Shortcomings of Phylogenetic Studies on Recent Radiated Insular Groups: A Meta-Analysis Using Cabo Verde Biodiversity

 ,
, 
 ,
,  ,
,  , and
, and
Abstract
:1. Background
2. Phylogenetic Inference and Divergence Time Estimation
3. Macaronesian Islands as Model Systems in Evolution
4. Phylogenetic Resolution and Divergence Time Estimation among Macaronesian Insular Groups–Case-Study Using A Meta-Analysis of Reptile Data
5. The Potential of Phylogenomics to Understand Evolutionary Relationships in Insular Lineages
6. Final Considerations
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- López-Fernández, H.; Duque, P.; Henriques, S.; Vázquez, N.; dez-Riverola, F.; Vieira, C.P.; Reboiro-Jato, M.; Viera, J. Bioinformatics Protocols for Quickly Obtaining Large-Scale Data Sets for Phylogenetic Inferences. Interdiscip. Sci. Comput. Life Sci. 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Roumpeka, D.D.; Wallace, R.J.; Escalettes, F.; Fotheringham, I.; Watson, M. A review of bioinformatics tools for bio-prospecting from metagenomic sequence data. Front. Genet. 2017, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [PubMed]
- Kulikova, T.; Akhtar, R.; Aldebert, F.; Althorpe, N.; Andersson, M.; Baldwin, A.; Bates, K.; Bhattacharyya, S.; Bower, L.; Browne, P.; et al. EMBL Nucleotide Sequence Database in 2006. Nucleic Acids Res. 2007, 35. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013, 41, D43–D47. [Google Scholar]
- Kodama, Y.; Mashima, J.; Kaminuma, E.; Gojobori, T.; Ogasawara, O.; Takagi, T.; Okubo, K.; Nakamura, Y. The DNA Data Bank of Japan Launches a New Resource, the DDBJ Omics Archive of Functional Genomics Experiments. Nucleic Acids Res. 2012, 40. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Andrew, P.F. The Cancer Genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Myers, N.; Mittermeier, R.A.; Mittermeier, C.G.; Da Fonseca, G.A.; Kent, J. Biodiversity hotspots for conservation priorities. Nature 2000, 403, 853. [Google Scholar] [CrossRef]
- Vasconcelos, R.; Brito, J.C.; Carranza, S.; Harris, D.J. Review of the distribution and conservation status of the terrestrial reptiles of the Cape Verde Islands. Oryx 2013, 47, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Romeiras, M.M.; Catarino, S.; Gomes, I.; Fernandes, C.; Costa, J.C.; Caujapé-Castells, J.; Duarte, M.C. IUCN Red List assessment of the Cape Verde endemic flora: Towards a global strategy for plant conservation in Macaronesia. Bot. J. Linn. Soc. 2016, 180, 413–425. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; Mcgettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Farris, J.S. Methods for Computing Wagner Trees. Syst. Biol. 1970, 19, 83–92. [Google Scholar] [CrossRef]
- Fitch, W.M. Toward defining the course of evolution: Minimum change for a specific tree topology. Syst. Biol. 1971, 20, 406–416. [Google Scholar] [CrossRef]
- Carine, M.A.; Francisco-Ortega, J.; Santos-Guerra, A.; Russell, S.J. Relationships of island and continental floras: Molecular evidence for multiple colonisations into Macaronesia and subsequent back-colonisation of the continent in Convolvulus L. Am. J. Bot. 2004, 91, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Francisco-Ortega, J.; Barber, J.; Santos-Guerra, A.; Febles-Hernández, R.; Jansen, R.K. Origin and evolution of the endemic genera of Gonosperminae (Asteraceae: Anthemideae) from the Canary Islands: Evidence from nucleotide sequences of the internal transcribed spacers of the nuclear ribosomal DNA. Am. J. Bot. 2001, 88, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Helfgott, D.M.; Franciso-Ortega, F.; Santos-Guerra, A.; Jansen, R.K.; Simpson, B.B. Biogeography and breeding system evolution of the woody Bencomia alliance (Rosaceae) in Macaronesia based on ITS sequence data. Syst. Bot. 2000, 25, 82–97. [Google Scholar] [CrossRef]
- Panero, J.L.; Francisco-Ortega, J.; Jansen, R.K.; Santos-Guerra, A. Molecular evidence for multiple origins of woodiness and a New World biogeographic connection of the Macaronesian Island endemic Pericallis (Asteraceae: Senecioneae). Proc. Natl. Acad. Sci. USA 1999, 96, 13886–13891. [Google Scholar] [CrossRef]
- Dias, E.F.; Kilian, N.; Silva, L.; Schaefer, H.; Carine, M.; Rudall, P.J.; Santos-Guerra, A.; Moura, M. Phylogeography of the Macaronesian Lettuce Species Lactuca watsoniana and L. palmensis (Asteraceae). Biochem. Genet. 2018, 56, 315. [Google Scholar] [CrossRef]
- Fernández-Mazuecos, M.; Vargas, P. Genetically depauperate in the continent but rich in oceanic islands: Cistus monspeliensis (Cistaceae) in the Canary Islands. PLoS ONE 2011, 6, e17172. [Google Scholar] [CrossRef] [PubMed]
- Menezes, T.; Romeiras, M.M.; Sequeira, M.M.; Moura, M. Phylogenetic relationships and phylogeography of relevant lineages within the complex Campanulaceae family in Macaronesia. Ecol. Evol. 2017, 8, 88–108. [Google Scholar] [CrossRef] [Green Version]
- Cox, S.C.; Carranza, S.; Brown, R.P. Divergence Times and Colonization of the Canary Islands by Gallotia Lizards. Mol. Phylogenet. Evol. 2010, 56, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Carranza, S.; Arnold, E.N. review of the geckos of the genus Hemidactylus (Squamata: Gekkonidae) from Oman based on morphology, mitochondrial and nuclear data, with descriptions of eight new species. Zootaxa 2012, 3378, 1–95. [Google Scholar] [CrossRef]
- Aldrich, J.R.A. Fisher and the Making of Maximum Likelihood 1912–1922. Stat. Sci. 1997, 12, 162–176. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F.; Nielsen, R.; Bollback, J.P. Bayesian inference of phylogeny and its impact on evolutionary biology. Science 2001, 294, 2310–2314. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Rannala, B. Molecular Phylogenetics: Principles and Practice. Nat. Rev. Genet. 2012, 13, 303–314. [Google Scholar] [CrossRef]
- Yang, Z.; Rannala, B. Bayesian phylogenetic inference using DNA sequences: A Markov chain Monte Carlo Method. Mol. Biol. Evol. 1997, 14, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Pearl, D.; Doss, H. Phylogenetic tree reconstruction using Markov chain Monte Carlo. J. Am. Stat. Assoc. 2000, 95, 493–508. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony; Version 4; Sinauer Associates: Sunderland, MA, USA, 2002. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzke, A.; Samani, B.S.; Neuffer, B.; Mummenhoff, K.; Hurka, H. Molecular evidence in Diplotaxis (Brassicaceae) suggests a Quaternary origin of the Cape Verdean flora. Plant Syst. Evol. 2017, 303, 467–479. [Google Scholar] [CrossRef]
- Romeiras, M.M.; Paulo, O.S.; Duarte, M.C.; Pina-Martins, F.; Cotrim, M.H.; Carine, M.A.; Pais, M.S. Origin and diversification of the genus Echium (Boraginaceae) in the Cape Verde archipelago. Taxon 2011, 60, 1375–1385. [Google Scholar] [CrossRef]
- Thiv, M.; Thulin, M.; Hjertson, M.; Kropf, M.; Linder, H.P. Evidence for a vicariant origin of Macaronesian-Eritreo/Arabian disjunctions in Campylanthus Roth (Plantaginaceae). Mol. Phylogenet. Evol. 2010, 54, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Vitales, D.; Garnatje, T.; Pellicer, J.; Vallès, J.; Santos-Guerra, A.; Sanmartín, I. The explosive radiation of Cheirolophus (Asteraceae, Cardueae) in Macaronesia. BMC Evol. Biol. 2014, 14, 118. [Google Scholar] [CrossRef] [PubMed]
- Carranza, S.; Arnold, E.N.; Geniez, Ph.; Roca, J.; Mateo, J.A. Radiation, Multiple Dispersal and Parallelism in the Skinks, Chalcides and Sphenops (Squamata: Scincidae), with Comments on Scincus and Scincopus and the Age of the Sahara Desert. Mol. Phylogenet. Evol. 2008, 46, 1071–1094. [Google Scholar] [CrossRef] [PubMed]
- Crottini, A.; Madsen, O.; Poux, C.; Strauss, A.; Vieites, D.R.; Vences, M. Vertebrate Time-Tree Elucidates the Biogeographic Pattern of a Major Biotic Change around the K-T Boundary in Madagascar. Proc. Natl. Acad. Sci. USA 2012, 109, 5358–5363. [Google Scholar] [CrossRef] [PubMed]
- Karin, B.R.; Metallinou, M.; Weinell, J.L.; Jackman, T.R.; Bauer, A.M. Resolving the Higher-Order Phylogenetic Relationships of the Circumtropical Mabuya Group (Squamata: Scincidae): An out-of-Asia Diversification. Mol. Phylogenet. Evol. 2016, 102, 220–232. [Google Scholar] [CrossRef]
- Metallinou, M.; Weinell, J.L.; Karin, B.R.; Conradie, W.; Wagner, P.; Schmitz, A.; Jackman, T.R.; Bauer, A.M. A Single Origin of Extreme Matrotrophy in African Mabuyine Skinks. Biol. Lett. 2016, 12, 20160430. [Google Scholar] [CrossRef]
- Miralles, A.; Vasconcelos, R.; Perera, A.; Harris, D.J.; Carranza, S. An Integrative Taxonomic Revision of the Cape Verdean Skinks (Squamata, Scincidae). Zoologica Scripta 2011, 40, 16–44. [Google Scholar] [CrossRef]
- Carranza, S.; Harris, D.J.; Arnold, E.N.; Batista, V.; de La Vega, J.P.G. Phylogeography of the Lacertid Lizard, Psammodromus algirus, in Iberia and across the Strait of Gibraltar. J. Biogeogr. 2006, 33, 1279–1288. [Google Scholar] [CrossRef]
- Agarwal, I.; Bauer, A.M.; Jackman, T.R.; Karanth, K.P. Insights into Himalayan Biogeography from Geckos: A Molecular Phylogeny of Cyrtodactylus (Squamata: Gekkonidae). Mol. Phylogenet. Evol. 2014, 80, 145–155. [Google Scholar] [CrossRef]
- Bansal, R.; Karanth, K.P. Phylogenetic Analysis and Molecular Dating Suggest That Hemidactylus anamallensis Is Not a Member of the Hemidactylus Radiation and Has an Ancient Late Cretaceous Origin. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Carranza, S.; Arnold, E.N. Systematics, Biogeography, and Evolution of Hemidactylus Geckos (Reptilia: Gekkonidae) Elucidated Using Mitochondrial DNA Sequences. Mol. Phylogenet. Evol. 2006, 38, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Gamble, T.; Bauer, A.M.; Colli, G.R.; Greenbaum, E.; Jackman, T.R.; Vitt, L.J.; Simons, A.M. Coming to America: Multiple Origins of New World Geckos: Origins of New World Geckos. J. Evol. Biol. 2011, 24, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Porta, J.; Morales, H.E.; Gómez-Díaz, E.; Sindaco, R.; Carranza, S. Patterns of Diversification in Islands: A Comparative Study across Three Gecko Genera in the Socotra Archipelago. Mol. Phylogenet. Evol. 2016, 98, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Šmíd, J.; Carranza, S.; Kratochvíl, L.; Gvoždík, V.; Nasher, A.K.; Moravec, J. Out of Arabia: A Complex Biogeographic History of Multiple Vicariance and Dispersal Events in the Gecko Genus Hemidactylus (Reptilia: Gekkonidae). PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Carranza, S.; Arnold, E.N.; Mateo, J.A.; Geniez, P. Relationships and Evolution of the North African Geckos, Geckonia and Tarentola (Reptilia: Gekkonidae), Based on Mitochondrial and Nuclear DNA Sequences. Mol. Phylogenet. Evol. 2002, 23, 244–256. [Google Scholar] [CrossRef]
- Rato, C.; Carranza, S.; Harris, D.J. Evolutionary History of the Genus Tarentola (Gekkota: Phyllodactylidae) from the Mediterranean Basin, Estimated Using Multilocus Sequence Data. BMC Evol. Biol. 2012, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, R.; Carranza, S.; Harris, D.J. Insight into an Island Radiation: The Tarentola Geckos of the Cape Verde Archipelago. J. Biogeogr. 2010, 37, 1047–1060. [Google Scholar] [CrossRef]
- Posada, D. JModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2016, msw260. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F.P. MRBAYES: Bayesian Inference of Phylogenetic Trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Delsuc, F.; Dufayard, J.F.; Gascuel, O. Estimating Maximum Likelihood Phylogenies with PhyML. Methods Mol. Biol. 2009, 537, 113–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwickl, D.J. Genetic Algorithm Approaches for the Phylogenetic Analysis of Large Biological Sequence Datasets under the Maximum Likelihood Criterion. Ph.D. Thesis, The University of Texas at Austin, Austin, TX, USA, 2006. [Google Scholar]
- Koutroumpa, K.; Theodoridis, S.; Warren, B.H.; Jiménez, A.; Celep, F.; Doğan, M.; Romeiras, M.M.; Santos-Guerra, A.; Fernández-Palacios, J.M.; Caujapé-Castells, J.; et al. An expanded molecular phylogeny of Plumbaginaceae, with emphasis on Limonium (sea lavenders): Taxonomic implications and biogeographic considerations. Ecol. Evol. 2018, 8, 12397–12424. [Google Scholar] [CrossRef] [PubMed]
- Bromham, L.; Duchêne, S.; Hua, X.; Ritchie, A.M.; Duchêne, D.A.; Ho, S.Y. Bayesian molecular dating: Opening up the black box. Biol. Rev. 2018, 93, 1165–1191. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Bouckaert, R.R. Bayesian Evolutionary Analysis with BEAST; Cambridge University Press: Cambridge, UK, 2015. [Google Scholar]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian Evolutionary Analysis by Sampling Trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.G.; Wu, C.-H.; Xie, D.; Suchard, M.; Rambaut, A.; Drummond, A.J. Beast2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2013, 10, 1003537. [Google Scholar] [CrossRef]
- Ogilvie, H.A.; Bouckaert, R.R.; Drummond, A.J. StarBEAST2 Brings Faster Species Tree Inference and Accurate Estimates of Substitution Rates. Mol. Biol. Evol. 2017, 34, 2101–2114. [Google Scholar] [CrossRef]
- Belfiore, N.M.; Liu, L.; Moritz, C. Multilocus Phylogenetics of a Rapid Radiation in the Genus Thomomys (Rodentia: Geomyidae). Syst. Biol. 2008, 57, 294–310. [Google Scholar] [CrossRef]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A Computer Program to Estimate Gene Genealogies. Mol. Ecol. 2010, 9, 1657–1659. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Romeiras, M.M.; Monteiro, F.; Duarte, M.C.; Schaefer, H.; Carine, M. Patterns of genetic diversity in three plant lineages endemic to the Cape Verde Islands. AoB Plants 2015, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, H.; Moura, M.; Belo Maciel, M.G.; Silva, L.; Rumsey, F.J.; Carine, M.A. The Linnean shortfall in oceanic island biogeography: A case study in the Azores. J. Biogeogr. 2011, 38, 1345–1355. [Google Scholar] [CrossRef]
- Romeiras, M.M.; Vieira, A.; Silva, D.N.; Moura, M.; Santos-Guerra, A.; Batista, D.; Duarte, M.C.; Paulo, O. Evolutionary and Biogeographic Insights on the Macaronesian Beta-Patellifolia Species (Amaranthaceae) from a Time-Scaled Molecular Phylogeny. PLoS ONE 2016, 11, e0152456. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, R.J.; Fernández-Palacios, J.M.; Matthews, T.J.; Borregaard, M.K.; Triantis, K.A. Island biogeography: Taking the long view of nature’s laboratories. Science 2017, 357, eaam8326. [Google Scholar] [CrossRef]
- Warren, B.H.; Simberloff, D.; Ricklefs, R.E.; Aguilee, R.; Condamine, F.L.; Gravel, D.; Morlon, H.; Mouquet, N.; Rosindell, J.; Casquet, J.; et al. Islands as model systems in ecology and evolution: Prospects fifty years after MacArthur-Wilson. Ecol. Lett. 2015, 18, 200–217. [Google Scholar] [CrossRef] [PubMed]
- Price, J.P.; Otto, R.; Menezes de Sequeira, M.; Kueffer, C.; Schaefer, H.; Caujapé-Castells, J.; Fernández-Palacios, J.M. Colonization and diversification shape species–area relationships in three Macaronesian archipelagos. J. Biogeogr. 2018, 45, 2027–2039. [Google Scholar] [CrossRef]
- Alarcón, M.; Roquet, C.; García-Fernández, A.; Vargas, P.; Aldasoro, J.J. Phylogenetic and phylogeographic evidence for a Pleistocene disjunction between Campanula jacobaea (Cape Verde Islands) and C. balfourii (Socotra). Mol. Phylogen. Evol. 2013, 69, 828–836. [Google Scholar] [CrossRef]
- Ojeda, D.I.; Santos-Guerra, A.; Oliva-Tejera, F.; Jaen-Molina, R.; Caujapé-Castells, J.; Marrero-Rodríguez, Á.; Cronk, Q. DNA barcodes successfully identified Macaronesian Lotus (Leguminosae) species within early diverged lineages of Cape Verde and mainland Africa. AoB Plants 2014, 6, plu050. [Google Scholar] [CrossRef]
- Ávila, S.P.; Melo, C.; Berning, B.; Cordeiro, R.; Landau, B.; Marques, C. Persististrombus Coronatus (Mollusca: Strombidae) in the Lower Pliocene of Santa Maria Island (Azores, NE Atlantic): Paleoecology, Paleoclimatology and Paleobiogeographic Implications. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2016, 441, 912–923. [Google Scholar] [CrossRef]
- Ramalho, R.S. Building the Cape Verde Islands, 1st ed.; Springer: Berlin, Germany, 2011; p. 207. [Google Scholar] [CrossRef]
- Torres, P.; Silva, L.; Serralheiro, A.; Tassinari, C.; Munhá, J. Enquadramento geocronológico pelo método K/Ar das principais sequências vulcano-estratigráficas da Ilha do Sal—Cabo Verde. Garcia de Orta Serviços Geológicos 2002, 18, 9–13. [Google Scholar]
- Fernández-Palacios, J.M.; Rijsdijk, K.F.; Norder, S.J.; Otto, R.; de Nascimento, L.; Fernández-Lugo, S.; Tjørve, E.; Whittaker, R.J. Towards a glacial-sensitive model of island biogeography. Global Ecol. Biogeogr. 2016, 25, 817–830. [Google Scholar] [CrossRef]
- Weigelt, P.; Steinbauer, M.J.; Cabral, J.S.; Kreft, H. Late Quaternary climate change shapes island biodiversity. Nature 2016, 532, 99. [Google Scholar] [CrossRef] [PubMed]
- Ávila, S.P.; Cordeiro, R.; Madeira, P.; Silva, L.; Medeiros, A.; Rebelo, A.C.; Melo, C.; Neto, A.I.; Haroun, R.; Monteiro, A.; et al. Global Change Impacts on Large-Scale Biogeographic Patterns of Marine Organisms on Atlantic Oceanic Islands. Mar. Pollut. Bull. 2018, 126, 101–112. [Google Scholar] [CrossRef]
- Shaffer, H.B.; Pauly, G.B.; Oliver, J.C.; Trenham, P.C. The molecular phylogenetics of endangerment: Cryptic variation and historical phylogeography of the California tiger salamander, Ambystoma californiense. Mol. Ecol. 2004, 13, 3033–3049. [Google Scholar] [CrossRef]
- Jin, Y.; Yang, Z.; Brown, R.P.; Liao, P.; Liu, N. Intraspecific lineages of the lizard Phrynocephalus putjatia from the Qinghai-Tibetan Plateau: Impact of physical events on divergence and discordance between morphology and molecular markers. Mol. Phylog. Evol. 2014, 71, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Liu, N.; Brown, R.P. The geography and timing of genetic divergence in the lizard Phrynocephalus theobaldi on the Qinghai-Tibetan plateau. Sci. Rep. 2017, 7, 2281. [Google Scholar] [CrossRef]
- Arnold, E.N.; Vasconcelos, R.; Harris, D.J.; Mateo, J.A.; Carranza, S. Systematics, biogeography and evolution of the endemic Hemidactylus geckos (Reptilia, Squamata, Gekkonidae) of the Cape Verde Islands: Based on morphology and mitochondrial and nuclear DNA sequences. Zool. Scr. 2008, 37, 619–636. [Google Scholar] [CrossRef]
- Vasconcelos, R.; Perera, A.; Geniez, P.; Harris, D.J.; Carranza, S. An integrative taxonomic revision of the Tarentola geckos (Squamata, Phyllodactylidae) of the Cape Verde Islands. Zool. J. Linn. Soc. 2002, 164, 328–360. [Google Scholar] [CrossRef]
- Pinho, C.; Santos, B.; Mata, V.; Seguro, M.; Romeiras, M.M.; Lopes, R.; Vasconcelos, R. What Is the Giant Wall Gecko Having for Dinner? Conservation Genetics for Guiding Reserve Management in Cabo Verde. Genes 2018, 9, 599. [Google Scholar] [CrossRef]
- Carranza, S.; Arnold, E.N. Lizards (Reptilia, Scincidae) crossed the Atlantic twice. Syst. Biol. 2003, 1, 275–282. [Google Scholar] [CrossRef]
- Datta-Roy, A.; Singh, M.; Srinivasulu, C.; Karanth, K.P. Phylogeny of the Asian Eutropis (Squamata: Scincidae) reveals an “into India” endemic Indian radiation. Mol. Phylog. Evol. 2012, 63, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Mausfeld, P.; Schmitz, A.; Böhme, W.; Misof, B.; Vrcibradic, D.; Rocha, C.F.D. Phylogenetic affinities of Mabuya atlantica Schmidt, 1945, endemic to the Atlantic Ocean archipelago of Fernando de Noronha (Brazil): Necessity of partitioning the genus Mabuya Fitzinger, 1826 (Scincidae: Lygosominae). Zool. Anz. 2002, 241, 281–293. [Google Scholar] [CrossRef]
- Ho, S.Y.W.; Phillips, M.J. Accounting for Calibration Uncertainty in Phylogenetic Estimation of Evolutionary Divergence Times. Syst. Biol. 2009, 58, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parham, J.F.; Donoghue, P.C.; Bell, C.J.; Calway, T.D.; Head, J.J.; Holroyd, P.A.; Inoue, J.G.; Irmis, R.B.; Jouce, W.G.; Ksepka, D.T.; et al. Best practices for justifying fossil calibrations. Syst. Biol. 2011, 61, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Walker, J.F.; Brown, J.; Walker-Hale, N. Nested phylogenetic conflicts, combinability, and deep phylogenomics in plants. bioRxiv 2018, 371930. [Google Scholar] [CrossRef]
- Wickett, N.J.; Mirarab, S.; Nguyen, N.; Warnow, T.; Carpenter, E.; Matasci, N.; Ayyampalayam, N. Phylotranscriptomic Analysis of the Origin and Early Diversification of Land Plants. Proc. Natl. Acad. Sci. USA 2014, 111, E4859–E4868. [Google Scholar] [CrossRef] [PubMed]
- Nater, A.; Burri, R.; Kawakami, T.; Smeds, L.; Ellegren, H. Resolving evolutionary relationships in closely related species with whole-genome sequencing data. Syst. Biol. 2015, 64, 1000–1017. [Google Scholar] [CrossRef] [PubMed]
- Mort, M.E.; Crawford, D.J.; Kelly, J.K.; Santos-Guerra, A.; Menezes de Sequeira, M.; Moura, M.; Caujapé-Castells, J. Multiplexed-shotgun-genotyping data resolve phylogeny within a very recently derived insular lineage. Am. J. Bot. 2015, 102, 634–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Wiens, J.J. Combining Phylogenomic and Supermatrix Approaches, and a Time-Calibrated Phylogeny for Squamate Reptiles (Lizards and Snakes) Based on 52 Genes and 4162 Species. Mol. Phylogenet. Evol. 2016, 94, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Zong, D.; Gan, P.; Zhou, A.; Zhang, Y.; Zou, X.; Duan, A.; Song, Y.; He, C. Plastome Sequences Help to Resolve Deep-Level Relationships of Populus in the Family Salicaceae. Front. Plant Sci. 2019, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Twyford, A.D.; Ness, R.W. Strategies for complete plastid genome sequencing. Mol. Ecol. Resour. 2017, 17, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.-J.; Hinsinger, D.D.; Elias, R.B.; Strijk, J.S. The plastome sequence of Laurus azorica (Seub.) Franco, an endemic tree species of the Azores islands. Mitochondrial DNA Part B 2019, 4, 363–365. [Google Scholar] [CrossRef]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the Power of RADseq for Ecological and Evolutionary Genomics. Nat. Rev. Genet. 2016, 2, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Catchen, J.M.; Hohenlohe, P.A.; Bernatchez, L.; Funk, W.C.; Andrews, K.R.; Allendorf, F.W. Unbroken: RADseq Remains a Powerful Tool for Understanding the Genetics of Adaptation in Natural Populations. Mol. Ecol. Resour. 2017, 17, 362–365. [Google Scholar] [CrossRef] [PubMed]
- McKinney, G.J.; Larson, W.A.; Seeb, L.W.; Seeb, J.E. RAD seq provides unprecedented insights into molecular ecology and evolutionary genetics: Comment on Breaking RAD by Lowry et al. Mol. Ecol. Resour. 2017, 17, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Ree, R.H.; Hipp, A.L. Inferring Phylogenetic History from Restriction Site Associated DNA (RADseq). In Next-Generation Sequencing in Plant Systematics, 1st ed.; Hörandl, E., Appelhans, M.S., Eds.; Koeltz Scientific Books: Oberreifenberg, Germany, 2015; p. 298. [Google Scholar] [CrossRef]
- Lowry, D.B.; Hoban, S.; Kelley, J.L.; Lotterhos, K.E.; Reed, L.K.; Antolin, M.F.; Storfer, A. Breaking RAD: An Evaluation of the Utility of Restriction Site-Associated DNA Sequencing for Genome Scans of Adaptation. Mol. Ecol. Resour. 2017, 17, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.S.B.; Silva, S.E.; Pina-Martins, F.; Loureiro, J.; Castro, M.; Gharbi, K.; Johnson, K.P.; Dietrich, C.H.; Borges, P.A.V.; Quartau, J.A.; et al. Assessing Genotype-Phenotype Associations in Three Dorsal Colour Morphs in the Meadow Spittlebug Philaenus Spumarius (L.) (Hemiptera: Aphrophoridae) Using Genomic and Transcriptomic Resources. BMC Genet. 2016, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Grewe, F.; Huang, J.-P.; Leavitt, S.D.; Lumbsch, H.T. Reference-Based RADseq Resolves Robust Relationships among Closely Related Species of Lichen-Forming Fungi Using Metagenomic DNA. Sci. Rep. 2017, 7, 9884. [Google Scholar] [CrossRef]
- Hohenlohe, P.A.; Bassham, S.; Etter, P.D.; Stiffler, N.; Johnson, E.A.; Cresko, W.A. Population Genomics of Parallel Adaptation in Threespine Stickleback Using Sequenced RAD Tags. PLoS Genet. 2010, 6, e1000862. [Google Scholar] [CrossRef]
- McCluskey, B.M.; Postlethwait, J.H. Phylogeny of Zebrafish, a “Model Species,” within Danio, a ‘Model Genus. Mol. Biol. Evol. 2015, 32, 635–652. [Google Scholar] [CrossRef] [PubMed]
- Tripp, E.A.; Tsai, Y.H.E.; Zhuang, Y.; Dexter, K.G. RADseq Dataset with 90% Missing Data Fully Resolves Recent Radiation of Petalidium (Acanthaceae) in the Ultra-Arid Deserts of Namibia. Ecol. Evol. 2017, 7, 7920–7936. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.P.; Paterson, S.; Risse, J. Genomic Signatures of Historical Allopatry and Ecological Divergence in an Island Lizard. Genome Biol. Evol. 2016, 8, 3618–3626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellegren, H. Genome sequencing and population genomics in non-model organisms. Trends Ecol. Evol. 2014, 29, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Barley, A.J.; Monnahan, P.J.; Thomson, R.C.; Grismer, L.L.; Brown, R.M. Sun skink landscape genomics: Assessing the roles of micro-evolutionary processes in shaping genetic and phenotypic diversity across a heterogeneous and fragmented landscape. Mol. Ecol. 2015, 24, 1696–1712. [Google Scholar] [CrossRef] [PubMed]
- Wessinger, C.A.; Freeman, C.C.; Mort, M.E.; Rausher, M.D.; Hileman, L.C. Multiplexed Shotgun Genotyping Resolves Species Relationships within the North American Genus Penstemon. Am. J. Bot. 2016, 103, 912–922. [Google Scholar] [CrossRef]
- Eaton, D.A.R.; Ree, R.H. Inferring Phylogeny and Introgression Using RADseq Data: An Example from Flowering Plants (Pedicularis: Orobanchaceae). Syst. Biol. 2013, 62, 689–706. [Google Scholar] [CrossRef] [PubMed]
- Curto, M.; Schachtler, C.; Puppo, P.; Meimberg, H. Using a new RAD-sequencing approach to study the evolution of Micromeria in the Canary Islands. Mol. Phylogenet. Evol. 2018, 119, 160–169. [Google Scholar] [CrossRef]
- Peñalba, J.V.; Smith, L.L.; Tonione, M.A.; Sass, C.; Hykin, S.M.; Skipwith, P.L.; Mcguire, J.A.; Bowie, R.C.K.; Moritz, C. Sequence capture using PCR-generated probes: A cost-effective method of targeted high-throughput sequencing for nonmodel organisms. Mol. Ecol. Resour. 2014, 14, 1000–1010. [Google Scholar] [CrossRef]
- Bragg, J.G.; Potter, S.; Bi, K.; Moritz, C. Exon capture phylogenomics: Efficacy across scales of divergence. Mol. Ecol. Resour. 2016, 16, 1059–1068. [Google Scholar] [CrossRef]
- Brandley, M.C.; Bragg, J.G.; Singhal, S.; Chapple, D.G.; Jennings, C.K.; Lemmon, A.R.; Lemmon, E.M.; Thompson, M.B.; Moritz, C. Evaluating the performance of anchored hybrid enrichment at the tips of the tree of life: A phylogenetic analysis of Australian Eugongylus group scincid lizards. BMC Evol. Biol. 2015, 15, 62. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
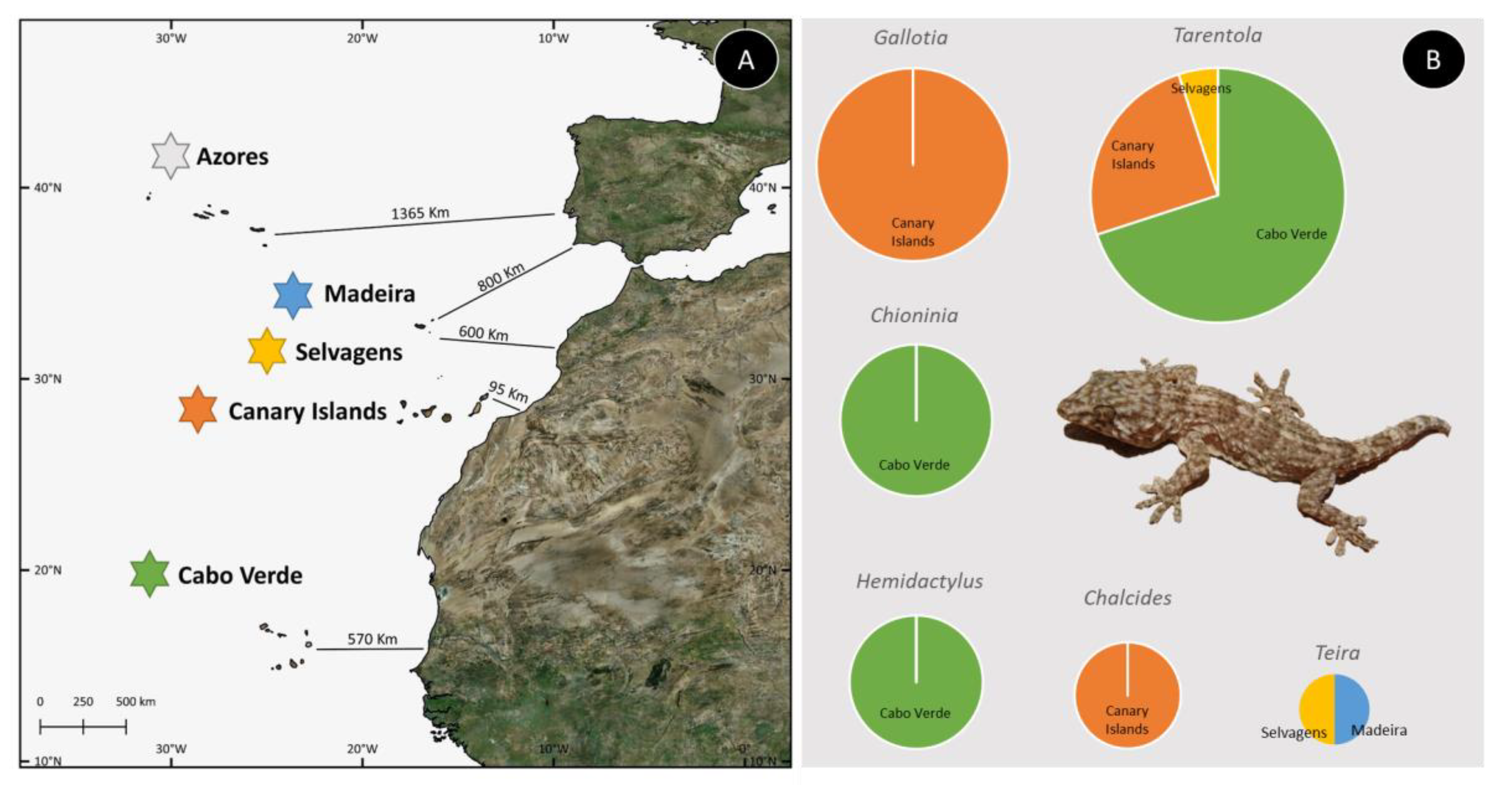
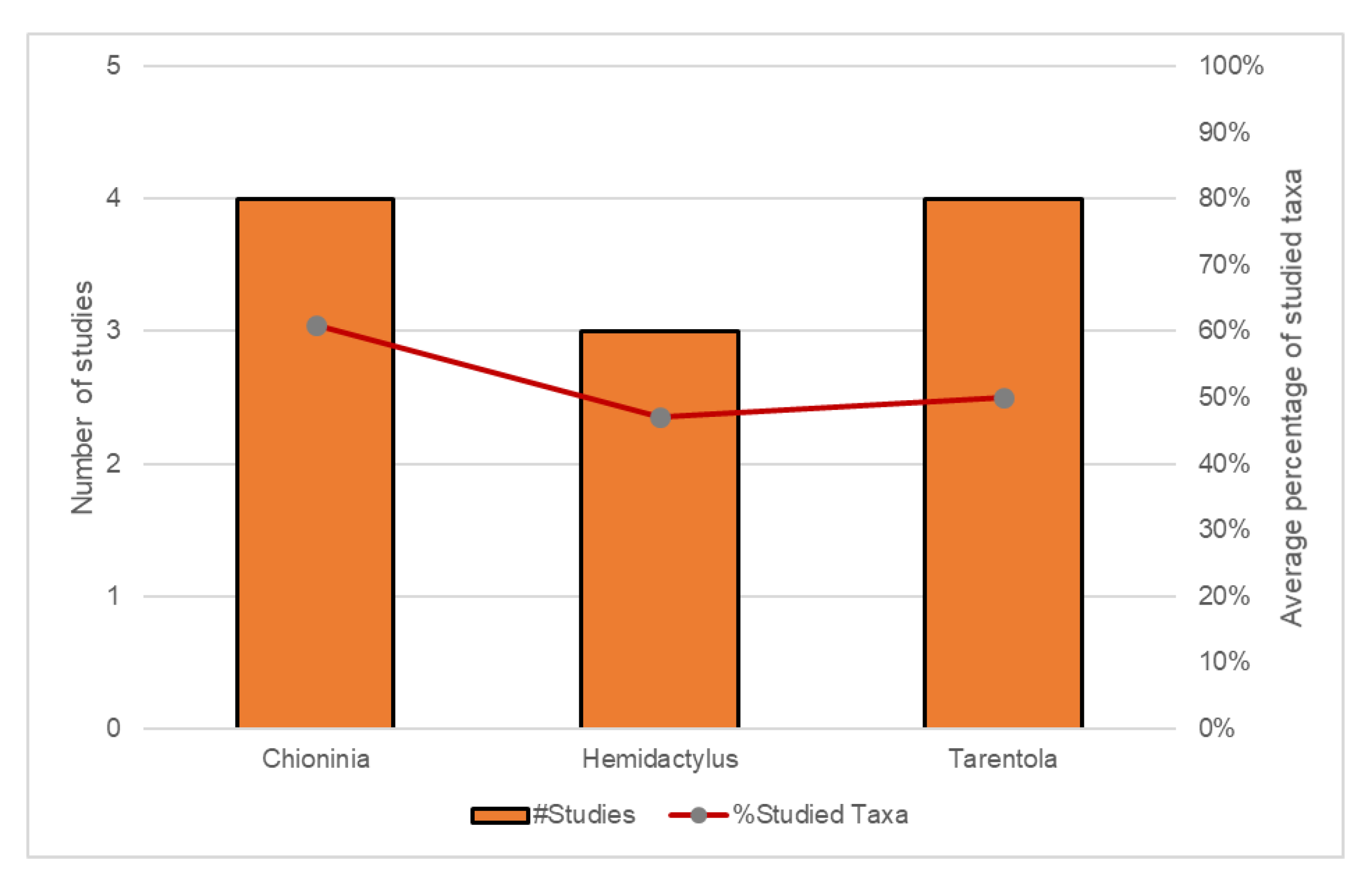
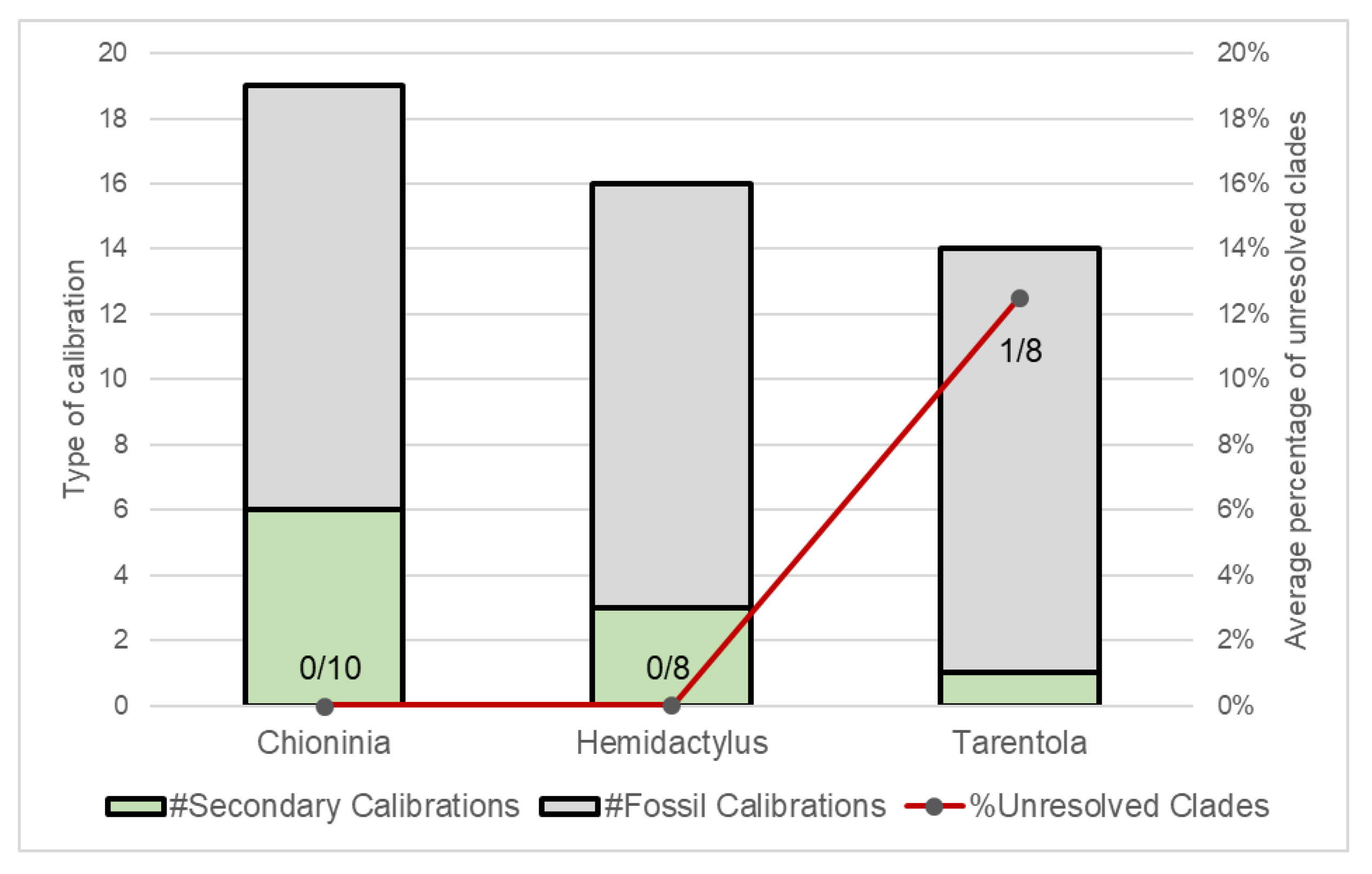
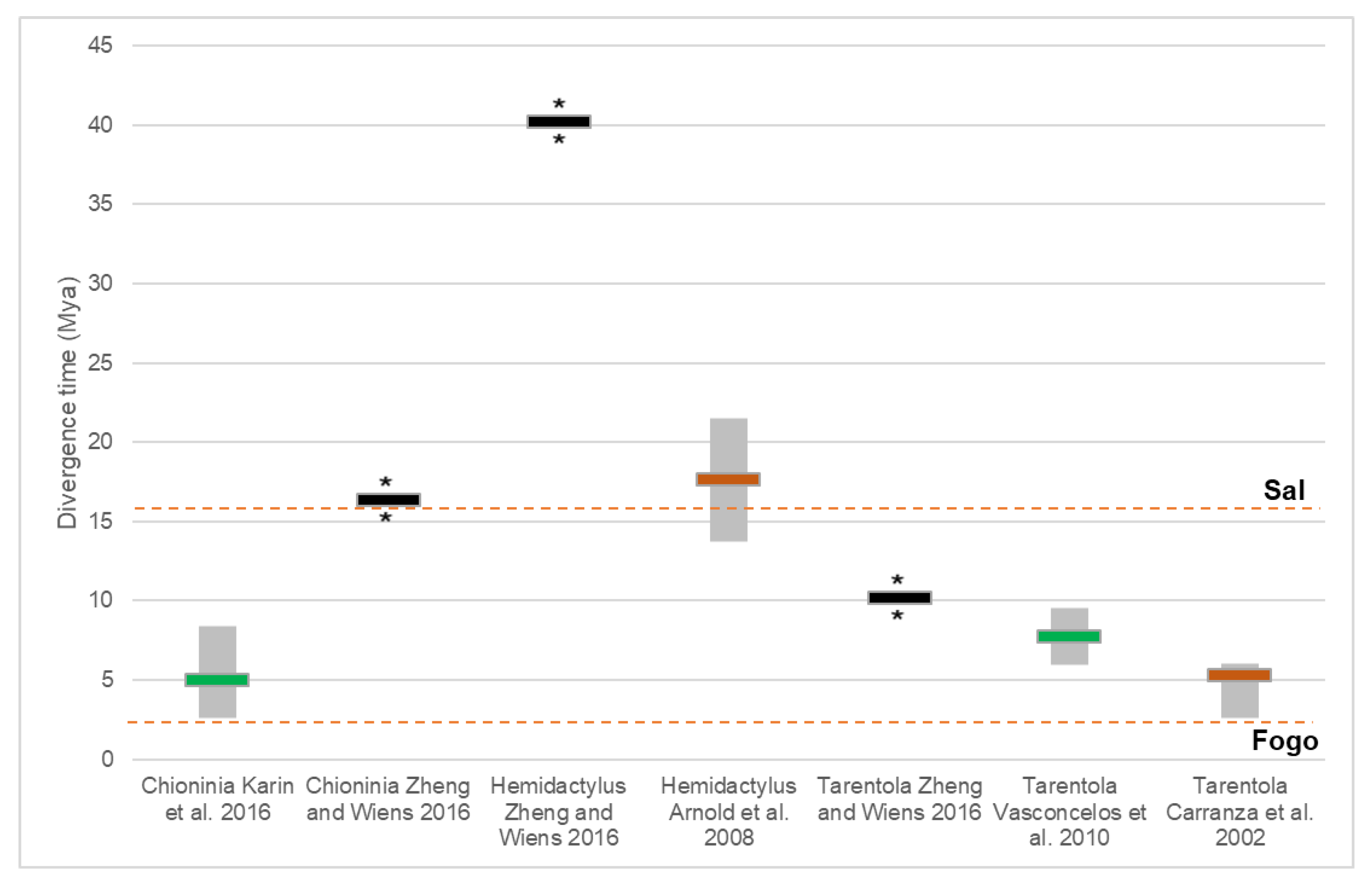
| Family/Genus | Markers | Analyses | Software | Dating | Year | Ref. |
|---|---|---|---|---|---|---|
| Scincidae | ||||||
| Chalcides | cyt b, 12S, 16S | ML+BI | ModelTest, MrBayes, PhyML | Mutation rate (12S, cyt b) + Geological | 2008 | [35] |
| Chalcides | RAG1, BDNF | BI | MrBayes, BEAST | Mutation rate + Geological | 2012 | [36] |
| Chioninia | 16S, ND2, BDNF, BRCA1, BRCA2, CMOS, EXPH5, KIF24, MC1R, MXRA5, RAG1 | ML+BI | PartitionFinder, RAxML, MrBayes | Mutation rate (16S, ND2) | 2016 | [37] |
| Chioninia | 16S, ND2, BDNF, BRCA1, BRCA2, CMOS, EXPH5, KIF24, MC1R, MXRA5, RAG1 | BI | BEAST | Mutation rate (previous works) | 2016 | [38] |
| Chioninia | cyt b, COI, 12S | ML+BI | jModelTest, PhyML, MrBayes | Mutation rate (12S, cyt b) | 2011 | [39] |
| Lacertidae | ||||||
| Gallotia | cyt b, 12S, 16S | ML, MP | ModelTest, PhyML, PAUP | Mutation rate (12S, cyt b) + Geological | 2006 | [40] |
| Gallotia | cyt b, 12S, 16S, COI | BI, MP | MrBayes, TNT, BEAST | Geological | 2010 | [22] |
| Gekkonidae | ||||||
| Hemidactylus | ND2, RAG1, PDC | ML+BI | PartitionFinder, RAxML, MrBayes, BEAST | Fossils | 2014 | [41] |
| Hemidactylus | Cmos, 12S, RAG1, PDC | ML+BI | ModelTest, Paup, BEAST | Fossils + Mutation rate | 2013 | [42] |
| Hemidactylus | cyt b, 12S | ML | ModelTest, Paup, PhyML, MrBayes | Mutation rate | 2006 | [43] |
| Hemidactylus | 12S, cyt b, cmos, ND4, MC1R, RAG2 | ML+BI | RAxML, MrBayes | Mutation rate (12S, cyt b) | 2012 | [23] |
| Hemidactylus | RAG1, RAG2, Cmos, ACM4, PDC | ML | RAxML | Fossils | 2011 | [44] |
| Hemidactylus | 12S, ACM4, cmos, RAG1, RAG2, PDC | BI | PartitionFinder, BEAST | Fossils + Geological | 2016 | [45] |
| Hemidactylus | 12S, cyt b, MC1R, cmos, RAG1, RAG2 | ML+BI | jModelTest, BEAST, RAxML, MrBayes | Mutation rate (12S, cyt b) | 2013 | [46] |
| Phyllodactylidae | ||||||
| Tarentola | cyt b, 12S, cmos | ML, MP | Paup | Geological | 2002 | [47] |
| Tarentola | 12S, 16S, PDC, ACM4, MC1R, RAG2 | ML+BI | jModelTest, RAxML, MrBayes | Mutation rate | 2012 | [48] |
| Tarentola | cyt b, 12S | ML+BI | jModelTest, MrBayes, PhyML | Mutation rate (previous works) | 2010 | [49] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romeiras, M.M.; Pena, A.R.; Menezes, T.; Vasconcelos, R.; Monteiro, F.; Paulo, O.S.; Moura, M. Shortcomings of Phylogenetic Studies on Recent Radiated Insular Groups: A Meta-Analysis Using Cabo Verde Biodiversity. Int. J. Mol. Sci. 2019, 20, 2782. https://doi.org/10.3390/ijms20112782
Romeiras MM, Pena AR, Menezes T, Vasconcelos R, Monteiro F, Paulo OS, Moura M. Shortcomings of Phylogenetic Studies on Recent Radiated Insular Groups: A Meta-Analysis Using Cabo Verde Biodiversity. International Journal of Molecular Sciences. 2019; 20(11):2782. https://doi.org/10.3390/ijms20112782
Chicago/Turabian StyleRomeiras, Maria M., Ana Rita Pena, Tiago Menezes, Raquel Vasconcelos, Filipa Monteiro, Octávio S. Paulo, and Mónica Moura. 2019. "Shortcomings of Phylogenetic Studies on Recent Radiated Insular Groups: A Meta-Analysis Using Cabo Verde Biodiversity" International Journal of Molecular Sciences 20, no. 11: 2782. https://doi.org/10.3390/ijms20112782
APA StyleRomeiras, M. M., Pena, A. R., Menezes, T., Vasconcelos, R., Monteiro, F., Paulo, O. S., & Moura, M. (2019). Shortcomings of Phylogenetic Studies on Recent Radiated Insular Groups: A Meta-Analysis Using Cabo Verde Biodiversity. International Journal of Molecular Sciences, 20(11), 2782. https://doi.org/10.3390/ijms20112782