Contribution of Impaired Insulin Signaling to the Pathogenesis of Diabetic Cardiomyopathy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Epidemiology and Pathophysiology of DCM
3. Insulin Resistance and the Pathogenesis of DCM
3.1. Molecular and Cellular Mechanisms of DCM
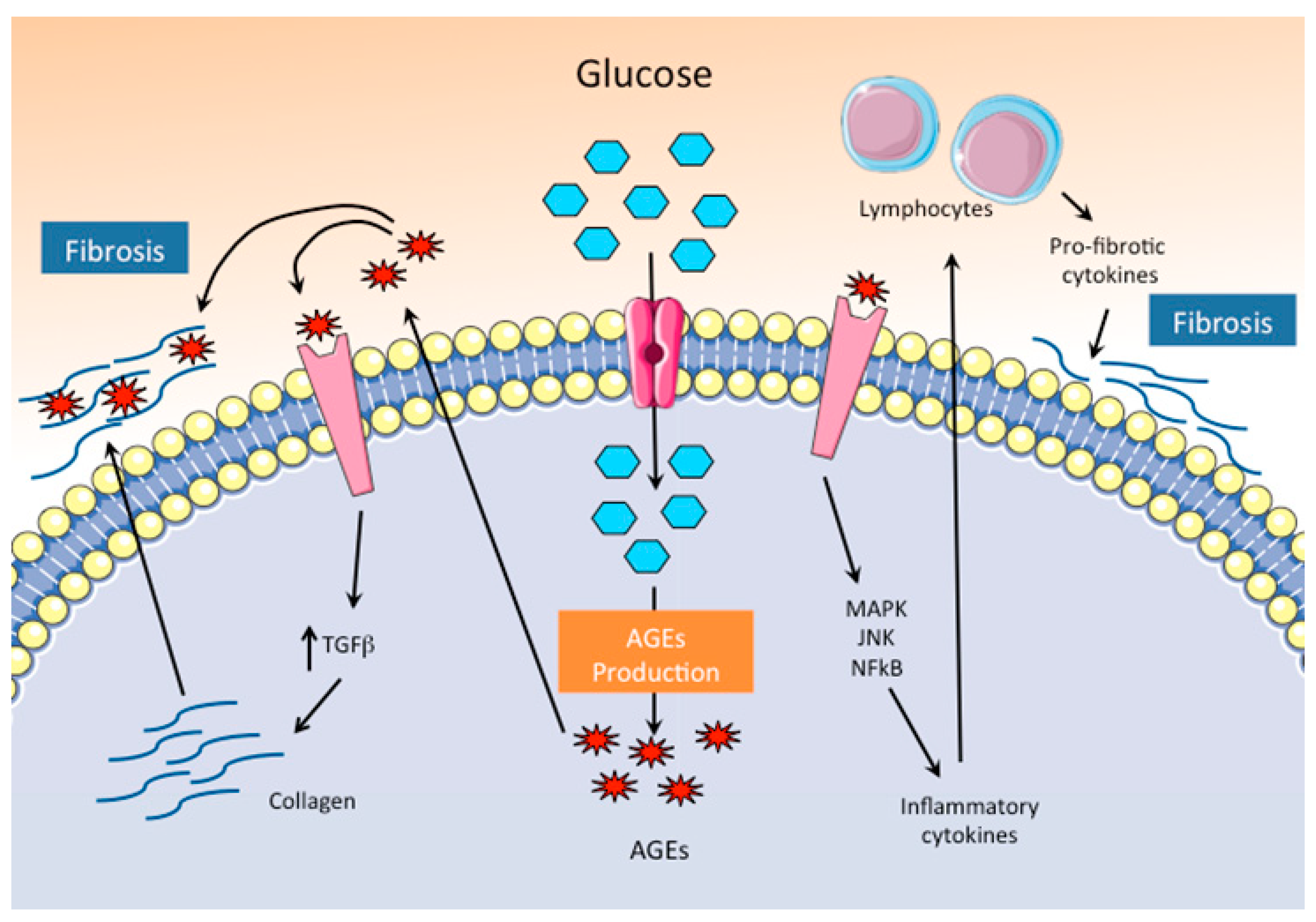
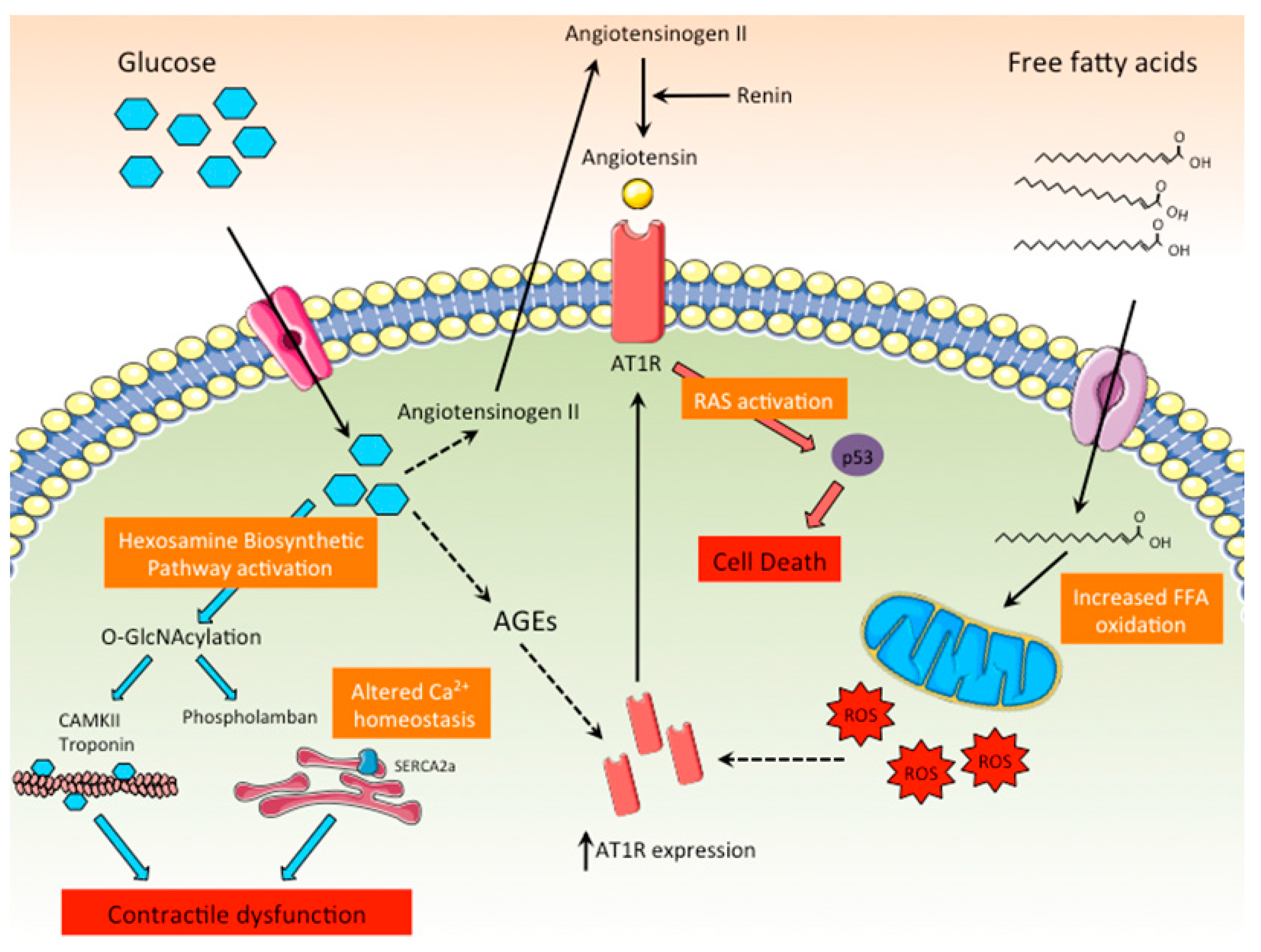
3.1.1. Increased Production of AGEs
3.1.2. Increased Substrate Flux through the Hexosamine Biosynthetic Pathway
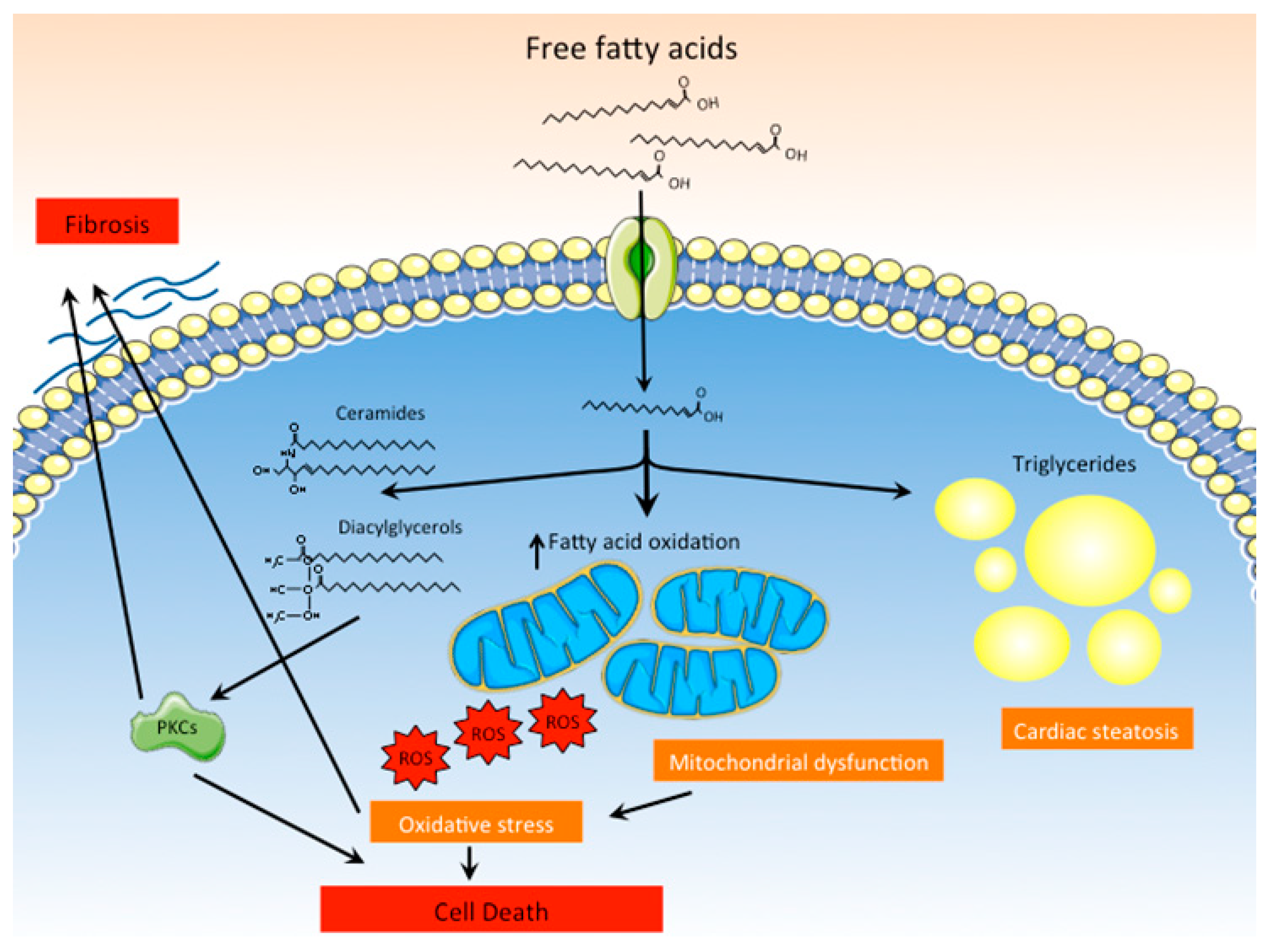
3.1.3. Lipotoxicity
3.1.4. ROS Production and Mitochondrial Dysfunction
3.1.5. Activation of RAS
3.1.6. Calcium Mishandling
3.2. Effects of Insulin Resistance on Metabolic Flexibility and its Implications on Cardiac Dysfunction
4. Rodent Models of Impaired Insulin Signaling for the Study of DCM
4.1. Genetic Models of Cardiac-Specific Disruption of Insulin Signaling for the Study of DCM
4.2. Rodent Models of Diabetes or Systemic Insulin Resistance for the Study of DCM
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Helath Organization. Global Report on Diabetes. 2016. Available online: www.who.int (accessed on 10 June 2019).
- Abbott, R.D.; Donahue, R.P.; Kannel, W.B.; Wilson, P.W. The impact of diabetes on survival following myocardial infarction in men vs women. The Framingham Study. JAMA 1988, 260, 3456–3460. [Google Scholar] [CrossRef] [PubMed]
- Iribarren, C.; Karter, A.J.; Go, A.S.; Ferrara, A.; Liu, J.Y.; Sidney, S.; Selby, J.V. Glycemic control and heart failure among adult patients with diabetes. Circulation 2001, 103, 2668–2673. [Google Scholar] [CrossRef] [PubMed]
- Lind, M.; Bounias, I.; Olsson, M.; Gudbjornsdottir, S.; Svensson, A.M.; Rosengren, A. Glycaemic control and incidence of heart failure in 20,985 patients with type 1 diabetes: an observational study. Lancet 2011, 378, 140–146. [Google Scholar] [CrossRef]
- Stratton, I.M.; Adler, A.I.; Neil, H.A.; Matthews, D.R.; Manley, S.E.; Cull, C.A.; Hadden, D.; Turner, R.C.; Holman, R.R. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): Prospective observational study. BMJ 2000, 321, 405–412. [Google Scholar] [CrossRef]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef]
- Di Bonito, P.; Cuomo, S.; Moio, N.; Sibilio, G.; Sabatini, D.; Quattrin, S.; Capaldo, B. Diastolic dysfunction in patients with non-insulin-dependent diabetes mellitus of short duration. Diabet. Med. 1996, 13, 321–324. [Google Scholar] [CrossRef]
- Nicolino, A.; Longobardi, G.; Furgi, G.; Rossi, M.; Zoccolillo, N.; Ferrara, N.; Rengo, F. Left ventricular diastolic filling in diabetes mellitus with and without hypertension. Am. J. Hypertens. 1995, 8, 382–389. [Google Scholar] [CrossRef]
- Poirier, P.; Bogaty, P.; Garneau, C.; Marois, L.; Dumesnil, J.G. Diastolic dysfunction in normotensive men with well-controlled type 2 diabetes: Importance of maneuvers in echocardiographic screening for preclinical diabetic cardiomyopathy. Diabetes Care 2001, 24, 5–10. [Google Scholar] [CrossRef]
- Di Bello, V.; Talarico, L.; Picano, E.; Di Muro, C.; Landini, L.; Paterni, M.; Matteucci, E.; Giusti, C.; Giampietro, O. Increased echodensity of myocardial wall in the diabetic heart: an ultrasound tissue characterization study. J. Am. Coll. Cardiol. 1995, 25, 1408–1415. [Google Scholar] [CrossRef]
- Regan, T.J.; Lyons, M.M.; Ahmed, S.S.; Levinson, G.E.; Oldewurtel, H.A.; Ahmad, M.R.; Haider, B. Evidence for cardiomyopathy in familial diabetes mellitus. J. Clin. Investig. 1977, 60, 884–899. [Google Scholar] [CrossRef]
- Jagasia, D.; Whiting, J.M.; Concato, J.; Pfau, S.; McNulty, P.H. Effect of non-insulin-dependent diabetes mellitus on myocardial insulin responsiveness in patients with ischemic heart disease. Circulation 2001, 103, 1734–1739. [Google Scholar] [CrossRef] [PubMed]
- Utriainen, T.; Takala, T.; Luotolahti, M.; Ronnemaa, T.; Laine, H.; Ruotsalainen, U.; Haaparanta, M.; Nuutila, P.; Yki-Jarvinen, H. Insulin resistance characterizes glucose uptake in skeletal muscle but not in the heart in NIDDM. Diabetologia 1998, 41, 555–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briat, A.; Slimani, L.; Perret, P.; Villemain, D.; Halimi, S.; Demongeot, J.; Fagret, D.; Ghezzi, C. In vivo assessment of cardiac insulin resistance by nuclear probes using an iodinated tracer of glucose transport. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1756–1764. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Abel, E.D. Rodent models of diabetic cardiomyopathy. Dis. Model. Mech. 2009, 2, 454–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, S.; Kim, J.K. New insights into insulin resistance in the diabetic heart. Trends Endocrinol. Metab. 2011, 22, 394–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodiga, V.L.; Eda, S.R.; Bodiga, S. Advanced glycation end products: Role in pathology of diabetic cardiomyopathy. Heart Fail. Rev. 2014, 19, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Vazquez, R.; Zabadi, S.; Watson, R.R.; Larson, D.F. T-lymphocytes mediate left ventricular fibrillar collagen cross-linking and diastolic dysfunction in mice. Matrix Biol. 2010, 29, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, J.M.; Kristiansen, S.B.; Norregaard, R.; Andersen, C.L.; Denner, L.; Nielsen, T.T.; Flyvbjerg, A.; Botker, H.E. Blockage of receptor for advanced glycation end products prevents development of cardiac dysfunction in db/db type 2 diabetic mice. Eur. J. Heart Fail. 2009, 11, 638–647. [Google Scholar] [CrossRef]
- Fricovsky, E.S.; Suarez, J.; Ihm, S.H.; Scott, B.T.; Suarez-Ramirez, J.A.; Banerjee, I.; Torres-Gonzalez, M.; Wang, H.; Ellrott, I.; Maya-Ramos, L.; et al. Excess protein O-GlcNAcylation and the progression of diabetic cardiomyopathy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R689–R699. [Google Scholar] [CrossRef]
- Hu, Y.; Belke, D.; Suarez, J.; Swanson, E.; Clark, R.; Hoshijima, M.; Dillmann, W.H. Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circ. Res. 2005, 96, 1006–1013. [Google Scholar] [CrossRef]
- Erickson, J.R.; Pereira, L.; Wang, L.; Han, G.; Ferguson, A.; Dao, K.; Copeland, R.J.; Despa, F.; Hart, G.W.; Ripplinger, C.M.; et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 2013, 502, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Yokoe, S.; Asahi, M.; Takeda, T.; Otsu, K.; Taniguchi, N.; Miyoshi, E.; Suzuki, K. Inhibition of phospholamban phosphorylation by O-GlcNAcylation: Implications for diabetic cardiomyopathy. Glycobiology 2010, 20, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Correa, G.A.; Ma, J.; Slawson, C.; Zeidan, Q.; Lugo-Fagundo, N.S.; Xu, M.; Shen, X.; Gao, W.D.; Caceres, V.; Chakir, K.; et al. Removal of Abnormal Myofilament O-GlcNAcylation Restores Ca2+ Sensitivity in Diabetic Cardiac Muscle. Diabetes 2015, 64, 3573–3587. [Google Scholar] [CrossRef] [PubMed]
- Ducheix, S.; Magre, J.; Cariou, B.; Prieur, X. Chronic O-GlcNAcylation and Diabetic Cardiomyopathy: The Bitterness of Glucose. Front. Endocrinol. 2018, 9, 642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, N.H.; Yu, S.; Tuinei, J.; Arai, K.; Hamai, H.; Homma, S.; Shulman, G.I.; Abel, E.D.; Goldberg, I.J. PPARγ-induced cardiolipotoxicity in mice is ameliorated by PPARα deficiency despite increases in fatty acid oxidation. J. Clin. Investig. 2010, 120, 3443–3454. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.T.; Grayburn, P.; Karim, A.; Shimabukuro, M.; Higa, M.; Baetens, D.; Orci, L.; Unger, R.H. Lipotoxic heart disease in obese rats: Implications for human obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 1784–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geraldes, P.; King, G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Belke, D.D.; Larsen, T.S.; Gibbs, E.M.; Severson, D.L. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E1104–E1113. [Google Scholar] [CrossRef]
- Boudina, S.; Sena, S.; O’Neill, B.T.; Tathireddy, P.; Young, M.E.; Abel, E.D. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 2005, 112, 2686–2695. [Google Scholar] [CrossRef]
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898. [Google Scholar] [CrossRef]
- Anderson, E.J.; Rodriguez, E.; Anderson, C.A.; Thayne, K.; Chitwood, W.R.; Kypson, A.P. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H118–H124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aragno, M.; Mastrocola, R.; Alloatti, G.; Vercellinatto, I.; Bardini, P.; Geuna, S.; Catalano, M.G.; Danni, O.; Boccuzzi, G. Oxidative stress triggers cardiac fibrosis in the heart of diabetic rats. Endocrinology 2008, 149, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Lashin, O.M.; Szweda, P.A.; Szweda, L.I.; Romani, A.M. Decreased complex II respiration and HNE-modified SDH subunit in diabetic heart. Free Radic. Biol. Med. 2006, 40, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zheng, S.; Thongboonkerd, V.; Xu, M.; Pierce, W.M., Jr.; Klein, J.B.; Epstein, P.N. Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E896–E905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turko, I.V.; Murad, F. Quantitative protein profiling in heart mitochondria from diabetic rats. J. Biol. Chem. 2003, 278, 35844–35849. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.; Metreveli, N.S.; Donthi, R.V.; Xia, S.; Xu, M.; Carlson, E.C.; Epstein, P.N. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes 2004, 53, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Zamora, M.; Villena, J.A. Targeting mitochondrial biogenesis to treat insulin resistance. Curr. Pharm. Des. 2014, 20, 5527–5557. [Google Scholar] [CrossRef] [PubMed]
- Metzler, B.; Schocke, M.F.; Steinboeck, P.; Wolf, C.; Judmaier, W.; Lechleitner, M.; Lukas, P.; Pachinger, O. Decreased high-energy phosphate ratios in the myocardium of men with diabetes mellitus type I. J. Cardiovasc. Magn. Reson. 2002, 4, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.H.; Moore, K.H.; Giacomelli, F.; Wiener, J. Defective oxidative metabolism of heart mitochondria from genetically diabetic mice. Diabetes 1983, 32, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Pierce, G.N.; Dhalla, N.S. Heart mitochondrial function in chronic experimental diabetes in rats. Can. J. Cardiol. 1985, 1, 48–54. [Google Scholar] [PubMed]
- Lasheras, J.; Vila, M.; Zamora, M.; Riu, E.; Pardo, R.; Poncelas, M.; Cases, I.; Ruiz-Meana, M.; Hernandez, C.; Feliu, J.E.; et al. Gene expression profiling in hearts of diabetic mice uncovers a potential role of estrogen-related receptor gamma in diabetic cardiomyopathy. Mol. Cell. Endocrinol. 2016, 430, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Sena, S.; Theobald, H.; Sheng, X.; Wright, J.J.; Hu, X.X.; Aziz, S.; Johnson, J.I.; Bugger, H.; Zaha, V.G.; et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 2007, 56, 2457–2466. [Google Scholar] [CrossRef] [PubMed]
- Giacchetti, G.; Sechi, L.A.; Rilli, S.; Carey, R.M. The renin-angiotensin-aldosterone system, glucose metabolism and diabetes. Trends Endocrinol. Metab. 2005, 16, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Le, B.; Khode, R.; Baker, K.M.; Kumar, R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes 2008, 57, 3297–3306. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Le, B.; Bhat, V.B.; Baker, K.M.; Kumar, R. High-glucose-induced regulation of intracellular ANG II synthesis and nuclear redistribution in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H939–H948. [Google Scholar] [CrossRef] [Green Version]
- Sechi, L.A.; Griffin, C.A.; Schambelan, M. The cardiac renin-angiotensin system in STZ-induced diabetes. Diabetes 1994, 43, 1180–1184. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, S.; Michelli, A.; Zuolo, G.; Candido, R.; Fabris, B. Update on RAAS Modulation for the Treatment of Diabetic Cardiovascular Disease. J. Diabetes Res. 2016, 2016, 8917578. [Google Scholar] [CrossRef]
- Shah, A.M.; Shin, S.H.; Takeuchi, M.; Skali, H.; Desai, A.S.; Kober, L.; Maggioni, A.P.; Rouleau, J.L.; Kelly, R.Y.; Hester, A.; et al. Left ventricular systolic and diastolic function, remodelling, and clinical outcomes among patients with diabetes following myocardial infarction and the influence of direct renin inhibition with aliskiren. Eur. J. Heart Fail. 2012, 14, 185–192. [Google Scholar] [CrossRef]
- Solomon, S.D.; Appelbaum, E.; Manning, W.J.; Verma, A.; Berglund, T.; Lukashevich, V.; Cherif Papst, C.; Smith, B.A.; Dahlof, B. Effect of the direct Renin inhibitor aliskiren, the Angiotensin receptor blocker losartan, or both on left ventricular mass in patients with hypertension and left ventricular hypertrophy. Circulation 2009, 119, 530–537. [Google Scholar] [CrossRef]
- Choi, K.M.; Zhong, Y.; Hoit, B.D.; Grupp, I.L.; Hahn, H.; Dilly, K.W.; Guatimosim, S.; Lederer, W.J.; Matlib, M.A. Defective intracellular Ca2+ signaling contributes to cardiomyopathy in Type 1 diabetic rats. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1398–H1408. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Tahiliani, A.G.; Vadlamudi, R.V.; Katz, S.; McNeill, J.H. Cardiac sarcoplasmic reticulum function in insulin- or carnitine-treated diabetic rats. Am. J. Physiol. 1983, 245, H969–H976. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Matthes, J.; Schuster, I.; Valdivia, H.H.; Herzig, S.; Richard, S.; Gomez, A.M. Mechanisms of [Ca2+]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes 2006, 55, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.Z.; Quamme, G.A.; McNeill, J.H. Altered [Ca2+]i mobilization in diabetic cardiomyocytes: responses to caffeine, KCl, ouabain, and ATP. Diabetes Res. Clin. Pract. 1995, 30, 9–20. [Google Scholar] [CrossRef]
- Belke, D.D.; Swanson, E.A.; Dillmann, W.H. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes 2004, 53, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Matsuda, N.; Kimura, J.; Ishitani, T.; Tamada, A.; Gando, S.; Kemmotsu, O.; Kanno, M. Diminished function and expression of the cardiac Na+-Ca2+ exchanger in diabetic rats: Implication in Ca2+ overload. J. Physiol. 2000, 527, 85–94. [Google Scholar] [CrossRef]
- Bertrand, L.; Horman, S.; Beauloye, C.; Vanoverschelde, J.L. Insulin signalling in the heart. Cardiovasc. Res. 2008, 79, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.K.; McGaffin, K.R.; Pastor-Soler, N.M.; Ahmad, F. SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc. Res. 2009, 84, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Wu, P.; Bowker-Kinley, M.M.; Harris, R.A. Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptor-alpha ligands, glucocorticoids, and insulin. Diabetes 2002, 51, 276–283. [Google Scholar] [CrossRef]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef]
- Lee, C.H.; Olson, P.; Evans, R.M. Minireview: Lipid metabolism, metabolic diseases, and peroxisome proliferator-activated receptors. Endocrinology 2003, 144, 2201–2207. [Google Scholar] [CrossRef]
- Villena, J.A.; Kralli, A. ERRα: A metabolic function for the oldest orphan. Trends Endocrinol. Metab. 2008, 19, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Burkart, E.M.; Sambandam, N.; Han, X.; Gross, R.W.; Courtois, M.; Gierasch, C.M.; Shoghi, K.; Welch, M.J.; Kelly, D.P. Nuclear receptors PPARβ/δ and PPARα direct distinct metabolic regulatory programs in the mouse heart. J. Clin. Investig. 2007, 117, 3930–3939. [Google Scholar] [CrossRef] [PubMed]
- Huss, J.M.; Torra, I.P.; Staels, B.; Giguere, V.; Kelly, D.P. Estrogen-related receptor α directs peroxisome proliferator-activated receptor α signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol. Cell. Biol. 2004, 24, 9079–9091. [Google Scholar] [CrossRef] [PubMed]
- Graneli, C.; Hicks, R.; Brolen, G.; Synnergren, J.; Sartipy, P. Diabetic cardiomyopathy modelling using induced pluripotent stem cell derived cardiomyocytes: Recent advances and emerging models. Stem Cell Rev. 2017, 15, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Pant, T.; Mishra, M.K.; Bai, X.; Ge, Z.D.; Bosnjak, Z.J.; Dhanasekaran, W.I. Microarray analysis of long non-coding RNA and mRNA expression profiles in diabetic cardiomyopathy using human induced pluripotent stem cell-derived cardiomyocytes. Diab. Vasc. Dis. Res. 2019, 16, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Geraets, I.M.E.; Chanda, D.; van Tienen, F.H.J.; van den Wijngaard, A.; Kamps, R.; Neumann, D.; Liu, Y.; Glatz, J.F.C.; Luiken, J.J.F.P.; Nabben, M. Human embryonic stem cell-derived cardiomyocytes as an in vitro model to study cardiac insulin resistance. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1984, 1960–1967. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Steinbusch, L.K.M.; Nabben, M.; Kapsokalyvas, D.; van Zandvoort, M.; Schonleitner, P.; Antoons, G.; Simons, P.J.; Coumans, W.A.; Geomini, A.; et al. Palmitate-induced vacuolar-type H+-ATPase inhibition feeds forward into insulin resistance and contractile dysfunction. Diabetes 2017, 66, 1521–1534. [Google Scholar] [CrossRef] [PubMed]
- Belke, D.D.; Betuing, S.; Tuttle, M.J.; Graveleau, C.; Young, M.E.; Pham, M.; Zhang, D.; Cooksey, R.C.; McClain, D.A.; Litwin, S.E.; et al. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J. Clin. Investig. 2002, 109, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Bugger, H.; Sena, S.; O’Neill, B.T.; Zaha, V.G.; Ilkun, O.; Wright, J.J.; Mazumder, P.K.; Palfreyman, E.; Tidwell, T.J.; et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation 2009, 119, 1272–1283. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Xu, Z.; Zhu, Q.; Thomas, C.; Kumar, R.; Feng, H.; Dostal, D.E.; White, M.F.; Baker, K.M.; Guo, S. Myocardial loss of IRS1 and IRS2 causes heart failure and is controlled by p38α MAPK during insulin resistance. Diabetes 2013, 62, 3887–3900. [Google Scholar] [CrossRef]
- Mora, A.; Davies, A.M.; Bertrand, L.; Sharif, I.; Budas, G.R.; Jovanovic, S.; Mouton, V.; Kahn, C.R.; Lucocq, J.M.; Gray, G.A.; et al. Deficiency of PDK1 in cardiac muscle results in heart failure and increased sensitivity to hypoxia. EMBO J. 2003, 22, 4666–4676. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Akazawa, H.; Tamagawa, M.; Furukawa, K.; Ogawa, W.; Yasuda, N.; Kudo, Y.; Liao, C.H.; Yamamoto, R.; Sato, T.; et al. PDK1 coordinates survival pathways and β-adrenergic response in the heart. Proc. Natl. Acad. Sci. USA 2009, 106, 8689–8694. [Google Scholar] [CrossRef] [PubMed]
- Crespo, M.J.; Zalacain, J.; Dunbar, D.C.; Cruz, N.; Arocho, L. Cardiac oxidative stress is elevated at the onset of dilated cardiomyopathy in streptozotocin-diabetic rats. J. Cardiovasc. Pharmacol. Ther. 2008, 13, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Flarsheim, C.E.; Grupp, I.L.; Matlib, M.A. Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. Am. J. Physiol. 1996, 271, H192–H202. [Google Scholar] [CrossRef] [PubMed]
- Glyn-Jones, S.; Song, S.; Black, M.A.; Phillips, A.R.; Choong, S.Y.; Cooper, G.J. Transcriptomic analysis of the cardiac left ventricle in a rodent model of diabetic cardiomyopathy: Molecular snapshot of a severe myocardial disease. Physiol. Genom. 2007, 28, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Han, X.; Courtois, M.; Aimond, F.; Nerbonne, J.M.; Kovacs, A.; Gross, R.W.; Kelly, D.P. A critical role for PPARα-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: Modulation by dietary fat content. Proc. Natl. Acad. Sci. USA 2003, 100, 1226–1231. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Hu, S.J.; Li, J.; Mou, Y.; Chen, B.P.; Xia, Q. Decreased cardiac sarcoplasmic reticulum Ca2+ -ATPase activity contributes to cardiac dysfunction in streptozotocin-induced diabetic rats. J. Physiol. Biochem. 2006, 62, 1–8. [Google Scholar] [CrossRef]
- Carley, A.N.; Semeniuk, L.M.; Shimoni, Y.; Aasum, E.; Larsen, T.S.; Berger, J.P.; Severson, D.L. Treatment of type 2 diabetic db/db mice with a novel PPARγ agonist improves cardiac metabolism but not contractile function. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E449–E455. [Google Scholar] [CrossRef]
- Barouch, L.A.; Berkowitz, D.E.; Harrison, R.W.; O’Donnell, C.P.; Hare, J.M. Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation 2003, 108, 754–759. [Google Scholar] [CrossRef]
- Daniels, A.; van Bilsen, M.; Janssen, B.J.; Brouns, A.E.; Cleutjens, J.P.; Roemen, T.H.; Schaart, G.; van der Velden, J.; van der Vusse, G.J.; van Nieuwenhoven, F.A. Impaired cardiac functional reserve in type 2 diabetic db/db mice is associated with metabolic, but not structural, remodelling. Acta Physiol. 2010, 200, 11–22. [Google Scholar] [CrossRef]
- Li, R.J.; Yang, J.; Yang, Y.; Ma, N.; Jiang, B.; Sun, Q.W.; Li, Y.J. Speckle tracking echocardiography in the diagnosis of early left ventricular systolic dysfunction in type II diabetic mice. BMC Cardiovasc. Disord. 2014, 14, 141. [Google Scholar] [CrossRef] [PubMed]
- Semeniuk, L.M.; Kryski, A.J.; Severson, D.L. Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db-hGLUT4 mice. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H976–H982. [Google Scholar] [CrossRef] [PubMed]
- Aasum, E.; Hafstad, A.D.; Severson, D.L.; Larsen, T.S. Age-dependent changes in metabolism, contractile function, and ischemic sensitivity in hearts from db/db mice. Diabetes 2003, 52, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, J.; Mazumder, P.K.; Hu, P.; Chakrabarti, G.; Roberts, M.W.; Yun, U.J.; Cooksey, R.C.; Litwin, S.E.; Abel, E.D. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 2005, 146, 5341–5349. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, P.K.; O’Neill, B.T.; Roberts, M.W.; Buchanan, J.; Yun, U.J.; Cooksey, R.C.; Boudina, S.; Abel, E.D. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes 2004, 53, 2366–2374. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zamora, M.; Villena, J.A. Contribution of Impaired Insulin Signaling to the Pathogenesis of Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2019, 20, 2833. https://doi.org/10.3390/ijms20112833
Zamora M, Villena JA. Contribution of Impaired Insulin Signaling to the Pathogenesis of Diabetic Cardiomyopathy. International Journal of Molecular Sciences. 2019; 20(11):2833. https://doi.org/10.3390/ijms20112833
Chicago/Turabian StyleZamora, Mònica, and Josep A. Villena. 2019. "Contribution of Impaired Insulin Signaling to the Pathogenesis of Diabetic Cardiomyopathy" International Journal of Molecular Sciences 20, no. 11: 2833. https://doi.org/10.3390/ijms20112833
APA StyleZamora, M., & Villena, J. A. (2019). Contribution of Impaired Insulin Signaling to the Pathogenesis of Diabetic Cardiomyopathy. International Journal of Molecular Sciences, 20(11), 2833. https://doi.org/10.3390/ijms20112833