Dysregulated Transcriptional Control in Prostate Cancer



Abstract
:1. Introduction
2. Promoters, Enhancers and Super-Enhancers
2.1. General Aspects
2.2. Non-Coding Cancer Driver Mutations
3. Dysregulated Transcription Control in Prostate Cancer
3.1. Early Events
3.1.1. Regulatory SNPs
3.1.2. ERG Translocation
3.1.3. Epithelial Splicing Regulating Protein 1 (ESRP1) Gene Duplication
3.1.4. Phosphatase and Tensin Homolog (PTEN) Inactivation
3.2. Advanced Prostate Cancer
3.2.1. Binding of BRD4 and Interacting Proteins at Gene Regulatory Elements
3.2.2. Acquired AR Enhancer and Androgen-Dependent Neo-Enhancers
3.2.3. Nuclear Enriched Abundant Transcript 1 (NEAT1) and FOXA1 Promoter Mutations
3.2.4. Reprogramming to Neuroendocrine Phenotype
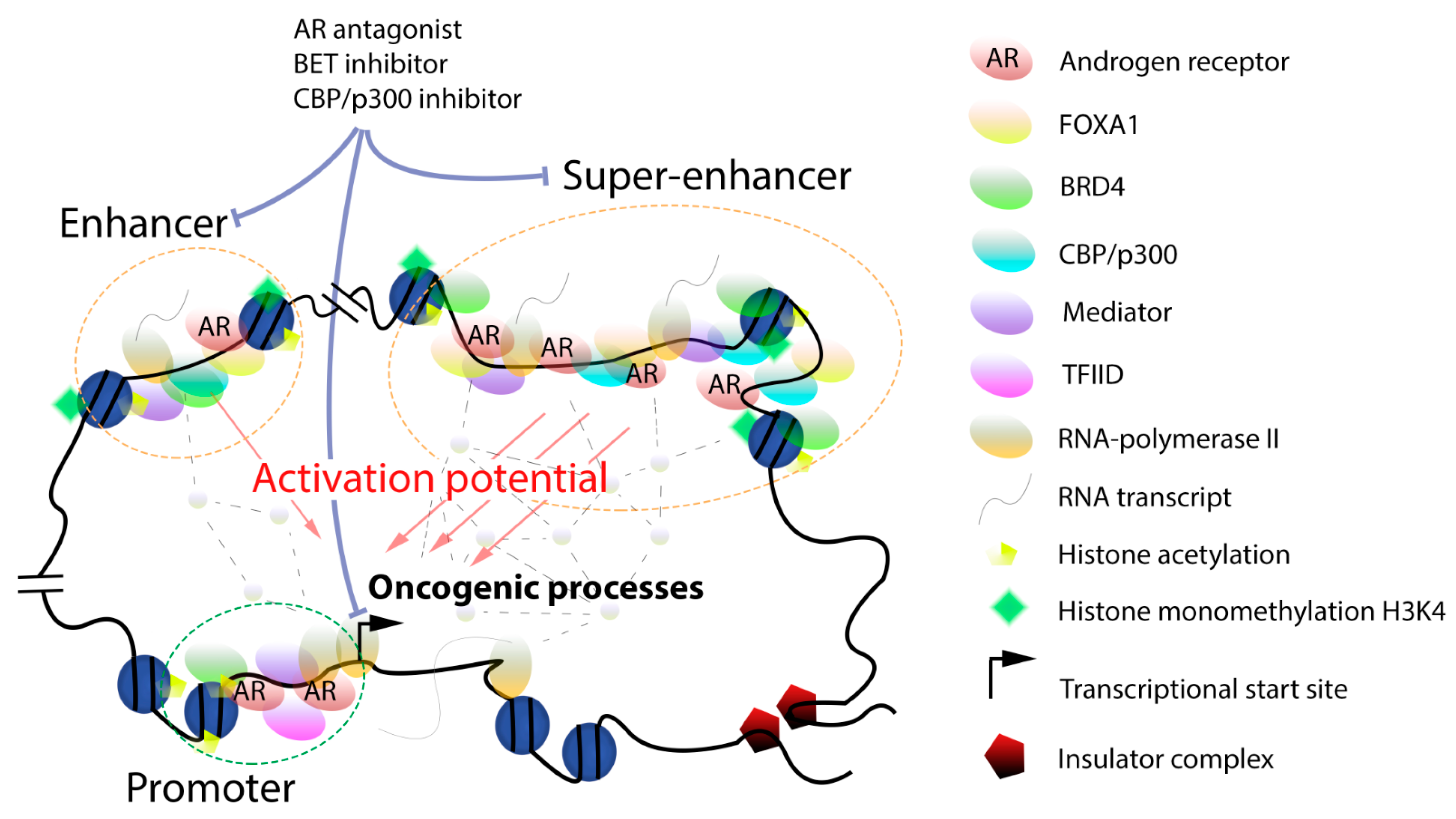
4. Targeting Dysregulated Gene Transcription
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angeles, A.K.; Bauer, S.; Ratz, L.; Klauck, S.M.; Sultmann, H. Genome-based classification and therapy of prostate cancer. Diagnostics 2018, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Wedge, D.C.; Gundem, G.; Mitchell, T.; Woodcock, D.J.; Martincorena, I.; Ghori, M.; Zamora, J.; Butler, A.; Whitaker, H.; Kote-Jarai, Z.; et al. Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat. Genet. 2018, 50, 682–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed]
- Spratt, D.E.; Zumsteg, Z.S.; Feng, F.Y.; Tomlins, S.A. Translational and clinical implications of the genetic landscape of prostate cancer. Nat. Rev. Clin. Oncol. 2016, 13, 597–610. [Google Scholar] [CrossRef]
- Linch, M.; Goh, G.; Hiley, C.; Shanmugabavan, Y.; McGranahan, N.; Rowan, A.; Wong, Y.N.S.; King, H.; Furness, A.; Freeman, A.; et al. Intratumoural evolutionary landscape of high-risk prostate cancer: The PROGENY study of genomic and immune parameters. Ann. Oncol. 2017, 28, 2472–2480. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 162, 1215–1228. [Google Scholar] [CrossRef]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: A multi-institutional prospective study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.H.; Beltran, H.; Zoubeidi, A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 2018, 15, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.H.; Petrovics, G.; Srivastava, S. Prostate cancer genomics: Recent advances and the prevailing underrepresentation from racial and ethnic minorities. Int. J. Mol. Sci. 2018, 19, 1255. [Google Scholar] [CrossRef] [PubMed]
- Bhagirath, D.; Yang, T.L.; Dahiya, R.; Saini, S. MicroRNAs as regulators of prostate cancer metastasis. Adv. Exp. Med. Biol. 2018, 1095, 83–100. [Google Scholar] [PubMed]
- Das, R.; Feng, F.Y.; Selth, L.A. Long non-coding RNAs in prostate cancer: Biological and clinical implications. Mol. Cell. Endocrinol. 2019, 480, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; Lajoie, B.R.; Jain, G.; Dekker, J. The long-range interaction landscape of gene promoters. Nature 2012, 489, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Harmston, N.; Lenhard, B. Chromatin and epigenetic features of long-range gene regulation. Nucleic Acids Res. 2013, 41, 7185–7199. [Google Scholar] [CrossRef]
- Matharu, N.; Ahituv, N. Minor loops in major folds: Enhancer-promoter looping, chromatin restructuring, and their association with transcriptional regulation and disease. PLoS Genet. 2015, 11, e1005640. [Google Scholar] [CrossRef]
- Stelloo, S.; Bergman, A.M.; Zwart, W. Androgen receptor enhancer usage and the chromatin regulatory landscape in human prostate cancers. Endocr.-Relat. Cancer 2019. [Google Scholar] [CrossRef]
- Hantsche, M.; Cramer, P. The structural basis of transcription: 10 years after the Nobel prize in chemistry. Angew. Chem. 2016, 55, 15972–15981. [Google Scholar] [CrossRef]
- Louder, R.K.; He, Y.; Lopez-Blanco, J.R.; Fang, J.; Chacon, P.; Nogales, E. Structure of promoter-bound TFIID and model of human pre-initiation complex assembly. Nature 2016, 531, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, M.H.; Kumarasiri, M.; Mekonnen, L.B.; Yu, M.; Diab, S.; Albrecht, H.; Milne, R.W.; Wang, S. Targeting CDK9: A promising therapeutic opportunity in prostate cancer. Endocr.-Relat. Cancer 2016, 23, T211–T226. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, A.S.; Li, C.H.; Zamudio, A.V.; Sigova, A.A.; Hannett, N.M.; Day, D.S.; Abraham, B.J.; Cohen, M.A.; Nabet, B.; Buckley, D.L.; et al. YY1 Is a structural regulator of enhancer-promoter loops. Cell 2017, 171, 1573–1588.e28. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.Y. The structural and functional roles of CTCF in the regulation of cell type-specific and human disease-associated super-enhancers. Genes Genom. 2019, 41, 257–265. [Google Scholar] [CrossRef]
- Meng, H.; Bartholomew, B. Emerging roles of transcriptional enhancers in chromatin looping and promoter-proximal pausing of RNA polymerase II. J. Biol. Chem. 2018, 293, 13786–13794. [Google Scholar] [CrossRef] [Green Version]
- Struhl, K. Transcriptional activation: Mediator can act after preinitiation complex formation. Mol. Cell 2005, 17, 752–754. [Google Scholar] [CrossRef]
- Catarino, R.R.; Stark, A. Assessing sufficiency and necessity of enhancer activities for gene expression and the mechanisms of transcription activation. Genes Dev. 2018, 32, 202–223. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Wang, Y.; Li, G. Dynamics of histone variant H3.3 and its coregulation with H2A.Z at enhancers and promoters. Nucleus 2014, 5, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Ramji, D.P.; Foka, P. CCAAT/enhancer-binding proteins: Structure, function and regulation. Biochem. J. 2002, 365, 561–575. [Google Scholar] [CrossRef]
- Kasper, L.H.; Qu, C.; Obenauer, J.C.; McGoldrick, D.J.; Brindle, P.K. Genome-wide and single-cell analyses reveal a context dependent relationship between CBP recruitment and gene expression. Nucleic Acids Res. 2014, 42, 11363–11382. [Google Scholar] [CrossRef]
- Wood, K.; Tellier, M.; Murphy, S. DOT1L and H3K79 methylation in transcription and genomic stability. Biomolecules 2018, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, S.; Hossan, T.; Alawi, M.; Najafova, Z.; Indenbirken, D.; Bedi, U.; Taipaleenmaki, H.; Ben-Batalla, I.; Scheller, M.; Loges, S.; et al. Bromodomain protein BRD4 is required for estrogen receptor-dependent enhancer activation and gene transcription. Cell Rep. 2014, 8, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Kannan, A.; Kern, M.; Moreno, M.A.; Vural, E.; Stack, B., Jr.; Suen, J.Y.; Tackett, A.J.; Gao, L. Disruption of BRD4 at H3K27Ac-enriched enhancer region correlates with decreased c-Myc expression in Merkel cell carcinoma. Epigenetics 2015, 10, 460–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tippens, N.D.; Vihervaara, A.; Lis, J.T. Enhancer transcription: What, where, when, and why? Genes Dev. 2018, 32, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Yoneda, M.; Higashi, M.; Ohkuma, Y.; Ito, T. Enhancer function regulated by combinations of transcription factors and cofactors. Genes Cells Devoted Mol. Cell. Mech. 2018, 23, 808–821. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, Y.I.; Pritchard, J.K. Trans Effects on Gene Expression Can Drive Omnigenic Inheritance. Cell 2019, 177, 1022.e6–1034.e6. [Google Scholar] [CrossRef]
- Lam, M.T.; Li, W.; Rosenfeld, M.G.; Glass, C.K. Enhancer RNAs and regulated transcriptional programs. Trends Biochem. Sci. 2014, 39, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina-Rivera, A.; Santiago-Algarra, D.; Puthier, D.; Spicuglia, S. Widespread enhancer activity from core promoters. Trends Biochem. Sci. 2018, 43, 452–468. [Google Scholar] [CrossRef] [PubMed]
- Nizovtseva, E.V.; Todolli, S.; Olson, W.K.; Studitsky, V.M. Towards quantitative analysis of gene regulation by enhancers. Epigenomics 2017, 9, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Taberlay, P.C.; Achinger-Kawecka, J.; Lun, A.T.; Buske, F.A.; Sabir, K.; Gould, C.M.; Zotenko, E.; Bert, S.A.; Giles, K.A.; Bauer, D.C.; et al. Three-dimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations. Genome Res. 2016, 26, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Jia, R.; Chai, P.; Zhang, H.; Fan, X. Novel insights into chromosomal conformations in cancer. Mol. Cancer 2017, 16, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krijger, P.H.; de Laat, W. Regulation of disease-associated gene expression in the 3D genome. Nat. Rev. Mol. Cell Biol. 2016, 17, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Schuijers, J.; Lin, C.Y.; Weintraub, A.S.; Abraham, B.J.; Lee, T.I.; Bradner, J.E.; Young, R.A. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell 2015, 58, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; George, R.E. Super-enhancer-driven transcriptional dependencies in cancer. Trends Cancer 2017, 3, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Choi, P.S.; Francis, J.M.; Imielinski, M.; Watanabe, H.; Cherniack, A.D.; Meyerson, M. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat. Genet. 2016, 48, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Hamdan, F.H.; Johnsen, S.A. Super enhancers—New analyses and perspectives on the low hanging fruit. Transcription 2018, 9, 123–130. [Google Scholar] [CrossRef]
- Gelato, K.A.; Schöckel, L.; Klingbeil, O.; Rückert, T.; Lesche, R.; Toedling, J.; Kalfon, E.; Héroult, M.; Lejeune, P.; Mönning, U.; et al. Super-enhancers define a proliferative PGC-1alpha-expressing melanoma subgroup sensitive to BET inhibition. Oncogene 2018, 37, 512–521. [Google Scholar] [CrossRef]
- Hnisz, D.; Shrinivas, K.; Young, R.A.; Chakraborty, A.K.; Sharp, P.A. A phase separation model for transcriptional control. Cell 2017, 169, 13–23. [Google Scholar] [CrossRef]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, W.K.; Spille, J.H.; Hecht, M.; Lee, C.; Li, C.; Grube, V.; Cisse, I.I. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 2018, 361, 412–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; Di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fontanals-Cirera, B.; Low, V.; Ntziachristos, P.; Yuen, S.K.; Lovell, C.D.; Dolgalev, I.; Yonekubo, Y.; Zhang, G.; Rusinova, E.; et al. Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell Rep. 2014, 9, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, M.; Chen, L.F.; Chen, R. P-TEFb: Finding its ways to release promoter-proximally paused RNA polymerase II. Transcription 2018, 9, 88–94. [Google Scholar] [CrossRef]
- Minzel, W.; Venkatachalam, A.; Fink, A.; Hung, E.; Brachya, G.; Burstain, I.; Shaham, M.; Rivlin, A.; Omer, I.; Zinger, A.; et al. Small molecules co-targeting CKIalpha and the transcriptional kinases CDK7/9 control AML in preclinical models. Cell 2018, 175, 171.e25–185.e25. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef] [PubMed]
- Christensen, C.L.; Kwiatkowski, N.; Abraham, B.J.; Carretero, J.; Al-Shahrour, F.; Zhang, T.; Chipumuro, E.; Herter-Sprie, G.S.; Akbay, E.A.; Altabef, A.; et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 2014, 26, 909–922. [Google Scholar] [CrossRef]
- Eliades, P.; Abraham, B.J.; Ji, Z.; Miller, D.M.; Christensen, C.L.; Kwiatkowski, N.; Kumar, R.; Njauw, C.N.; Taylor, M.; Miao, B.; et al. High MITF expression is associated with super-enhancers and suppressed by CDK7 inhibition in melanoma. J. Investig. Dermatol. 2018, 138, 1582–1590. [Google Scholar] [CrossRef]
- Sharifnia, T.; Wawer, M.J.; Chen, T.; Huang, Q.Y.; Weir, B.A.; Sizemore, A.; Lawlor, M.A.; Goodale, A.; Cowley, G.S.; Vazquez, F.; et al. Small-molecule targeting of brachyury transcription factor addiction in chordoma. Nat. Med. 2019, 25, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kwiatkowski, N.; Olson, C.M.; Dixon-Clarke, S.E.; Abraham, B.J.; Greifenberg, A.K.; Ficarro, S.B.; Elkins, J.M.; Liang, Y.; Hannett, N.M.; et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat. Chem. Biol. 2016, 12, 876–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, A.S.; Cattoglio, C.; Darzacq, X.; Tjian, R. Recent evidence that TADs and chromatin loops are dynamic structures. Nucleus 2018, 9, 20–32. [Google Scholar] [CrossRef]
- Oomen, M.E.; Hansen, A.S.; Liu, Y.; Darzacq, X.; Dekker, J. CTCF sites display cell cycle-dependent dynamics in factor binding and nucleosome positioning. Genome Res. 2019, 29, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Lazaris, C.; Sakellaropoulos, T.; Lozano, A.; Kambadur, P.; Ntziachristos, P.; Aifantis, I.; Tsirigos, A. Stratification of TAD boundaries reveals preferential insulation of super-enhancers by strong boundaries. Nat. Commun. 2018, 9, 542. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.Y.; Oh, S.; Yoo, K.H. Functional enhancers as master regulators of tissue-specific gene regulation and cancer development. Mol. Cells 2017, 40, 169–177. [Google Scholar] [PubMed]
- Kim, Y.J.; Xie, P.; Cao, L.; Zhang, M.Q.; Kim, T.H. Global transcriptional activity dynamics reveal functional enhancer RNAs. Genome Res. 2018, 28, 1799–1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Chen, Y.; Yang, M.; Guo, A.; Xu, Y.; Xu, L.; Koeffler, H.P. dbCoRC: A database of core transcriptional regulatory circuitries modeled by H3K27ac ChIP-seq signals. Nucleic Acids Res. 2018, 46, D71–D77. [Google Scholar] [CrossRef]
- Jiang, Y.; Qian, F.; Bai, X.; Liu, Y.; Wang, Q.; Ai, B.; Han, X.; Shi, S.; Zhang, J.; Li, X.; et al. SEdb: A comprehensive human super-enhancer database. Nucleic Acids Res. 2019, 47, D235–D243. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, K.; Cai, W.; Liu, X.; Zhang, Y.; Orkin, S.H.; Xu, J.; Yuan, G.C. Dissecting super-enhancer hierarchy based on chromatin interactions. Nat. Commun. 2018, 9, 943. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M.R.; Abraham, B.J.; Anders, L.; Berezovskaya, A.; Gutierrez, A.; Durbin, A.D.; Etchin, J.; Lawton, L.; Sallan, S.E.; Silverman, L.B.; et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014, 346, 1373–1377. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, B.; Kumar, R. Altered TERT promoter and other genomic regulatory elements: Occurrence and impact. Int. J. Cancer 2017, 141, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Griewank, K.G.; Murali, R.; Puig-Butille, J.A.; Schilling, B.; Livingstone, E.; Potrony, M.; Carrera, C.; Schimming, T.; Moller, I.; Schwamborn, M.; et al. TERT promoter mutation status as an independent prognostic factor in cutaneous melanoma. J. Natl. Cancer Inst. 2014, 106, dju246. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S.; Alsaadi, R.; Murugan, A.K.; Sadiq, B.B. TERT promoter mutations in thyroid cancer. Horm. Cancer 2016, 7, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.Y. Targeting super-enhancers for disease treatment and diagnosis. Mol. Cells 2018, 41, 506–514. [Google Scholar] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional addiction in cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Achinger-Kawecka, J.; Taberlay, P.C.; Clark, S.J. Alterations in three-dimensional organization of the cancer genome and epigenome. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 41–51. [Google Scholar] [CrossRef]
- Zhang, X.; Choi, P.S.; Francis, J.M.; Gao, G.F.; Campbell, J.D.; Ramachandran, A.; Mitsuishi, Y.; Ha, G.; Shih, J.; Vazquez, F.; et al. Somatic superenhancer duplications and hotspot mutations lead to oncogenic activation of the KLF5 transcription factor. Cancer Discov. 2018, 8, 108–125. [Google Scholar] [CrossRef]
- Mack, S.C.; Pajtler, K.W.; Chavez, L.; Okonechnikov, K.; Bertrand, K.C.; Wang, X.; Erkek, S.; Federation, A.; Song, A.; Lee, C.; et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 2018, 553, 101–105. [Google Scholar] [CrossRef]
- Glodzik, D.; Morganella, S.; Davies, H.; Simpson, P.T.; Li, Y.; Zou, X.; Diez-Perez, J.; Staaf, J.; Alexandrov, L.B.; Smid, M.; et al. A somatic-mutational process recurrently duplicates germline susceptibility loci and tissue-specific super-enhancers in breast cancers. Nat. Genet. 2017, 49, 341–348. [Google Scholar] [CrossRef]
- Viswanathan, S.R.; Ha, G.; Hoff, A.M.; Wala, J.A.; Carrot-Zhang, J.; Whelan, C.W.; Haradhvala, N.J.; Freeman, S.S.; Reed, S.C.; Rhoades, J.; et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell 2018, 174, 433.e19–447.e19. [Google Scholar] [CrossRef] [PubMed]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.D.; Bailey, A.M.; et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 2018, 175, 889. [Google Scholar] [CrossRef] [PubMed]
- Takeda, D.Y.; Spisak, S.; Seo, J.H.; Bell, C.; O’Connor, E.; Korthauer, K.; Ribli, D.; Csabai, I.; Solymosi, N.; Szallasi, Z.; et al. A somatically acquired enhancer of the androgen receptor is a noncoding driver in advanced prostate cancer. Cell 2018, 174, 422.e13–432.e13. [Google Scholar] [CrossRef] [PubMed]
- Ahmadiyeh, N.; Pomerantz, M.M.; Grisanzio, C.; Herman, P.; Jia, L.; Almendro, V.; He, H.H.; Brown, M.; Liu, X.S.; Davis, M.; et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc. Natl. Acad. Sci. USA 2010, 107, 9742–9746. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Tillmans, L.; Gao, J.; Gao, P.; Yuan, T.; Dittmar, R.L.; Song, W.; Yang, Y.; Sahr, N.; Wang, T.; et al. Chromatin interactions and candidate genes at ten prostate cancer risk loci. Sci. Rep. 2016, 6, 23202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, M.; Kim, S.; Wang, K.; Farnham, P.J.; Coetzee, G.A.; Lu, W. 4C-seq revealed long-range interactions of a functional enhancer at the 8q24 prostate cancer risk locus. Sci. Rep. 2016, 6, 22462. [Google Scholar] [CrossRef] [Green Version]
- Lynch, H.T.; Kosoko-Lasaki, O.; Leslie, S.W.; Rendell, M.; Shaw, T.; Snyder, C.; D’Amico, A.V.; Buxbaum, S.; Isaacs, W.B.; Loeb, S.; et al. Screening for familial and hereditary prostate cancer. Int. J. Cancer 2016, 138, 2579–2591. [Google Scholar] [CrossRef]
- Nowinski, S.; Santaolalla, A.; O’Leary, B.; Loda, M.; Mirchandani, A.; Emberton, M.; Van Hemelrijck, M.; Grigoriadis, A. Systematic identification of functionally relevant risk alleles to stratify aggressive versus indolent prostate cancer. Oncotarget 2018, 9, 12812–12824. [Google Scholar] [CrossRef] [Green Version]
- Benafif, S.; Eeles, R. Genetic predisposition to prostate cancer. Br. Med. Bull. 2016, 120, 75–89. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Li, F.; Takeda, D.Y.; Lenci, R.; Chonkar, A.; Chabot, M.; Cejas, P.; Vazquez, F.; Cook, J.; Shivdasani, R.A.; et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat. Genet. 2015, 47, 1346–1351. [Google Scholar] [CrossRef]
- Stelloo, S.; Nevedomskaya, E.; Kim, Y.; Schuurman, K.; Valle-Encinas, E.; Lobo, J.; Krijgsman, O.; Peeper, D.S.; Chang, S.L.; Feng, F.Y.; et al. Integrative epigenetic taxonomy of primary prostate cancer. Nat. Commun. 2018, 9, 4900. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, D.; Spring, D.J.; DePinho, R.A. Genetics and biology of prostate cancer. Genes Dev. 2018, 32, 1105–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazelett, D.J.; Rhie, S.K.; Gaddis, M.; Yan, C.; Lakeland, D.L.; Coetzee, S.G.; Ellipse, G.-O.N.C.; Practical, C.; Henderson, B.E.; Noushmehr, H.; et al. Comprehensive functional annotation of 77 prostate cancer risk loci. PLoS Genet. 2014, 10, e1004102. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.L.; Cramer, S.D.; Wiklund, F.; Isaacs, S.D.; Stevens, V.L.; Sun, J.; Smith, S.; Pruett, K.; Romero, L.M.; Wiley, K.E.; et al. Fine mapping association study and functional analysis implicate a SNP in MSMB at 10q11 as a causal variant for prostate cancer risk. Hum. Mol. Genet. 2009, 18, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Yeager, M.; Li, H.; Bosquet, J.G.; Hayes, R.B.; Orr, N.; Yu, K.; Hutchinson, A.; Jacobs, K.B.; Kraft, P.; et al. Fine mapping and functional analysis of a common variant in MSMB on chromosome 10q11.2 associated with prostate cancer susceptibility. Proc. Natl. Acad. Sci. USA 2009, 106, 7933–7938. [Google Scholar] [CrossRef] [PubMed]
- Krätzschmar, J.; Haendler, B.; Eberspaecher, U.; Roosterman, D.; Donner, P.; Schleuning, W.D. The human cysteine-rich secretory protein (CRISP) family. Primary structure and tissue distribution of CRISP-1, CRISP-2 and CRISP-3. Eur. J. Biochem. 1996, 236, 827–836. [Google Scholar]
- Anklesaria, J.H.; Mhatre, D.R.; Mahale, S.D. Structural and molecular biology of PSP94: Its significance in prostate pathophysiology. Front. Biosci. 2018, 23, 535–562. [Google Scholar]
- Zhang, X.; Cowper-Sal lari, R.; Bailey, S.D.; Moore, J.H.; Lupien, M. Integrative functional genomics identifies an enhancer looping to the SOX9 gene disrupted by the 17q24.3 prostate cancer risk locus. Genome Res. 2012, 22, 1437–1446. [Google Scholar] [CrossRef]
- Jin, H.J.; Jung, S.; DebRoy, A.R.; Davuluri, R.V. Identification and validation of regulatory SNPs that modulate transcription factor chromatin binding and gene expression in prostate cancer. Oncotarget 2016, 7, 54616–54626. [Google Scholar] [CrossRef] [Green Version]
- Romanel, A.; Garritano, S.; Stringa, B.; Blattner, M.; Dalfovo, D.; Chakravarty, D.; Soong, D.; Cotter, K.A.; Petris, G.; Dhingra, P.; et al. Inherited determinants of early recurrent somatic mutations in prostate cancer. Nat. Commun. 2017, 8, 48. [Google Scholar] [CrossRef]
- Rebbeck, T.R. Prostate cancer disparities by race and ethnicity: From nucleotide to neighborhood. Cold Spring Harb. Perspect. Med. 2018, 8, a030387. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Rhie, S.K.; Lay, F.D.; Farnham, P.J. A prostate cancer risk element functions as a repressive loop that regulates HOXA13. Cell Rep. 2017, 21, 1411–1417. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Whitington, T.; Gao, P.; Lindberg, J.F.; Yang, Y.; Sun, J.; Vaisanen, M.R.; Szulkin, R.; Annala, M.; Yan, J.; et al. A prostate cancer susceptibility allele at 6q22 increases RFX6 expression by modulating HOXB13 chromatin binding. Nat. Genet. 2014, 46, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Brechka, H.; Bhanvadia, R.R.; VanOpstall, C.; Vander Griend, D.J. HOXB13 mutations and binding partners in prostate development and cancer: Function, clinical significance, and future directions. Genes Dis. 2017, 4, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wu, D.; Thomas-Ahner, J.M.; Lu, C.; Zhao, P.; Zhang, Q.; Geraghty, C.; Yan, P.S.; Hankey, W.; Sunkel, B.; et al. Diverse AR-V7 cistromes in castration-resistant prostate cancer are governed by HoxB13. Proc. Natl. Acad. Sci. USA 2018, 115, 6810–6815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Ahmed, M.; Zhang, F.; Yao, C.Q.; Li, S.; Liang, Y.; Hua, J.; Soares, F.; Sun, Y.; Langstein, J.; et al. Modulation of long noncoding RNAs by risk SNPs underlying genetic predispositions to prostate cancer. Nat. Genet. 2016, 48, 1142–1150. [Google Scholar] [CrossRef]
- Rotinen, M.; You, S.; Yang, J.; Coetzee, S.G.; Reis-Sobreiro, M.; Huang, W.C.; Huang, F.; Pan, X.; Yanez, A.; Hazelett, D.J.; et al. ONECUT2 is a targetable master regulator of lethal prostate cancer that suppresses the androgen axis. Nat. Med. 2018, 24, 1887–1898. [Google Scholar] [CrossRef]
- Gao, P.; Xia, J.H.; Sipeky, C.; Dong, X.M.; Zhang, Q.; Yang, Y.; Zhang, P.; Cruz, S.P.; Zhang, K.; Zhu, J.; et al. Biology and clinical implications of the 19q13 aggressive prostate cancer susceptibility locus. Cell 2018, 174, 576.e18–589.e18. [Google Scholar] [CrossRef]
- Hua, J.T.; Ahmed, M.; Guo, H.; Zhang, Y.; Chen, S.; Soares, F.; Lu, J.; Zhou, S.; Wang, M.; Li, H.; et al. Risk SNP-mediated promoter-enhancer switching drives prostate cancer through lncRNA PCAT19. Cell 2018, 174, 564.e18–575.e18. [Google Scholar] [CrossRef]
- Guo, Y.; Perez, A.A.; Hazelett, D.J.; Coetzee, G.A.; Rhie, S.K.; Farnham, P.J. CRISPR-mediated deletion of prostate cancer risk-associated CTCF loop anchors identifies repressive chromatin loops. Genome Biol. 2018, 19, 160. [Google Scholar] [CrossRef]
- Smith, A.J.P.; Deloukas, P.; Munroe, P.B. Emerging applications of genome-editing technology to examine functionality of GWAS-associated variants for complex traits. Physiol. Genom. 2018, 50, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Kron, K.J.; Murison, A.; Zhou, S.; Huang, V.; Yamaguchi, T.N.; Shiah, Y.J.; Fraser, M.; van der Kwast, T.; Boutros, P.C.; Bristow, R.G.; et al. TMPRSS2-ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer. Nat. Genet. 2017, 49, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Rickman, D.S.; Soong, T.D.; Moss, B.; Mosquera, J.M.; Dlabal, J.; Terry, S.; MacDonald, T.Y.; Tripodi, J.; Bunting, K.; Najfeld, V.; et al. Oncogene-mediated alterations in chromatin conformation. Proc. Natl. Acad. Sci. USA 2012, 109, 9083–9088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toropainen, S.; Niskanen, E.A.; Malinen, M.; Sutinen, P.; Kaikkonen, M.U.; Palvimo, J.J. Global analysis of transcription in castration-resistant prostate cancer cells uncovers active enhancers and direct androgen receptor targets. Sci. Rep. 2016, 6, 33510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chng, K.R.; Lingadahalli, S.; Chen, Z.; Liu, M.H.; Do, H.H.; Cai, S.; Rinaldi, N.; Poh, H.M.; Li, G.; et al. An AR-ERG transcriptional signature defined by long-range chromatin interactomes in prostate cancer cells. Genome Res. 2019, 29, 223–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blee, A.M.; Liu, S.; Wang, L.; Huang, H. BET bromodomain-mediated interaction between ERG and BRD4 promotes prostate cancer cell invasion. Oncotarget 2016, 7, 38319–38332. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.H.; Lee, G.Y.; Lee, M.; Kang, J.; Shin, H.W.; Chun, Y.S.; Park, J.W. Aberrant expression of CITED2 promotes prostate cancer metastasis by activating the nucleolin-AKT pathway. Nat. Commun. 2018, 9, 4113. [Google Scholar] [CrossRef] [PubMed]
- Gerhauser, C.; Favero, F.; Risch, T.; Simon, R.; Feuerbach, L.; Assenov, Y.; Heckmann, D.; Sidiropoulos, N.; Waszak, S.M.; Hubschmann, D.; et al. Molecular evolution of early-onset prostate cancer identifies molecular risk markers and clinical trajectories. Cancer Cell 2018, 34, 996–1011.e8. [Google Scholar] [CrossRef]
- Rajan, P.; Elliott, D.J.; Robson, C.N.; Leung, H.Y. Alternative splicing and biological heterogeneity in prostate cancer. Nat. Rev. Urol. 2009, 6, 454–460. [Google Scholar] [CrossRef]
- Whang, Y.E.; Wu, X.; Suzuki, H.; Reiter, R.E.; Tran, C.; Vessella, R.L.; Said, J.W.; Isaacs, W.B.; Sawyers, C.L. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc. Natl. Acad. Sci. USA 1998, 95, 5246–5250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Poluri, R.T.K.; Audet-Walsh, E. Genomic deletion at 10q23 in prostate cancer: More than PTEN loss? Front. Oncol. 2018, 8, 246. [Google Scholar] [CrossRef]
- Park, S.; Kim, Y.S.; Kim, D.Y.; So, I.; Jeon, J.H. PI3K pathway in prostate cancer: All resistant roads lead to PI3K. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 198–206. [Google Scholar] [CrossRef]
- Crumbaker, M.; Khoja, L.; Joshua, A.M. AR signaling and the PI3K pathway in prostate cancer. Cancers 2017, 9, 34. [Google Scholar] [CrossRef]
- Valdes-Mora, F.; Gould, C.M.; Colino-Sanguino, Y.; Qu, W.; Song, J.Z.; Taylor, K.M.; Buske, F.A.; Statham, A.L.; Nair, S.S.; Armstrong, N.J.; et al. Acetylated histone variant H2A.Z is involved in the activation of neo-enhancers in prostate cancer. Nat. Commun. 2017, 8, 1346. [Google Scholar] [CrossRef] [PubMed]
- Gallenkamp, D.; Gelato, K.A.; Haendler, B.; Weinmann, H. Bromodomains and their pharmacological inhibitors. ChemMedChem 2014, 9, 438–464. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Zuber, V.; Bettella, F.; Witoelar, A.; Consortium, P.; Cruk, G.; Consortium, B.; Consortium, T.; Andreassen, O.A.; Mills, I.G.; Urbanucci, A. Bromodomain protein 4 discriminates tissue-specific super-enhancers containing disease-specific susceptibility loci in prostate and breast cancer. BMC Genom. 2017, 18, 270. [Google Scholar] [CrossRef]
- Urbanucci, A.; Barfeld, S.J.; Kytola, V.; Itkonen, H.M.; Coleman, I.M.; Vodak, D.; Sjoblom, L.; Sheng, X.; Tolonen, T.; Minner, S.; et al. Androgen receptor deregulation drives bromodomain-mediated chromatin alterations in prostate cancer. Cell Rep. 2017, 19, 2045–2059. [Google Scholar] [CrossRef]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.X.; Smith, E.R.; Shilatifard, A. Born to run: Control of transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2018, 19, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, L.; Ren, S.; Wang, L.; Blackburn, P.R.; McNulty, M.S.; Gao, X.; Qiao, M.; Vessella, R.L.; Kohli, M.; et al. Activation of P-TEFb by androgen receptor-regulated enhancer RNAs in castration-resistant prostate cancer. Cell Rep. 2016, 15, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Kuser-Abali, G.; Alptekin, A.; Lewis, M.; Garraway, I.P.; Cinar, B. YAP1 and AR interactions contribute to the switch from androgen-dependent to castration-resistant growth in prostate cancer. Nat. Commun. 2015, 6, 8126. [Google Scholar] [CrossRef] [PubMed]
- Seo, W.I.; Park, S.; Gwak, J.; Ju, B.G.; Chung, J.I.; Kang, P.M.; Oh, S. Wnt signaling promotes androgen-independent prostate cancer cell proliferation through up-regulation of the hippo pathway effector YAP. Biochem. Biophys. Res. Commun. 2017, 486, 1034–1039. [Google Scholar] [CrossRef]
- Bai, S.; Cao, S.; Jin, L.; Kobelski, M.; Schouest, B.; Wang, X.; Ungerleider, N.; Baddoo, M.; Zhang, W.; Corey, E.; et al. A positive role of c-Myc in regulating androgen receptor and its splice variants in prostate cancer. Oncogene 2019. [Google Scholar] [CrossRef]
- Gao, L.; Schwartzman, J.; Gibbs, A.; Lisac, R.; Kleinschmidt, R.; Wilmot, B.; Bottomly, D.; Coleman, I.; Nelson, P.; McWeeney, S.; et al. Androgen receptor promotes ligand-independent prostate cancer progression through c-Myc upregulation. PLoS ONE 2013, 8, e63563. [Google Scholar] [CrossRef]
- Shah, N.; Wang, P.; Wongvipat, J.; Karthaus, W.R.; Abida, W.; Armenia, J.; Rockowitz, S.; Drier, Y.; Bernstein, B.E.; Long, H.W.; et al. Regulation of the glucocorticoid receptor via a BET-dependent enhancer drives antiandrogen resistance in prostate cancer. eLife 2017, 6, e27861. [Google Scholar] [CrossRef]
- Akamatsu, S.; Inoue, T.; Ogawa, O.; Gleave, M.E. Clinical and molecular features of treatment-related neuroendocrine prostate cancer. Int. J. Urol. 2018, 25, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Bakht, M.K.; Derecichei, I.; Li, Y.; Ferraiuolo, R.M.; Dunning, M.; Oh, S.W.; Hussein, A.; Youn, H.; Stringer, K.F.; Jeong, C.W.; et al. Neuroendocrine differentiation of prostate cancer leads to PSMA suppression. Endocr.-Relat. Cancer 2018, 26, 131–146. [Google Scholar] [CrossRef]
- Eapen, R.S.; Nzenza, T.C.; Murphy, D.G.; Hofman, M.S.; Cooperberg, M.; Lawrentschuk, N. PSMA PET applications in the prostate cancer journey: From diagnosis to theranostics. World J. Urol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Lee, J.K.; Witte, O.N.; Huang, J. FOXA2 is a sensitive and specific marker for small cell neuroendocrine carcinoma of the prostate. Mod. Pathol. 2017, 30, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Zeng, T.; Wen, Y.C.; Yeh, H.L.; Jiang, K.C.; Chen, W.H.; Zhang, Q.; Huang, J.; Liu, Y.N. Androgen deprivation-induced ZBTB46-PTGS1 signaling promotes neuroendocrine differentiation of prostate cancer. Cancer Lett. 2019, 440–441, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Venalainen, E.; Ci, X.; Cheng, H.; Pikor, L.; Parolia, A.; Xue, H.; Nur Saidy, N.R.; Lin, D.; Lam, W.; et al. The role of epigenetics and long noncoding RNA MIAT in neuroendocrine prostate cancer. Epigenomics 2016, 8, 721–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clermont, P.L.; Lin, D.; Crea, F.; Wu, R.; Xue, H.; Wang, Y.; Thu, K.L.; Lam, W.L.; Collins, C.C.; Wang, Y.; et al. Polycomb-mediated silencing in neuroendocrine prostate cancer. Clin. Epigenet. 2015, 7, 40. [Google Scholar] [CrossRef]
- Yang, Y.A.; Yu, J. EZH2, an epigenetic driver of prostate cancer. Protein Cell 2013, 4, 331–341. [Google Scholar] [CrossRef]
- Donaldson-Collier, M.C.; Sungalee, S.; Zufferey, M.; Tavernari, D.; Katanayeva, N.; Battistello, E.; Mina, M.; Douglass, K.M.; Rey, T.; Raynaud, F.; et al. EZH2 oncogenic mutations drive epigenetic, transcriptional, and structural changes within chromatin domains. Nat. Genet. 2019, 51, 517–528. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, D.; Zhou, T.; Song, H.; Hulsurkar, M.; Su, N.; Liu, Y.; Wang, Z.; Shao, L.; Ittmann, M.; et al. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat. Commun. 2018, 9, 4080. [Google Scholar] [CrossRef]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef]
- Chang, Y.T.; Lin, T.P.; Campbell, M.; Pan, C.C.; Lee, S.H.; Lee, H.C.; Yang, M.H.; Kung, H.J.; Chang, P.C. REST is a crucial regulator for acquiring EMT-like and stemness phenotypes in hormone-refractory prostate cancer. Sci. Rep. 2017, 7, 42795. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.T.; Lin, T.P.; Tang, J.T.; Campbell, M.; Luo, Y.L.; Lu, S.Y.; Yang, C.P.; Cheng, T.Y.; Chang, C.H.; Liu, T.T.; et al. HOTAIR is a REST-regulated lncRNA that promotes neuroendocrine differentiation in castration resistant prostate cancer. Cancer Lett. 2018, 433, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Flores-Morales, A.; Bergmann, T.B.; Lavallee, C.; Batth, T.S.; Lin, D.; Lerdrup, M.; Friis, S.; Bartels, A.; Kristensen, G.; Krzyzanowska, A.; et al. Proteogenomic characterization of patient-derived xenografts highlights the role of REST in neuroendocrine differentiation of castration-resistant prostate cancer. Clin. Cancer Res. 2019, 25, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Donmez, N.; Sahinalp, C.; Xie, N.; Wang, Y.; Xue, H.; Mo, F.; Beltran, H.; Gleave, M.; Wang, Y.; et al. SRRM4 drives neuroendocrine transdifferentiation of prostate adenocarcinoma under androgen receptor pathway inhibition. Eur. Urol. 2017, 71, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Q.; Lovnicki, J.; Chen, R.; Fazli, L.; Wang, Y.; Gleave, M.; Huang, J.; Dong, X. SRRM4 gene expression correlates with neuroendocrine prostate cancer. Prostate 2019, 79, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Ramnarine, V.R.; Alshalalfa, M.; Mo, F.; Nabavi, N.; Erho, N.; Takhar, M.; Shukin, R.; Brahmbhatt, S.; Gawronski, A.; Kobelev, M.; et al. The long noncoding RNA landscape of neuroendocrine prostate cancer and its clinical implications. GigaScience 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Oromendia, C.; Danila, D.C.; Montgomery, B.; Hoimes, C.; Szmulewitz, R.Z.; Vaishampayan, U.; Armstrong, A.J.; Stein, M.; Pinski, J.; et al. A phase II trial of the aurora kinase A inhibitor Alisertib for patients with castration-resistant and neuroendocrine prostate cancer: Efficacy and biomarkers. Clin. Cancer Res. 2019, 25, 43–51. [Google Scholar] [CrossRef]
- Copeland, B.T.; Pal, S.K.; Bolton, E.C.; Jones, J.O. The androgen receptor malignancy shift in prostate cancer. Prostate 2018, 78, 521–531. [Google Scholar] [CrossRef]
- Alpajaro, S.I.R.; Harris, J.A.K.; Evans, C.P. Non-metastatic castration resistant prostate cancer: A review of current and emerging medical therapies. Prostate Cancer Prostatic Dis. 2019, 22, 16–23. [Google Scholar] [CrossRef]
- Dellis, A.E.; Papatsoris, A.G. Perspectives on the current and emerging chemical androgen receptor antagonists for the treatment of prostate cancer. Expert Opin. Pharmacother. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Labbe, D.P.; Brown, M. Transcriptional regulation in prostate cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030437. [Google Scholar] [CrossRef]
- Nevedomskaya, E.; Baumgart, S.J.; Haendler, B. Recent advances in prostate cancer treatment and drug discovery. Int. J. Mol. Sci. 2018, 19, 1359. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D.; Schellhammer, P.F.; McLeod, D.G.; Moul, J.W.; Higano, C.S.; Shore, N.; Denis, L.; Iversen, P.; Eisenberger, M.A.; Labrie, F. Androgen receptor targeted treatments of prostate cancer: 35 years of progress with antiandrogens. J. Urol. 2018, 200, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Lee, S.O.; Liang, L.; Luo, J.; Huang, C.K.; Li, L.; Niu, Y.; Chang, C. Targeting the unique methylation pattern of androgen receptor (AR) promoter in prostate stem/progenitor cells with 5-aza-2’-deoxycytidine (5-AZA) leads to suppressed prostate tumorigenesis. J. Biol. Chem. 2012, 287, 39954–39966. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Shao, G.; Zhang, H.T.; Li, C.; Zhang, D.; Cheng, L.; Elzey, B.D.; Pili, R.; Ratliff, T.L.; Huang, J.; et al. Protein arginine methyltransferase 5 functions as an epigenetic activator of the androgen receptor to promote prostate cancer cell growth. Oncogene 2017, 36, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, T.; Waer, C.; Itakura, K. AT-rich interactive domain 5B regulates androgen receptor transcription in human prostate cancer cells. Prostate 2018, 78, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Jernberg, E.; Bergh, A.; Wikstrom, P. Clinical relevance of androgen receptor alterations in prostate cancer. Endocr. Connect. 2017, 6, R146–R161. [Google Scholar] [CrossRef] [Green Version]
- Paschalis, A.; Sharp, A.; Welti, J.C.; Neeb, A.; Raj, G.V.; Luo, J.; Plymate, S.R.; de Bono, J.S. Alternative splicing in prostate cancer. Nat. Rev. Clin. Oncol. 2018, 15, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z.; Santer, F.R. Studies on steroid receptor coactivators in prostate cancer. Methods Mol. Biol. 2018, 1786, 259–262. [Google Scholar] [PubMed]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in nonmetastatic, castration-resistant prostate cancer. N. Engl. J. Med. 2019, 380, 1235–1246. [Google Scholar] [CrossRef]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef]
- Clegg, N.J.; Wongvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. ARN-509: A novel antiandrogen for prostate cancer treatment. Cancer Res. 2012, 72, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Moilanen, A.M.; Riikonen, R.; Oksala, R.; Ravanti, L.; Aho, E.; Wohlfahrt, G.; Nykanen, P.S.; Tormakangas, O.P.; Palvimo, J.J.; Kallio, P.J. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci. Rep. 2015, 5, 12007. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Baumgart, S.J.; Nevedomskaya, E.; Reichert, K.; Steuber, H.; Lejeune, P.; Mumberg, D.; Haendler, B. Darolutamide is a potent androgen receptor antagonist with strong efficacy in prostate cancer models. Int. J. Cancer 2019. [Google Scholar] [CrossRef]
- Peltola, K.J.; Bono, P.; Jones, R.H.; Vjaters, E.; Nykanen, P.; Vuorela, A.; Oksala, R.; Pohjanjousi, P.; Mustonen, M.V.J.; Fizazi, K.; et al. ODM-204, a novel dual inhibitor of CYP17A1 and androgen receptor: Early results from phase I dose escalation in men with castration-resistant prostate cancer. Eur. Urol. Focus 2018. [Google Scholar] [CrossRef] [PubMed]
- Salami, J.; Alabi, S.; Willard, R.R.; Vitale, N.J.; Wang, J.; Dong, H.; Jin, M.; McDonnell, D.P.; Crew, A.P.; Neklesa, T.K.; et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun. Biol. 2018, 1, 100. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Tian, J.; Chou, F.; Lin, C.; Xing, E.Z.; Zuo, L.; Niu, Y.; Yeh, S.; Chang, C. Targeting the androgen receptor (AR) with AR degradation enhancer ASC-J9(R) led to increase docetaxel sensitivity via suppressing the p21 expression. Cancer Lett. 2019, 444, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, C.; Qin, C.; Xiang, W.; Fernandez-Salas, E.; Yang, C.Y.; Wang, M.; Zhao, L.; Xu, T.; Chinnaswamy, K.; et al. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J. Med. Chem. 2019, 62, 941–964. [Google Scholar] [CrossRef]
- Yang, Y.C.; Banuelos, C.A.; Mawji, N.R.; Wang, J.; Kato, M.; Haile, S.; McEwan, I.J.; Plymate, S.; Sadar, M.D. Targeting androgen receptor activation function-1 with EPI to overcome resistance mechanisms in castration-resistant prostate cancer. Clin. Cancer Res. 2016, 22, 4466–4477. [Google Scholar] [CrossRef]
- Bianchini, D.; Omlin, A.; Pezaro, C.; Lorente, D.; Ferraldeschi, R.; Mukherji, D.; Crespo, M.; Figueiredo, I.; Miranda, S.; Riisnaes, R.; et al. First-in-human Phase I study of EZN-4176, a locked nucleic acid antisense oligonucleotide to exon 4 of the androgen receptor mRNA in patients with castration-resistant prostate cancer. Br. J. Cancer 2013, 109, 2579–2586. [Google Scholar] [CrossRef] [Green Version]
- De Las Pozas, A.; Reiner, T.; De Cesare, V.; Trost, M.; Perez-Stable, C. Inhibiting multiple deubiquitinases to reduce androgen receptor expression in prostate cancer cells. Sci. Rep. 2018, 8, 13146. [Google Scholar] [CrossRef]
- Winters, B.; Brown, L.; Coleman, I.; Nguyen, H.; Minas, T.Z.; Kollath, L.; Vasioukhin, V.; Nelson, P.; Corey, E.; Uren, A.; et al. Inhibition of ERG activity in patient-derived prostate cancer xenografts by YK-4-279. Anticancer Res. 2017, 37, 3385–3396. [Google Scholar] [PubMed]
- Mohamed, A.A.; Xavier, C.P.; Sukumar, G.; Tan, S.H.; Ravindranath, L.; Seraj, N.; Kumar, V.; Sreenath, T.; McLeod, D.G.; Petrovics, G.; et al. Identification of a small molecule that selectively inhibits ERG-positive cancer cell growth. Cancer Res. 2018, 78, 3659–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyce, A.; Degenhardt, Y.; Bai, Y.; Le, B.; Korenchuk, S.; Crouthame, M.C.; McHugh, C.F.; Vessella, R.; Creasy, C.L.; Tummino, P.J.; et al. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget 2013, 4, 2419–2429. [Google Scholar] [CrossRef] [PubMed]
- Markowski, M.C.; De Marzo, A.M.; Antonarakis, E.S. BET inhibitors in metastatic prostate cancer: Therapeutic implications and rational drug combinations. Expert Opin. Investig. Drugs 2017, 26, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Lucking, U.; Scholz, A.; Lienau, P.; Siemeister, G.; Kosemund, D.; Bohlmann, R.; Briem, H.; Terebesi, I.; Meyer, K.; Prelle, K.; et al. Identification of atuveciclib (BAY 1143572), the first highly selective, clinical PTEFb/CDK9 inhibitor for the treatment of cancer. ChemMedChem 2017, 12, 1776–1793. [Google Scholar] [CrossRef]
- Zhang, H.; Pandey, S.; Travers, M.; Sun, H.; Morton, G.; Madzo, J.; Chung, W.; Khowsathit, J.; Perez-Leal, O.; Barrero, C.A.; et al. Targeting CDK9 reactivates epigenetically silenced genes in cancer. Cell 2018, 175, 1244.e26–1258.e26. [Google Scholar] [CrossRef]
- Raisner, R.; Kharbanda, S.; Jin, L.; Jeng, E.; Chan, E.; Merchant, M.; Haverty, P.M.; Bainer, R.; Cheung, T.; Arnott, D.; et al. Enhancer activity requires CBP/P300 bromodomain-dependent histone H3K27 acetylation. Cell Rep. 2018, 24, 1722–1729. [Google Scholar] [CrossRef]
- Santer, F.R.; Hoschele, P.P.; Oh, S.J.; Erb, H.H.; Bouchal, J.; Cavarretta, I.T.; Parson, W.; Meyers, D.J.; Cole, P.A.; Culig, Z. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol. Cancer 2011, 10, 1644–1655. [Google Scholar] [CrossRef]
- Ianculescu, I.; Wu, D.Y.; Siegmund, K.D.; Stallcup, M.R. Selective roles for cAMP response element-binding protein binding protein and p300 protein as coregulators for androgen-regulated gene expression in advanced prostate cancer cells. J. Biol. Chem. 2012, 287, 4000–4013. [Google Scholar] [CrossRef]
- Jin, L.; Garcia, J.; Chan, E.; de la Cruz, C.; Segal, E.; Merchant, M.; Kharbanda, S.; Raisner, R.; Haverty, P.M.; Modrusan, Z.; et al. Therapeutic targeting of the CBP/p300 bromodomain blocks the growth of castration-resistant prostate cancer. Cancer Res. 2017, 77, 5564–5575. [Google Scholar] [CrossRef]
- Xiang, Q.; Wang, C.; Zhang, Y.; Xue, X.; Song, M.; Zhang, C.; Li, C.; Wu, C.; Li, K.; Hui, X.; et al. Discovery and optimization of 1-(1H-indol-1-yl)ethanone derivatives as CBP/EP300 bromodomain inhibitors for the treatment of castration-resistant prostate cancer. Eur. J. Med. Chem. 2018, 147, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Pegg, N.; Worthington, J.; Young, B.; Prosser, A.; Gaughan, L.; Spencer, G.; Somervaille, T.; Burns, J.; Knowles, M.; Brooks, N. Novel small molecule inhibitors of p300/CBP down-regulate androgen receptor (AR) and c-Myc for the treatment of prostate cancer and beyond. Cancer Res. 2018, 78, 3991. [Google Scholar]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, Y.; Lu, X.; Song, B.; Fong, K.W.; Cao, Q.; Licht, J.D.; Zhao, J.C.; Yu, J. Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep. 2018, 25, 2808.e4–2820.e4. [Google Scholar] [CrossRef]
- Wu, C.; Jin, X.; Yang, J.; Yang, Y.; He, Y.; Ding, L.; Pan, Y.; Chen, S.; Jiang, J.; Huang, H. Inhibition of EZH2 by chemo- and radiotherapy agents and small molecule inhibitors induces cell death in castration-resistant prostate cancer. Oncotarget 2016, 7, 3440–3452. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Wang, W.; Li, B.; Cheng, B.; Lin, K.; Bai, J.; Li, H.; Yang, G. Targeting Ezh2 could overcome docetaxel resistance in prostate cancer cells. BMC Cancer 2019, 19, 27. [Google Scholar] [CrossRef]
- Fioravanti, R.; Stazi, G.; Zwergel, C.; Valente, S.; Mai, A. Six Years (2012-2018) of researches on catalytic EZH2 inhibitors: The boom of the 2-pyridone compounds. Chem. Rec. 2018, 18, 1818–1832. [Google Scholar] [CrossRef]
- Taplin, M.E.; Hussain, A.; Shore, N.D.; Bradley, B.; Trojer, P.; Lebedinsky, C.; Senderowicz, A.M.; Antonarakis, E.S. A phase 1b/2 study of CPI-1205, a small molecule inhibitor of EZH2, combined with enzalutamide (E) or abiraterone/prednisone (A/P) in patients with metastatic castration resistant prostate cancer (mCRPC). J. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Jain, P.; Di Croce, L. Mutations and deletions of PRC2 in prostate cancer. Bioessays 2016, 38, 446–454. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, J.; Yu, Z.; Zhang, H.; Wang, Y.; Lingel, A.; Qi, W.; Gu, J.; Zhao, K.; Shultz, M.D.; et al. Discovery of first-in-class, potent, and orally bioavailable embryonic ectoderm development (EED) inhibitor with robust anticancer efficacy. J. Med. Chem. 2017, 60, 2215–2226. [Google Scholar] [CrossRef]
- Qi, W.; Zhao, K.; Gu, J.; Huang, Y.; Wang, Y.; Zhang, H.; Zhang, M.; Zhang, J.; Yu, Z.; Li, L.; et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat. Chem. Biol. 2017, 13, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Sodir, N.M.; Evan, G.I. Finding cancer’s weakest link. Oncotarget 2011, 2, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.G.; Gonda, T.J. MYB function in normal and cancer cells. Nat. Rev. Cancer 2008, 8, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, S.J.; Haendler, B. Exploiting epigenetic alterations in prostate cancer. Int. J. Mol. Sci. 2017, 18, 1017. [Google Scholar] [CrossRef] [PubMed]
- Scheer, S.; Ackloo, S.; Medina, T.S.; Schapira, M.; Li, F.; Ward, J.A.; Lewis, A.M.; Northrop, J.P.; Richardson, P.L.; Kaniskan, H.U.; et al. A chemical biology toolbox to study protein methyltransferases and epigenetic signaling. Nat. Commun. 2019, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Janiaud, P.; Serghiou, S.; Ioannidis, J.P.A. New clinical trial designs in the era of precision medicine: An overview of definitions, strengths, weaknesses, and current use in oncology. Cancer Treat. Rev. 2018, 73, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Chen, N.; Lee, J.J. Bayesian adaptive randomization and trial monitoring with predictive probability for time-to-event endpoint. Stat. Biosci. 2018, 10, 420–438. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Target | Target Function | Compound | Mode of Action | Status | Identifier |
|---|---|---|---|---|---|
| AR | Transcription factor | Enzalutamide Apalutamide Darolutamide | Competitive antagonists | FDA-approved FDA-approved Phase 3 active Phase 3 active | NCT00974311 NCT01212991 NCT02003924 NCT01946204 NCT02200614 NCT02799602 |
| ARV-110 ARD-69 ASC-J9 | Degraders | Phase 1 recruiting Preclinical Preclinical | NCT03888612 | ||
| EPI-506 | N-terminal domain binder | Phase 1/2 terminated | NCT02606123 | ||
| EZN-4176 AZD-5312 | Antisense oligonucleotides | Phase 1a/1b suspended Phase 1 completed | NCT01337518 NCT02144051 | ||
| AR/CYP 17A | Transcription factor/cytochrome | ODM-204 | Dual inhibitor | Phase 2 ongoing | NCT02344017 |
| ERG | Transcription factor | YK-4-279 | Helicase interaction inhibitor | Preclinical | |
| NSC139021 | Ribosomal biogenesis regulator | Preclinical | |||
| BET/BRD4 | Acetylated lysine reader | GSK525762 ABBV-075 ABBV-744 GS-5829 ZEN003694 ZEN003694 | Bromodomain inhibitors | Phase 1B ongoing Phase 1 active Phase 1 ongoing Phase 1 ongoing Phase 1 completed Phase 1 active | NCT03150056 NCT02391480 NCT03360006 NCT02607228 NCT02705469 NCT02711956 |
| CDK7 | Part of transcription factor II complex | THZ1 | Kinase inhibitor | Preclinical | |
| CDK9 | Part of P-TEFb complex | Atuveciclib MC180295 | Kinase inhibitors | Phase 1 completed Preclinical | NCT02345382 |
| CBP/p300 | Transcriptional coactivator | CCS1477 GNE-049 32h | Bromodomain inhibitors | Phase 1/2 ongoing Preclinical Preclinical | NCT03568656 |
| A-485 | Acetyl-transferase inhibitor | Preclinical | |||
| EZH2 | H3 lysine 27 methyl-transferase | CPI-1205 GSK126 | Methyl-transferase inhibitors | Phase 1b/2 ongoing Preclinical | NCT03480646 |
| EED | EZH2 interactor | MAK683 | Inhibits H3K27me3 binding | Phase 1/2 ongoing | NCT02900651 |
| Aurora A | Serine/threonine kinase | Alisertib | Kinase inhibitor | Phase 2 completed | NCT01799278 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baumgart, S.J.; Nevedomskaya, E.; Haendler, B. Dysregulated Transcriptional Control in Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 2883. https://doi.org/10.3390/ijms20122883
Baumgart SJ, Nevedomskaya E, Haendler B. Dysregulated Transcriptional Control in Prostate Cancer. International Journal of Molecular Sciences. 2019; 20(12):2883. https://doi.org/10.3390/ijms20122883
Chicago/Turabian StyleBaumgart, Simon J., Ekaterina Nevedomskaya, and Bernard Haendler. 2019. "Dysregulated Transcriptional Control in Prostate Cancer" International Journal of Molecular Sciences 20, no. 12: 2883. https://doi.org/10.3390/ijms20122883
APA StyleBaumgart, S. J., Nevedomskaya, E., & Haendler, B. (2019). Dysregulated Transcriptional Control in Prostate Cancer. International Journal of Molecular Sciences, 20(12), 2883. https://doi.org/10.3390/ijms20122883