherens Junctions. Science 2018, 359, 1161–1166. [Google Scholar] [CrossRef]
- Nighot, P.; Young, K.; Nighot, M.; Rawat, M.; Sung, E.J.; Maharshak, N.; Plevy, S.E.; Ma, T.; Blikslager, A. Chloride Channel ClC-2 Is a Key Factor in the Development of DSS-Induced Murine Colitis. Inflamm. Bowel Dis. 2013, 19, 2867–2877. [Google Scholar] [CrossRef]
- Jin, Y.; Ibrahim, D.; Magness, S.T.; Blikslager, A.T. Knockout of ClC-2 Reveals Critical Functions of Adherens Junctions in Colonic Homeostasis and Tumorigenicity. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G966–G979. [Google Scholar] [CrossRef]
- Muise, A.; Rotin, D. Apical Junction Complex Proteins and Ulcerative Colitis: A Focus on the PTPRS Gene. Expert Rev. Mol. Diagn. 2008, 8, 465–477. [Google Scholar] [CrossRef]
- Wheeler, J.M.D. Hypermethylation of the Promoter Region of the E-Cadherin Gene (CDH1) in Sporadic and Ulcerative Colitis Associated Colorectal Cancer. Gut 2001, 48, 367–371. [Google Scholar] [CrossRef]
- Azarschab, P.; Porschen, R.; Gregor, M.; Blin, N.; Holzmann, K. Epigenetic Control of the E-Cadherin Gene (CDH1) by CpG Methylation in Colectomy Samples of Patients with Ulcerative Colitis. Genes. Chromosomes Cancer 2002, 35, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Ihara, S.; Hirata, Y.; Hikiba, Y.; Yamashita, A.; Tsuboi, M.; Hata, M.; Konishi, M.; Suzuki, N.; Sakitani, K.; Kinoshita, H.; et al. Adhesive Interactions between Mononuclear Phagocytes and Intestinal Epithelium Perturb Normal Epithelial Differentiation and Serve as a Therapeutic Target in Inflammatory Bowel Disease. J. Crohns Colitis 2018, 12, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Brazil, J.C.; Parkos, C.A. Pathobiology of Neutrophil-Epithelial Interactions. Immunol. Rev. 2016, 273, 94–111. [Google Scholar] [CrossRef]
- Gordon, M.H.; Chauvin, A.; Boisvert, F.-M.; MacNaughton, W.K. Proteolytic Processing of the Epithelial Adherens Junction Molecule E-Cadherin by Neutrophil Elastase Generates Short Peptides With Novel Wound-Healing Bioactivity. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 483–486.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
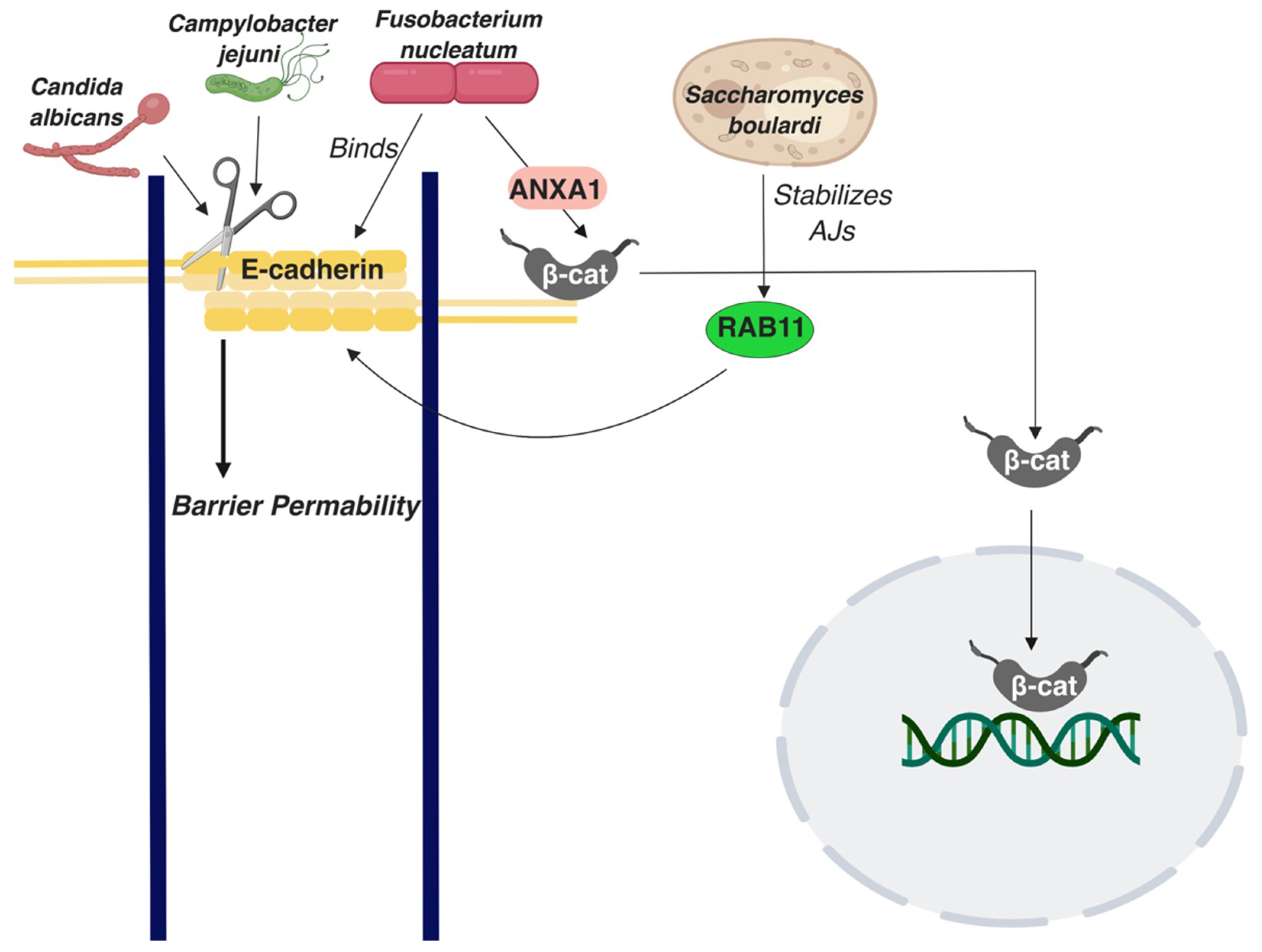
- Terciolo, C.; Dobric, A.; Ouaissi, M.; Siret, C.; Breuzard, G.; Silvy, F.; Marchiori, B.; Germain, S.; Bonier, R.; Hama, A.; et al. Saccharomyces Boulardii CNCM I-745 Restores Intestinal Barrier Integrity by Regulation of E-Cadherin Recycling. J. Crohns Colitis 2017, 11, 999–1010. [Google Scholar] [CrossRef]
- Demetter, P.; Cuvelier, C.A.; Bullock, G.; De Vos, M.; Van Damme, N.; Baeten, D.; Elewaut, D.; Veys, E.M.; De Keyser, F.; Verbruggen, G.; et al. Focal Up-Regulation of E-Cadherin-Catenin Complex in Inflamed Bowel Mucosa but Reduced Expression in Ulcer-Associated Cell Lineage. Am. J. Clin. Pathol. 2000, 114, 364–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smalley-Freed, W.G.; Efimov, A.; Burnett, P.E.; Short, S.P.; Davis, M.A.; Gumucio, D.L.; Washington, M.K.; Coffey, R.J.; Reynolds, A.B. P120-Catenin Is Essential for Maintenance of Barrier Function and Intestinal Homeostasis in Mice. J. Clin. Invest. 2010, 120, 1824–1835. [Google Scholar] [CrossRef]
- Ahmed, I.; Roy, B.C.; Raach, R.-M.T.; Owens, S.M.; Xia, L.; Anant, S.; Sampath, V.; Umar, S. Enteric Infection Coupled with Chronic Notch Pathway Inhibition Alters Colonic Mucus Composition Leading to Dysbiosis, Barrier Disruption and Colitis. PLoS ONE 2018, 13, e0206701. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, L.; Tian, Y.; Ye, J.; Liu, Y.; Song, L.; Pan, Q.; He, Y.; Chen, W.; Peng, Z.; et al. Numb Modulates the Paracellular Permeability of Intestinal Epithelial Cells through Regulating Apical Junctional Complex Assembly and Myosin Light Chain Phosphorylation. Exp. Cell Res. 2013, 319, 3214–3225. [Google Scholar] [CrossRef]
- Mishra, J.; Verma, R.K.; Alpini, G.; Meng, F.; Kumar, N. Role of Janus Kinase 3 in Mucosal Differentiation and Predisposition to Colitis. J. Biol. Chem. 2013, 288, 31795–31806. [Google Scholar] [CrossRef] [Green Version]
- Glover, L.E.; Bowers, B.E.; Saeedi, B.; Ehrentraut, S.F.; Campbell, E.L.; Bayless, A.J.; Dobrinskikh, E.; Kendrick, A.A.; Kelly, C.J.; Burgess, A.; et al. Control of Creatine Metabolism by HIF Is an Endogenous Mechanism of Barrier Regulation in Colitis. Proc. Natl. Acad. Sci. 2013, 110, 19820–19825. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Xia, J.; Wu, X.; Zhang, L.; Wang, P.; Wang, H.; Li, H.; Wang, X.; Chen, Y.; Agnetti, J.; et al. Deficiency in Class III PI3-Kinase Confers Postnatal Lethality with IBD-like Features in Zebrafish. Nat. Commun. 2018, 9, 2639. [Google Scholar] [CrossRef] [PubMed]
- Ardesia, M.; Ferlazzo, G.; Fries, W. Vitamin D and Inflammatory Bowel Disease. BioMed Res. Int. 2015, 2015, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Zhang, Z.; Musch, M.W.; Ning, G.; Sun, J.; Hart, J.; Bissonnette, M.; Li, Y.C. Novel Role of the Vitamin D Receptor in Maintaining the Integrity of the Intestinal Mucosal Barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G208–G216. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.-N.; Xu, K.-Q.; Zhang, W.; Wang, Q.; Wu, J.; Wang, X.-Y. Associations between Vitamin D Receptor Polymorphisms and Susceptibility to Ulcerative Colitis and Crohn’s Disease: A Meta-Analysis. Inflamm. Bowel Dis. 2013, 19, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Chen, G. The Role of the Gut Microbiome in Colorectal Cancer. Clin. Colon Rectal Surg. 2018, 31, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prindiville, T. Bacteroides Fragilis Enterotoxin Gene Sequences in Patients with Inflammatory Bowel Disease. Emerg. Infect. Dis. 2000, 6, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lim, K.-C.; Huang, J.; Saidi, R.F.; Sears, C.L. Bacteroides Fragilis Enterotoxin Cleaves the Zonula Adherens Protein, E-Cadherin. Proc. Natl. Acad. Sci. 1998, 95, 14979–14984. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Morin, P.J.; Maouyo, D.; Sears, C.L. Bacteroides Fragilis Enterotoxin Induces C-Myc Expression and Cellular Proliferation. Gastroenterology 2003, 124, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rhee, K.-J.; Zhang, M.; Franco, A.; Sears, C.L. Bacteroides Fragilis Toxin Stimulates Intestinal Epithelial Cell Shedding and -Secretase-Dependent E-Cadherin Cleavage. J. Cell Sci. 2007, 120, 1944–1952. [Google Scholar] [CrossRef] [PubMed]
- Dejea, C.M.; Wick, E.C.; Hechenbleikner, E.M.; White, J.R.; Welch, J.L.M.; Rossetti, B.J.; Peterson, S.N.; Snesrud, E.C.; Borisy, G.G.; Lazarev, M.; et al. Microbiota Organization Is a Distinct Feature of Proximal Colorectal Cancers. Proc. Natl. Acad. Sci. 2014, 111, 18321–18326. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.F.; Hostetter, M.K. Cleavage of E-Cadherin: A Mechanism for Disruption of the Intestinal Epithelial Barrier by Candida Albicans. Transl. Res. 2007, 149, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Sitaraman, S.V.; Babbin, B.A.; Gerner-Smidt, P.; Ribot, E.M.; Garrett, N.; Alpern, J.A.; Akyildiz, A.; Theiss, A.L.; Nusrat, A.; et al. Invasive Escherichia Coli Are a Feature of Crohn’s Disease. Lab. Invest. 2007, 87, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium Nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via Its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.R.; Baik, J.E.; Lagana, S.M.; Han, R.P.; Raab, W.J.; Sahoo, D.; Dalerba, P.; Wang, T.C.; Han, Y.W. Fusobacterium Nucleatum Promotes Colorectal Cancer by Inducing Wnt/Β-catenin Modulator Annexin A1. EMBO Rep. 2019, 20, e47638. [Google Scholar] [CrossRef] [PubMed]
- Hummel, S.; Veltman, K.; Cichon, C.; Sonnenborn, U.; Schmidt, M.A. Differential Targeting of the E-Cadherin/β-Catenin Complex by Gram-Positive Probiotic Lactobacilli Improves Epithelial Barrier Function. Appl. Environ. Microbiol. 2012, 78, 1140–1147. [Google Scholar] [CrossRef] [PubMed]
- Elmi, A.; Nasher, F.; Jagatia, H.; Gundogdu, O.; Bajaj-Elliott, M.; Wren, B.; Dorrell, N. Campylobacter Jejuni Outer Membrane Vesicle-Associated Proteolytic Activity Promotes Bacterial Invasion by Mediating Cleavage of Intestinal Epithelial Cell E-Cadherin and Occludin: Campylobacter Jejuni OMV-Associated Proteolytic Activity. Cell. Microbiol. 2016, 18, 561–572. [Google Scholar] [CrossRef]
- Crotti, S.; Piccoli, M.; Rizzolio, F.; Giordano, A.; Nitti, D.; Agostini, M. Extracellular Matrix and Colorectal Cancer: How Surrounding Microenvironment Affects Cancer Cell Behavior? J. Cell. Physiol. 2017, 232, 967–975. [Google Scholar] [CrossRef]
- Rieder, F.; Fiocchi, C. Intestinal Fibrosis in Inflammatory Bowel Disease—Current Knowledge and Future Perspectives. J. Crohns Colitis 2008, 2, 279–290. [Google Scholar] [CrossRef]
- Basson, M.D.; Turowski, G.; Emenaker, N.J. Regulation of Human (Caco-2) Intestinal Epithelial Cell Differentiation by Extracellular Matrix Proteins. Exp. Cell Res. 1996, 225, 301–305. [Google Scholar] [CrossRef]
- Basson, M.D.; Li, G.D.; Hong, F.; Han, O.; Sumpio, B.E. Amplitude-Dependent Modulation of Brush Border Enzymes and Proliferation by Cyclic Strain in Human Intestinal Caco-2 Monolayers. J. Cell. Physiol. 1996, 168, 476–488. [Google Scholar] [CrossRef]
- Tilghman, R.W.; Cowan, C.R.; Mih, J.D.; Koryakina, Y.; Gioeli, D.; Slack-Davis, J.K.; Blackman, B.R.; Tschumperlin, D.J.; Parsons, J.T. Matrix Rigidity Regulates Cancer Cell Growth and Cellular Phenotype. PLoS ONE 2010, 5, e12905. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wright, S.; Zhang, J.; Brekken, R.A. Getting a Grip on Adhesion: Cadherin Switching and Collagen Signaling. Biochim. Biophys. Acta BBA Mol. Cell Res. 2019, 12, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.Y.; Saif, M.T.A. Substrate Stiffness Mediated Metastasis Like Phenotype of Colon Cancer Cells Is Independent of Cell to Gel Adhesion. Cell. Mol. Bioeng. 2014, 7, 532–543. [Google Scholar] [CrossRef]
- Tang, X.; Kuhlenschmidt, T.B.; Zhou, J.; Bell, P.; Wang, F.; Kuhlenschmidt, M.S.; Saif, T.A. Mechanical Force Affects Expression of an In Vitro Metastasis-Like Phenotype in HCT-8 Cells. Biophys. J. 2010, 99, 2460–2469. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, J.; Vignjevic, D.; Fütterer, C.; Beaurepaire, E.; Robine, S.; Farge, E. Mechanical Factors Activate β-catenin-dependent Oncogene Expression in APC 1638N/+ Mouse Colon. HFSP J. 2008, 2, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sánchez, M.E.; Barbier, S.; Whitehead, J.; Béalle, G.; Michel, A.; Latorre-Ossa, H.; Rey, C.; Fouassier, L.; Claperon, A.; Brullé, L.; et al. Mechanical Induction of the Tumorigenic β-Catenin Pathway by Tumour Growth Pressure. Nature 2015, 523, 92–95. [Google Scholar] [CrossRef]
- Röper, J.-C.; Mitrossilis, D.; Stirnemann, G.; Waharte, F.; Brito, I.; Fernandez-Sanchez, M.-E.; Baaden, M.; Salamero, J.; Farge, E. The Major β-Catenin/E-Cadherin Junctional Binding Site Is a Primary Molecular Mechano-Transductor of Differentiation in Vivo. eLife 2018, 7, e33381. [Google Scholar] [CrossRef]
- Broders-Bondon, F.; Nguyen Ho-Bouldoires, T.H.; Fernandez-Sanchez, M.-E.; Farge, E. Mechanotransduction in Tumor Progression: The Dark Side of the Force. J. Cell Biol. 2018, 217, 1571–1587. [Google Scholar] [CrossRef]
- Canel, M.; Serrels, A.; Frame, M.C.; Brunton, V.G. E-Cadherin-Integrin Crosstalk in Cancer Invasion and Metastasis. J. Cell Sci. 2013, 126, 393–401. [Google Scholar] [CrossRef]
- Huveneers, S.; Danen, E.H.J. Adhesion Signaling - Crosstalk between Integrins, Src and Rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Samak, G.; Gangwar, R.; Crosby, L.M.; Desai, L.P.; Wilhelm, K.; Waters, C.M.; Rao, R. Cyclic Stretch Disrupts Apical Junctional Complexes in Caco-2 Cell Monolayers by a JNK-2-, c-Src-, and MLCK-Dependent Mechanism. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G947–G958. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Radjendirane, V.; Wary, K.K.; Chakrabarty, S. Transforming Growth Factor β Regulates Cell–Cell Adhesion through Extracellular Matrix Remodeling and Activation of Focal Adhesion Kinase in Human Colon Carcinoma Moser Cells. Oncogene 2004, 23, 5558–5561. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daulagala, A.C.; Bridges, M.C.; Kourtidis, A. E-cadherin Beyond Structure: A Signaling Hub in Colon Homeostasis and Disease. Int. J. Mol. Sci. 2019, 20, 2756. https://doi.org/10.3390/ijms20112756
Daulagala AC, Bridges MC, Kourtidis A. E-cadherin Beyond Structure: A Signaling Hub in Colon Homeostasis and Disease. International Journal of Molecular Sciences. 2019; 20(11):2756. https://doi.org/10.3390/ijms20112756
Chicago/Turabian StyleDaulagala, Amanda C., Mary Catherine Bridges, and Antonis Kourtidis. 2019. "E-cadherin Beyond Structure: A Signaling Hub in Colon Homeostasis and Disease" International Journal of Molecular Sciences 20, no. 11: 2756. https://doi.org/10.3390/ijms20112756
APA StyleDaulagala, A. C., Bridges, M. C., & Kourtidis, A. (2019). E-cadherin Beyond Structure: A Signaling Hub in Colon Homeostasis and Disease. International Journal of Molecular Sciences, 20(11), 2756. https://doi.org/10.3390/ijms20112756