Inhibition of Karyopherin-α2 Augments Radiation-Induced Cell Death by Perturbing BRCA1-Mediated DNA Repair

 , ,
, ,  and
and 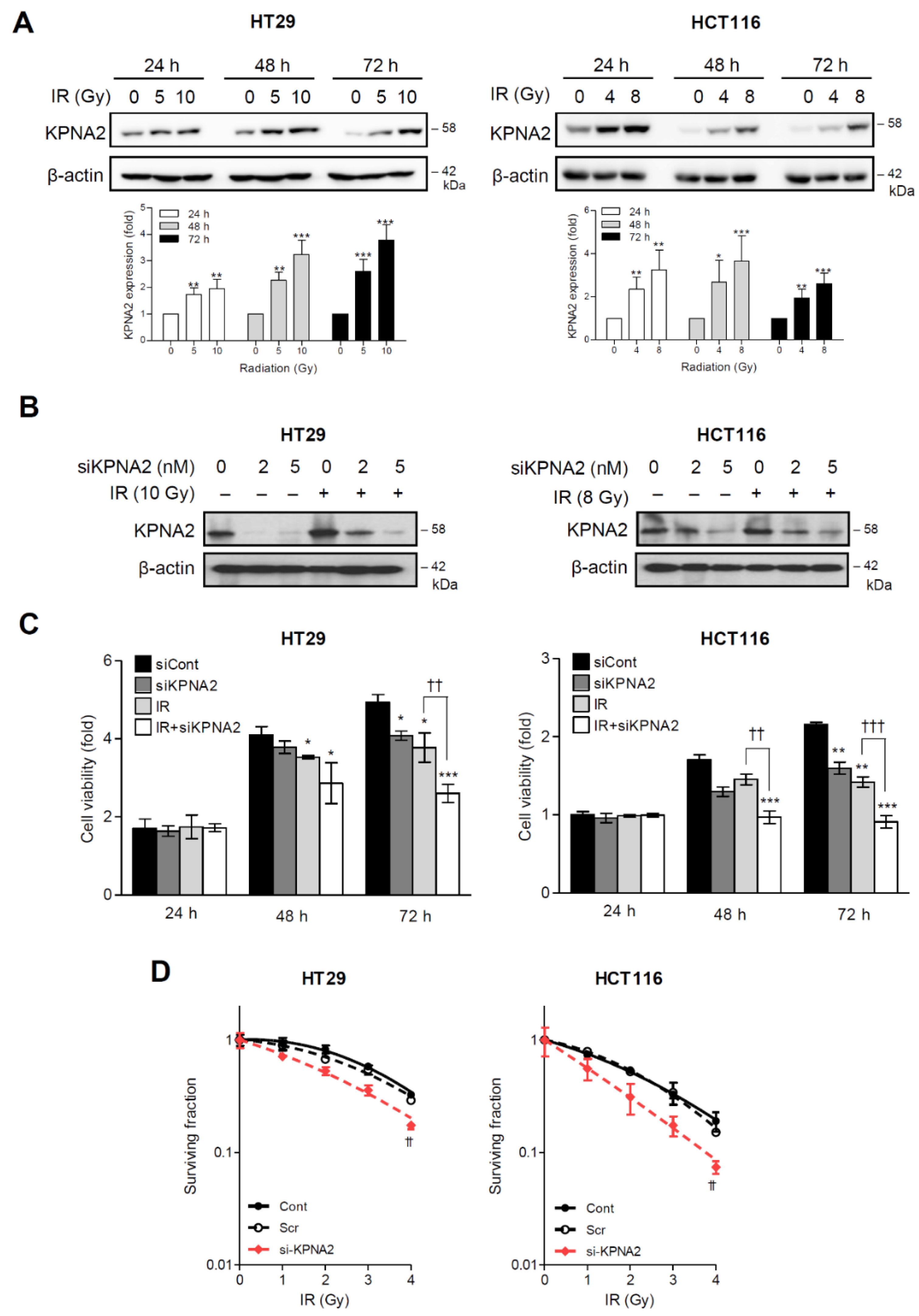{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
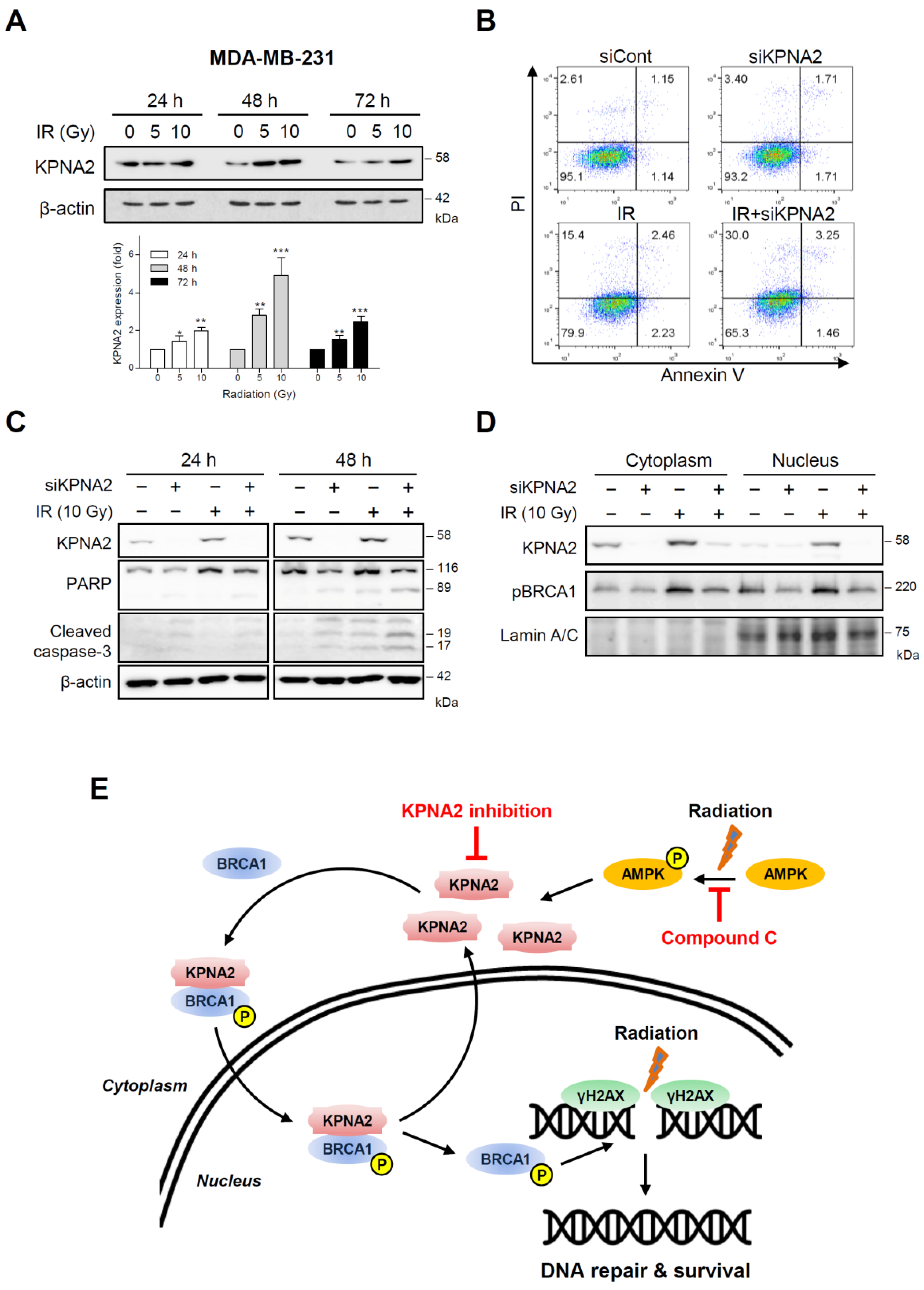
2.1. KPNA2 is Associated with Radioresistance in Human Colorectal Cancer Cells
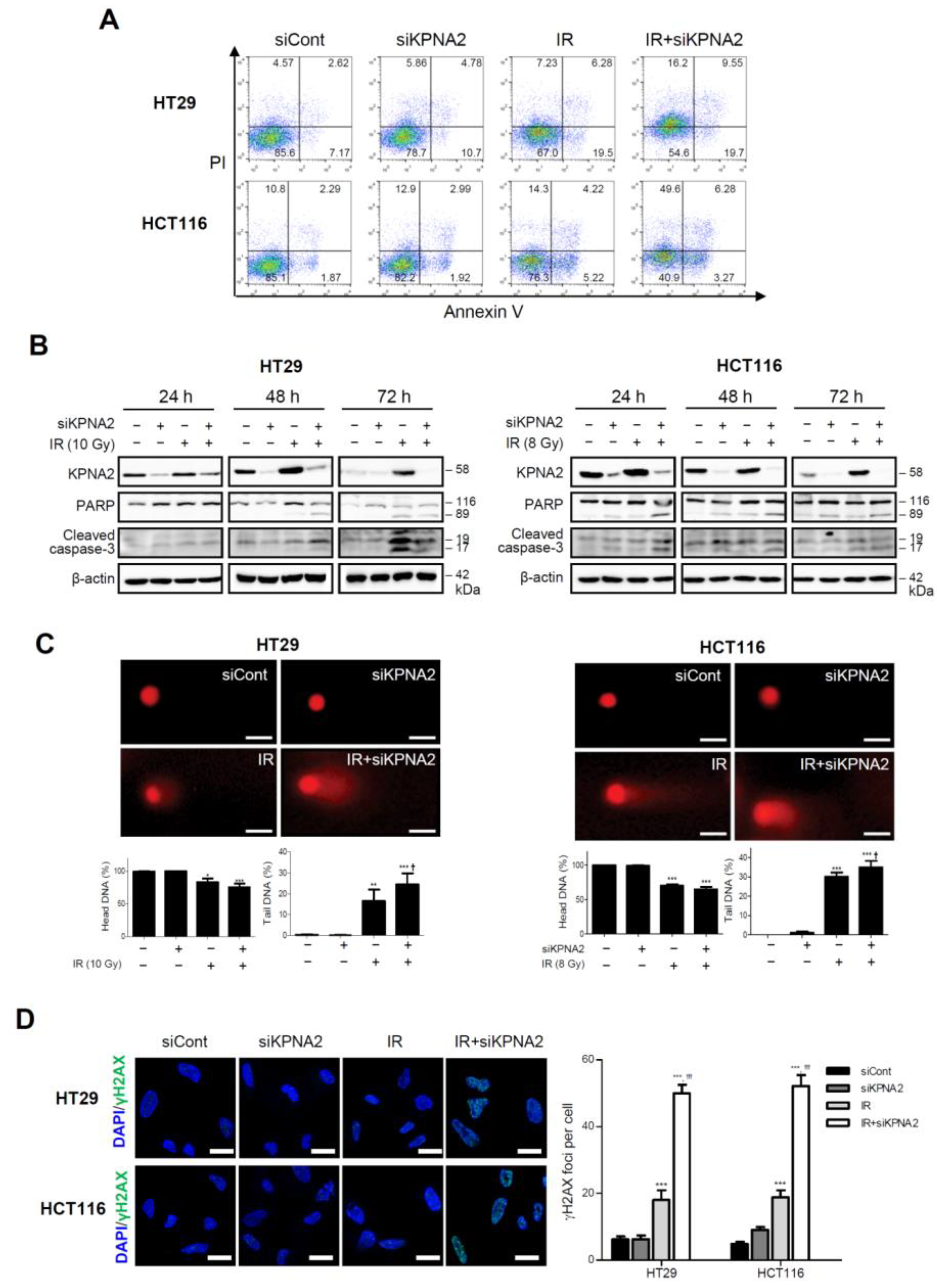
2.2. Knockdown of KPNA2 Increases Radiation-Induced Apoptosis
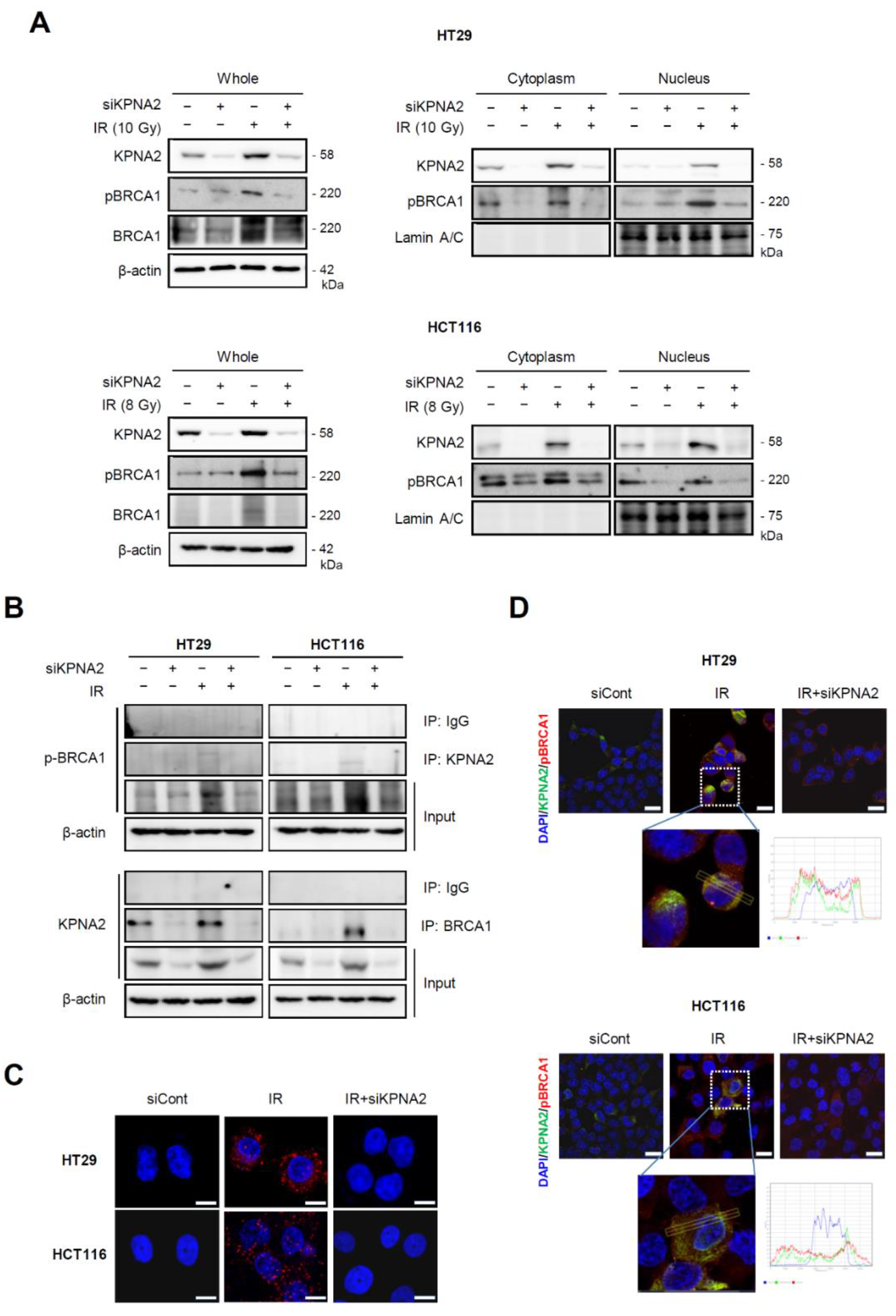
2.3. Knockdown of KPNA2 Prevents BRCA1 Activation
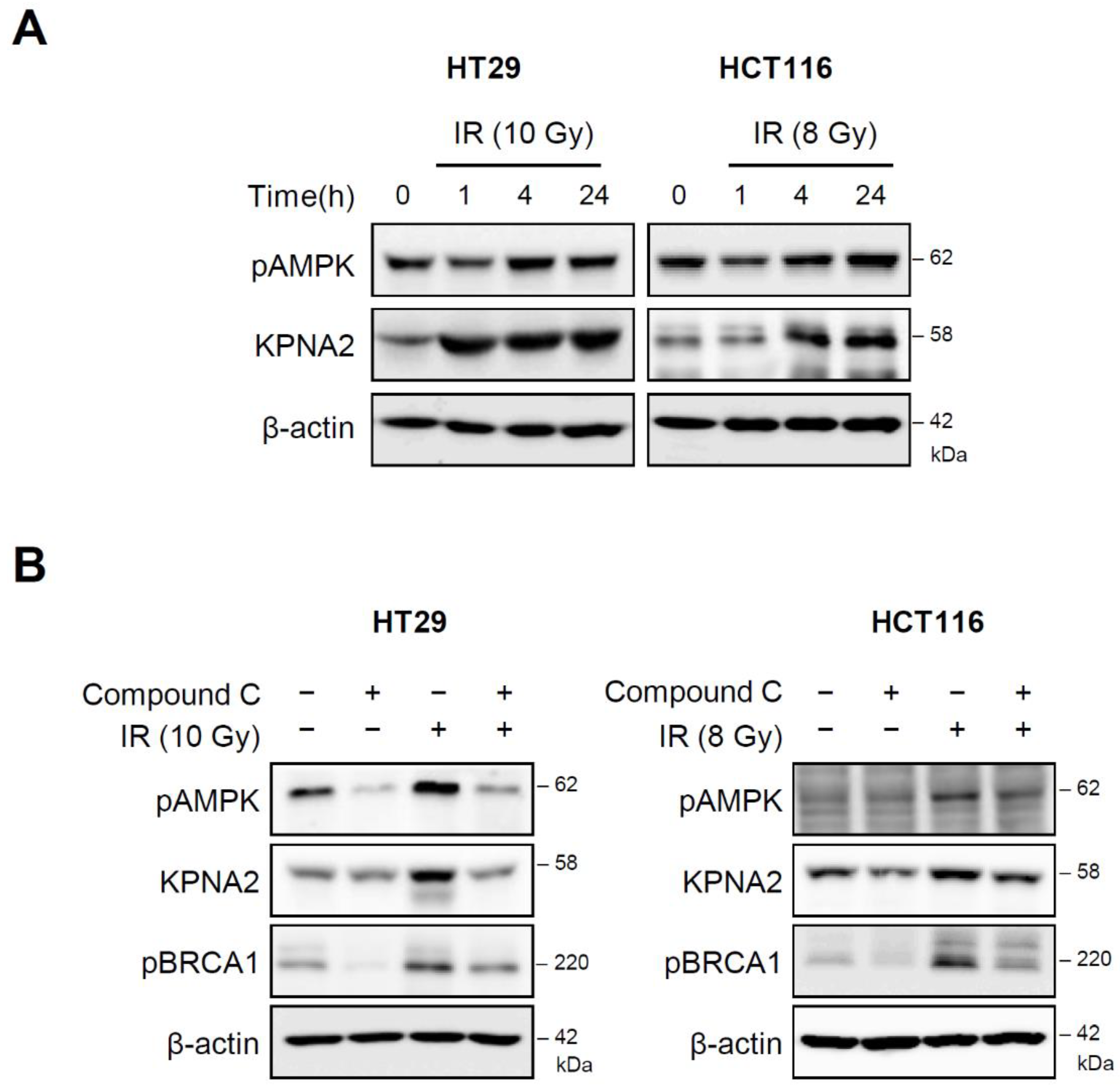
2.4. AMP-Activated Protein Kinase (AMPK) Regulates KPNA2 Expression
2.5. KPNA2-Depleted Breast Cancer Cells Exhibit Radiosensitivity
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. siRNA Transfection
4.3. Western Blot Analysis
4.4. Cell Viability Assay
4.5. Clonogenic Assay
4.6. Annexin V/Propidium Iodide Staining
4.7. Preparation of Cytoplasmic and Nuclear Fractions
4.8. Comet Assay
4.9. Immunoprecipitation Assay
4.10. In Situ Proximity Ligation Assay
4.11. Immunofluorescence Staining
4.12. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Harrington, K.J.; Billingham, L.J.; Brunner, T.B.; Burnet, N.G.; Chan, C.S.; Hoskin, P.; Mackay, R.I.; Maughan, T.S.; Macdougall, J.; McKenna, W.G.; et al. Guidelines for preclinical and early phase clinical assessment of novel radiosensitisers. Br. J. Cancer 2011, 105, 628–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, H.C.; Tsai, C.Y.; Tsai, M.M.; Lin, K.H. Impact of DNA and RNA Methylation on Radiobiology and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 555. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.M.; Hong, Y.; Lee, S.; Liu, P.; Lim, J.H.; Lee, Y.H.; Lee, T.H.; Chang, K.T.; Hong, Y. Therapeutic Implications for Overcoming Radiation Resistance in Cancer Therapy. Int. J. Mol. Sci. 2015, 16, 26880–26913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoya, N.; Miyagawa, K. Targeting DNA damage response in cancer therapy. Cancer Sci. 2014, 105, 370–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhang, L.; Liu, Y.; Sun, C.; Zhang, H.; Miao, G.; Di, C.X.; Zhou, X.; Zhou, R.; Wang, Z. DNA-PKcs deficiency inhibits glioblastoma cell-derived angiogenesis after ionizing radiation. J. Cell. Physiol. 2015, 230, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Lin, S.Y. Exploiting the homologous recombination DNA repair network for targeted cancer therapy. World J. Clin. Oncol. 2011, 2, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Song, K.-H.; Jung, S.-Y.; Kang, S.-M.; Kim, M.-H.; Ahn, J.; Hwang, S.-G.; Lee, J.-H.; Lim, D.-S.; Nam, S.Y.; Song, J.-Y. Induction of immunogenic cell death by radiation-upregulated karyopherin alpha 2 in vitro. Eur. J. Cell Biol. 2016, 95, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Kelley, J.B.; Talley, A.M.; Spencer, A.; Gioeli, D.; Paschal, B.M. Karyopherin alpha7 (KPNA7), a divergent member of the importin alpha family of nuclear import receptors. BMC Cell Biol. 2010, 11, 63. [Google Scholar] [CrossRef]
- Moroianu, J.; Hijikata, M.; Blobel, G.; Radu, A. Mammalian karyopherin alpha 1 beta and alpha 2 beta heterodimers: Alpha 1 or alpha 2 subunit binds nuclear localization signal and beta subunit interacts with peptide repeat-containing nucleoporins. Proc. Natl. Acad. Sci. USA 1995, 92, 6532–6536. [Google Scholar] [CrossRef]
- Christiansen, A.; Dyrskjot, L. The functional role of the novel biomarker karyopherin alpha 2 (KPNA2) in cancer. Cancer Lett. 2013, 331, 18–23. [Google Scholar] [CrossRef]
- Kosyna, F.K.; Depping, R. Controlling the Gatekeeper: Therapeutic Targeting of Nuclear Transport. Cells 2018, 7, 221. [Google Scholar] [CrossRef] [PubMed]
- Alshareeda, A.T.; Negm, O.H.; Green, A.R.; Nolan, C.C.; Tighe, P.; Albarakati, N.; Sultana, R.; Madhusudan, S.; Ellis, I.O.; Rakha, E.A. KPNA2 is a nuclear export protein that contributes to aberrant localisation of key proteins and poor prognosis of breast cancer. Br. J. Cancer 2015, 112, 1929–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachidi, S.M.; Qin, T.; Sun, S.; Zheng, W.J.; Li, Z. Molecular profiling of multiple human cancers defines an inflammatory cancer-associated molecular pattern and uncovers KPNA2 as a uniform poor prognostic cancer marker. PLoS ONE 2013, 8, e57911. [Google Scholar] [CrossRef] [PubMed]
- Altan, B.; Yokobori, T.; Mochiki, E.; Ohno, T.; Ogata, K.; Ogawa, A.; Yanai, M.; Kobayashi, T.; Luvsandagva, B.; Asao, T.; et al. Nuclear karyopherin-alpha2 expression in primary lesions and metastatic lymph nodes was associated with poor prognosis and progression in gastric cancer. Carcinogenesis 2013, 34, 2314–2321. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Huang, J.; Jiang, M.; Chen, Q.; Jiang, Z.; Feng, H. CAMK1 phosphoinositide signal-mediated protein sorting and transport network in human hepatocellular carcinoma (HCC) by biocomputation. Cell Biochem. Biophys. 2014, 70, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Jia, L.L.; Shi, M.M.; Li, X.; Li, Z.H.; Li, H.F.; Wang, E.H.; Jia, X.S. Downregulation of KPNA2 in non-small-cell lung cancer is associated with Oct4 expression. J. Transl. Med. 2013, 11, 232. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhou, Y.; Cao, X.P.; Lin, J.X.; Zhang, L.; Huang, S.T.; Zheng, M. KPNA2 promotes migration and invasion in epithelial ovarian cancer cells by inducing epithelial-mesenchymal transition via Akt/GSK-3beta/Snail activation. J. Cancer 2018, 9, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Winnepenninckx, V.; Lazar, V.; Michiels, S.; Dessen, P.; Stas, M.; Alonso, S.R.; Avril, M.F.; Ortiz Romero, P.L.; Robert, T.; Balacescu, O.; et al. Gene expression profiling of primary cutaneous melanoma and clinical outcome. J. Natl. Cancer Inst. 2006, 98, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.I.; Wang, C.L.; Wang, C.W.; Chen, C.D.; Wu, C.C.; Liang, Y.; Tsai, Y.H.; Chang, Y.S.; Yu, J.S.; Yu, C.J. Importin subunit alpha-2 is identified as a potential biomarker for non-small cell lung cancer by integration of the cancer cell secretome and tissue transcriptome. Int. J. Cancer 2011, 128, 2364–2372. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wang, H.Y.; Li, J.D.; Wang, J.H.; Zhou, Y.; Luo, R.Z.; Yun, J.P.; Zhang, Y.; Jia, W.H.; Zheng, M. KPNA2 promotes cell proliferation and tumorigenicity in epithelial ovarian carcinoma through upregulation of c-Myc and downregulation of FOXO3a. Cell Death Dis. 2013, 4, e745. [Google Scholar] [CrossRef]
- Takada, T.; Tsutsumi, S.; Takahashi, R.; Ohsone, K.; Tatsuki, H.; Suto, T.; Kato, T.; Fujii, T.; Yokobori, T.; Kuwano, H. KPNA2 over-expression is a potential marker of prognosis and therapeutic sensitivity in colorectal cancer patients. J. Surg. Oncol. 2016, 113, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, A.; Bajpayee, M.; Parmar, D. Comet assay: A reliable tool for the assessment of DNA damage in different models. Cell Biol. Toxicol. 2009, 25, 5–32. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Lim, D.S. The many substrates and functions of ATM. Nat. Rev. Mol. Cell Biol. 2000, 1, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Collins, K.M.; Brown, A.L.; Lee, C.H.; Chung, J.H. hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature 2000, 404, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Sanli, T.; Rashid, A.; Liu, C.; Harding, S.; Bristow, R.G.; Cutz, J.-C.; Singh, G.; Wright, J.; Tsakiridis, T. Ionizing radiation activates AMP-activated kinase (AMPK): A target for radiosensitization of human cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Hair, J.M.; Terzoudi, G.I.; Hatzi, V.I.; Lehockey, K.A.; Srivastava, D.; Wang, W.; Pantelias, G.E.; Georgakilas, A.G. BRCA1 role in the mitigation of radiotoxicity and chromosomal instability through repair of clustered DNA lesions. Chem. Biol. Interact. 2010, 188, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Singleton, B.K.; Griffin, C.S.; Thacker, J. Clustered DNA damage leads to complex genetic changes in irradiated human cells. Cancer Res. 2002, 62, 6263–6269. [Google Scholar]
- Burma, S.; Chen, B.P.; Chen, D.J. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair 2006, 5, 1042–1048. [Google Scholar] [CrossRef]
- Iliakis, G. The role of DNA double strand breaks in ionizing radiation-induced killing of eukaryotic cells. Bioessays 1991, 13, 641–648. [Google Scholar]
- Nowsheen, S.; Wukovich, R.L.; Aziz, K.; Kalogerinis, P.T.; Richardson, C.C.; Panayiotidis, M.I.; Bonner, W.M.; Sedelnikova, O.A.; Georgakilas, A.G. Accumulation of oxidatively induced clustered DNA lesions in human tumor tissues. Mutat. Res. 2009, 674, 131–136. [Google Scholar] [CrossRef]
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Fertil, B.; Malaise, E.P. Intrinsic radiosensitivity of human cell lines is correlated with radioresponsiveness of human tumors: Analysis of 101 published survival curves. Int. J. Radiat. Oncol. Biol. Phys. 1985, 11, 1699–1707. [Google Scholar] [CrossRef]
- Sharma, R.A.; Plummer, R.; Stock, J.K.; Greenhalgh, T.A.; Ataman, O.; Kelly, S.; Clay, R.; Adams, R.A.; Baird, R.D.; Billingham, L.; et al. Clinical development of new drug-radiotherapy combinations. Nat. Rev. Clin. Oncol. 2016, 13, 627–642. [Google Scholar] [CrossRef] [PubMed]
- Stochaj, U.; Rassadi, R.; Chiu, J. Stress-mediated inhibition of the classical nuclear protein import pathway and nuclear accumulation of the small GTPase Gsp1p. FASEB J. 2000, 14, 2130–2132. [Google Scholar] [CrossRef] [PubMed]
- Furuta, M.; Kose, S.; Koike, M.; Shimi, T.; Hiraoka, Y.; Yoneda, Y.; Haraguchi, T.; Imamoto, N. Heat-shock induced nuclear retention and recycling inhibition of importin alpha. Genes Cells 2004, 9, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, Y.; Miyamoto, Y.; Yamashiro, T.; Asally, M.; Masui, A.; Wong, C.; Loveland, K.L.; Yoneda, Y. Nuclear retention of importin alpha coordinates cell fate through changes in gene expression. EMBO J. 2012, 31, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Shen, Y.; Gao, L.; Chen, M.; Xiao, M.; Huang, Z.; Zhang, D. Karyopherin Alpha 2 Promotes the Inflammatory Response in Rat Pancreatic Acinar Cells Via Facilitating NF-κB Activation. Dig. Dis. Sci. 2016, 61, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xiao, L.; Liu, Y.; Wang, H.; Li, H.; Zhou, Q.; Pan, J.; Lei, B.; Huang, A.; Qi, S. MIR517C inhibits autophagy and the epithelial-to-mesenchymal (-like) transition phenotype in human glioblastoma through KPNA2-dependent disruption of TP53 nuclear translocation. Autophagy 2015, 11, 2213–2232. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Xu, X.; Sun, C.; Wang, Y.; Wang, S.; Liu, Z.; Zhai, L.; Cheng, H.; Xiao, M.; Zhang, D. KPNA2 interacts with P65 to modulate catabolic events in osteoarthritis. Exp. Mol. Pathol. 2015, 99, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Tseng, S.F.; Chang, C.Y.; Wu, K.J.; Teng, S.C. Importin KPNA2 is required for proper nuclear localization and multiple functions of NBS1. J. Biol. Chem. 2005, 280, 39594–39600. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-I.; Chien, K.-Y.; Wang, C.-L.; Liu, H.-P.; Cheng, C.-C.; Chang, Y.-S.; Yu, J.-S.; Yu, C.-J. Quantitative proteomics reveals regulation of karyopherin subunit alpha-2 (KPNA2) and its potential novel cargo proteins in nonsmall cell lung cancer. Mol. Cell. Proteom. MCP 2012, 11, 1105–1122. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.F.; Li, S.; Chen, Y.; Chen, P.L.; Sharp, Z.D.; Lee, W.H. The nuclear localization sequences of the BRCA1 protein interact with the importin-alpha subunit of the nuclear transport signal receptor. J. Biol. Chem. 1996, 271, 32863–32868. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Powell, S.N. The role of the BRCA1 tumor suppressor in DNA double-strand break repair. Mol. Cancer Res. 2005, 3, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.S.; Xia, F. BRCA1 16 years later: DNA damage-induced BRCA1 shuttling. FEBS J. 2010, 277, 3079–3085. [Google Scholar] [CrossRef]
- Feng, Z.; Kachnic, L.; Zhang, J.; Powell, S.N.; Xia, F. DNA damage induces p53-dependent BRCA1 nuclear export. J. Biol. Chem. 2004, 279, 28574–28584. [Google Scholar] [CrossRef]
- Li, M.; Yu, X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 2013, 23, 693–704. [Google Scholar] [CrossRef]
- Takaoka, M.; Miki, Y. BRCA1 gene: Function and deficiency. Int. J. Clin. Oncol. 2018, 23, 36–44. [Google Scholar] [CrossRef]
- Wang, B.; Matsuoka, S.; Ballif, B.A.; Zhang, D.; Smogorzewska, A.; Gygi, S.P.; Elledge, S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007, 316, 1194–1198. [Google Scholar] [CrossRef]
- Cantor, S.B.; Bell, D.W.; Ganesan, S.; Kass, E.M.; Drapkin, R.; Grossman, S.; Wahrer, D.C.; Sgroi, D.C.; Lane, W.S.; Haber, D.A.; et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 2001, 105, 149–160. [Google Scholar] [CrossRef]
- Zhong, Q.; Chen, C.F.; Li, S.; Chen, Y.; Wang, C.C.; Xiao, J.; Chen, P.L.; Sharp, Z.D.; Lee, W.H. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science 1999, 285, 747–750. [Google Scholar] [CrossRef]
- Zou, T.; Liu, L.; Rao, J.N.; Marasa, B.S.; Chen, J.; Xiao, L.; Zhou, H.; Gorospe, M.; Wang, J.Y. Polyamines modulate the subcellular localization of RNA-binding protein HuR through AMP-activated protein kinase-regulated phosphorylation and acetylation of importin alpha1. Biochem. J. 2008, 409, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Cook, P.R.; Brazell, I.A. Detection and repair of single-strand breaks in nuclear DNA. Nature 1976, 263, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Ostling, O.; Johanson, K.J. Microelectrophoretic study of radiation-induced DNA damages in individual mammalian cells. Biochem. Biophys. Res. Commun. 1984, 123, 291–298. [Google Scholar] [CrossRef]
- Koos, B.; Andersson, L.; Clausson, C.-M.; Grannas, K.; Klaesson, A.; Cane, G.; Söderberg, O. Analysis of protein interactions in situ by proximity ligation assays. Curr. Top. Microbiol. Immunol. 2014, 377, 111–126. [Google Scholar] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, K.-H.; Jung, S.-Y.; Park, J.-I.; Ahn, J.; Park, J.K.; Um, H.-D.; Park, I.-C.; Hwang, S.-G.; Ha, H.; Song, J.-Y. Inhibition of Karyopherin-α2 Augments Radiation-Induced Cell Death by Perturbing BRCA1-Mediated DNA Repair. Int. J. Mol. Sci. 2019, 20, 2843. https://doi.org/10.3390/ijms20112843
Song K-H, Jung S-Y, Park J-I, Ahn J, Park JK, Um H-D, Park I-C, Hwang S-G, Ha H, Song J-Y. Inhibition of Karyopherin-α2 Augments Radiation-Induced Cell Death by Perturbing BRCA1-Mediated DNA Repair. International Journal of Molecular Sciences. 2019; 20(11):2843. https://doi.org/10.3390/ijms20112843
Chicago/Turabian StyleSong, Kyung-Hee, Seung-Youn Jung, Jeong-In Park, Jiyeon Ahn, Jong Kuk Park, Hong-Duck Um, In-Chul Park, Sang-Gu Hwang, Hunjoo Ha, and Jie-Young Song. 2019. "Inhibition of Karyopherin-α2 Augments Radiation-Induced Cell Death by Perturbing BRCA1-Mediated DNA Repair" International Journal of Molecular Sciences 20, no. 11: 2843. https://doi.org/10.3390/ijms20112843
APA StyleSong, K.-H., Jung, S.-Y., Park, J.-I., Ahn, J., Park, J. K., Um, H.-D., Park, I.-C., Hwang, S.-G., Ha, H., & Song, J.-Y. (2019). Inhibition of Karyopherin-α2 Augments Radiation-Induced Cell Death by Perturbing BRCA1-Mediated DNA Repair. International Journal of Molecular Sciences, 20(11), 2843. https://doi.org/10.3390/ijms20112843